Introduction
Sepsis is a clinical syndrome that occurs following
infection or injury (1). If not
timely and properly controlled, sepsis can develop into systemic
inflammatory response syndrome and ultimately result in multiple
organ dysfunction syndrome (MODS) (2). Cardiac dysfunction is a severe
sepsis-related complication characterized by left ventricular
dilatation, decreased ejection fraction and recovery in 7-10 days
(3). The molecular mechanisms of
cardiac tissue damage during sepsis remain elusive. In previous
studies, we examined the role of inflammation, oxidative stress,
apoptosis and autophagy in sepsis-related myocardial injury
(4,5). Recently, pyroptosis, a specific type
of programmed cell death, was reported as a common cause of
sepsis-induced tissue damage (6-9).
During the development of sepsis, pyroptosis can destroy the
integrity of cell membranes, resulting in inflammatory cytokine
secretion and augmented inflammatory responses (10,11).
Thus, selective suppression of genes and proteins involved in
pyroptosis may be a potential therapeutic strategy for sepsis or
sepsis-induced cardiomyopathy.
The NOD-like receptor protein 3 (NLRP3) inflammasome
is a multiprotein heteromeric complex that detected a variety of
danger signals that originate not only from microorganisms but also
from metabolic disorders. The assembly of the NLRP3 inflammasome
contributes to the self-shearing of caspase-1, leading to the
generation of activated caspase-1 fragments. Activated caspase-1
induces the maturation of the proinflammatory cytokines, pro-IL-1β
and pro-IL-18. In addition, activated caspase-1 shears gasdermin D
(GSDMD); the cleaved N-terminal domain of GSDMD translocates to the
plasma membrane and forms pores, thereby facilitating the
extracellular secretion of inflammatory cytokines into the
circulation system and triggering the classical pathway of
pyroptosis (12). Increasing
evidence suggest that the NLRP3/caspase-1/GSDMD signaling pathway
is involved in several pathophysiological mechanisms, such as
innate immunity, myeloid proliferation, tumorigenesis and
Alzheimer's disease (13-19).
It has also been reported that NLRP3 inflammasome activation
requires two signals, a priming signal and an activating signal.
First, the priming signal provided by pathogen- and
danger-associated molecular patterns activates the NF-κB signaling
pathway and subsequently upregulates the expression levels of NLRP3
and pro-IL-1β (20). The activating
signal is then provided by a variety of molecular or cellular
events, including reactive oxygen species (ROS) production, ionic
flux and lysosomal damage (21).
Compared with the latter two models, the ROS model was considered
one of the most crucial signaling pathways for the activation of
the NLRP3 inflammasome (22).
Mechanistically, increased intracellular ROS levels can lead to the
detachment of TXNIP from the TXNIP-Trx protein complex, then free
TXNIP can bind to NLRP3, resulting in NLRP3 activation and
promoting the assembly of the NLRP3 inflammasome (23).
DDX3X, an ATPase/RNA helicase of the DEAD-box
family, participates in several RNA metabolic processes (24). This protein is also involved in cell
cycle progression, apoptosis, antiviral innate immunity and cancer
development (25-28).
A recent study demonstrated that DDX3X is crucial for NLRP3
inflammasome assembly due to its direct binding interaction with
NLRP3, and Ddx3x knockdown in peritoneal macrophages
suppresses NLRP3 inflammasome activation and reduces pyroptosis
(29). The binding site for DDX3X
is in the NACHT region of NLRP3, which exerts ATPase activity
required for NLRP3 oligomerization following activation. This
suggests that DDX3X plays an indispensable role in facilitating the
oligomerization of NLRP3 (30-32).
To the best of our knowledge, the role of DDX3X in
lipopolysaccharide (LPS)-induced cardiomyocyte stress response has
not yet been investigated. Thus, the present study aimed to
determine whether DDX3X participates in LPS-induced cardiomyocyte
injury by regulating NLRP3 inflammasome formation and subsequent
pyroptosis.
Materials and methods
Cell culture and treatment
The H9c2 rat myocardial cell line was purchased from
the National Collection of Authenticated Cell Culture (https://cellbank.org.cn). H9c2 cells were maintained
in DMEM (cat. no. 12800017) supplemented with 1.5 g/l
NaHCO3, 10% fetal bovine serum (all purchased from
Gibco; Thermo Fisher Scientific, Inc.) and 1% antibiotics (100 U/ml
penicillin and 100 mg/ml streptomycin; Beijing Solarbio Science
& Technology Co., Ltd.), at 37˚C with 5% CO2.
LPS was purchased from MedChemExpress (cat. no.
HY-D1056) and reconstituted in DMSO. When the cells reached 70%
confluence, LPS (1 µg/ml) was added to the culture medium to mimic
sepsis-induced cardiomyocyte pyroptosis in vitro; the
control group was treated with DMSO.
Cell transfection
Small interfering (si)RNA against rat Ddx3x
and scramble siRNA were purchased from Shanghai GenePharma Co.,
Ltd. Scramble siRNA was used as the negative control for siRNA
silencing. When H9c2 cells reached 50-60% confluence, siRNA was
transfected into H9c2 cells using Lipofectamine® 3000
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions. Briefly, 50 nM DDX3X siRNA or scramble
siRNA in combination with 5 µl of Lipofectamine® 3000
was added to each well. Following incubation with serum-free DMEM
at 37˚C for 6 h, the medium was replaced with DMEM containing
serum. After additional incubation at 37˚C for 18 h, H9c2 cells
were treated with LPS (1 µg/ml). The sense and antisense siRNA
sequences are listed in Table
I.
 | Table ISense and antisense siRNA
sequences. |
Table I
Sense and antisense siRNA
sequences.
| Gene | Sequence
(5'-3') |
|---|
| DDX3X siRNA sense
strand |
GGAGGAUUUCUUAUACCAUTT |
| DDX3X siRNA
antisense strand |
AUGGUAUAAGAAAUCCUCCTT |
| Control siRNA sense
strand |
UUCUCCGAACGUGUCACGUTT |
| Control siRNA
antisense strand |
ACGUGACACGUUCGGAGAATT |
Western blotting
Total protein extraction was performed following
treatment with LPS for 24 h. RIPA lysis buffer supplemented with 1
mM phenylmethylsulfonyl fluoride (Beyotime Institute of
Biotechnology) was added dropwise to each well to lyse the cells.
The supernatant of the cell lysates was collected following
centrifugation at 11,588 x g for 15 min at 4˚C. For secretory
protein extraction, cell medium supernatant was collected and
centrifuged at 600 x g for 5 min at 4˚C and 11,588 x g for 15 min
at 4˚C. The bicinchoninic acid assay kit (Beyotime Institute of
Biotechnology) was used to quantify protein samples, according to
the manufacturer's instructions. Equal amounts of protein samples
(30 µg) were separated by 10, 12.5 and 15% SDS-PAGE (EpiZyme),
transferred onto PVDF membranes and blocked with 5% non-fat milk at
room temperature for 1 h. The membranes were incubated with primary
antibodies against rabbit anti-GAPDH (cat. no. 5174; 1:8,000; Cell
Signaling Technology, Inc.), rabbit anti-DDX3X (cat. no.
11115-1-AP; 1:1,000; ProteinTech Group, Inc.), rabbit
anti-Caspase1/P20/P10 (cat. no. 22915-1-AP; 1:1,500; ProteinTech
Group, Inc.), rabbit anti-NLRP3 (cat. no. 19771-1-AP; 1:1,000;
ProteinTech Group, Inc.), rabbit anti-IL-1β (cat. no. AF5103;
1:1,000; Affinity Biosciences) and rabbit anti-Cleaved-IL-1β (cat.
no. AF4006; 1:1,000; Affinity Biosciences) overnight at 4˚C. GAPDH
was used as the internal control. The PVDF membranes were washed
three times with Tris-buffered saline with 1% Tween-20 (Beyotime
Institute of Biotechnology) and subsequently incubated with
Anti-rabbit IgG, HRP-linked Antibody (cat. no. 7074; 1:10,000; Cell
Signaling Technology, Inc.) at room temperature for 1 h. Coomassie
blue staining was performed by incubating the gels with Coomassie
blue staining solution (Beyotime Institute of Biotechnology) at
room temperature for 30 min and washing them in Coomassie blue
eluent (methanol:glacial acetic acid : distilled water = 3:1:6)
until clear bands appeared. The immunoblots were detected using
chemiluminescence reagents (MilliporeSigma) and the blots were
scanned using a chemiluminescent analyzer (ProteinSimple). Relative
immunoblot intensities were analyzed using ImageJ v1.8.0.112
software (National Institutes of Health).
Reverse transcription-quantitative
(RT-q)PCR
Following treatment with LPS for 24 h,
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) was used to extract total RNA from H9c2 cells. Equal amounts
of total RNA (1-2 µg) were reverse transcribed into cDNA using the
HiScript III RT SuperMix for qPCR (+gDNA wiper; Vazyme Biotech Co.,
Ltd.). The following temperature protocol was used for reverse
transcription: 42˚C for 2 min, followed by 37˚C for 15 min and 85˚C
for 5 sec. Primer sequences were purchased from BGI (https://www.bgi.com/). qPCR was performed in a 20-µl
reaction volume consisting of ChamQ Universal SYBR qPCR Master Mix
(Vazyme Biotech Co., Ltd.). RT-qPCR was performed on a CFX96
Real-Time PCR Detection System (Bio-Rad Laboratories, Inc.). The
following thermocycling conditions were used for qPCR: 95˚C for 30
sec, followed by 40 cycles of 95˚C for 10 sec, 60˚C for 30 sec and
65˚C for 5 sec. Relative expression levels were calculated using
the 2-ΔΔCq method (33)
and normalized to the internal reference gene GAPDH. The primer
sequences used for qPCR are listed in Table II.
| Table IIPrimer sequences used for
quantitative PCR. |
Table II
Primer sequences used for
quantitative PCR.
| Gene | Sequence | Product size (bp)
(5'-3') | TM (˚C) |
|---|
| DDX3X-Rat | F:
AAACCTTGGTCTTGCCACCTC | 21 | 57.57 |
| | R:
CCACGGCTGCTACCCTTATAG | 21 | 59.52 |
| NLRP3-Rat | F:
GCTAAGAAGGACCAGCCAGA | 20 | 57.45 |
| | R:
TCCCAGCAAACCTATCCACT | 19 | 55.40 |
| IL-1β-Rat | F:
CACCTCTCAAGCAGAGCACAG | 21 | 59.76 |
| | R:
GGGTTCCATGGTGAAGTCAAC | 21 | 57.80 |
| GAPDH-Rat | F:
GTATTGGGCGCCTGGTCACC | 20 | 61.55 |
| | R:
CGCTCCTGGAAGATGGTGATGG | 22 | 61.85 |
Measurement of caspase-1 activity
Caspase-1 activity was measured using the caspase-1
activity assay kit (Beyotime Institute of Biotechnology), according
to the manufacturer's instructions. Absorbance was measured using
an enzyme-labeled instrument (BioTek Instruments, Inc.) at a
wavelength of 405 nm.
Determination of ROS levels
Following treatment with LPS, the culture medium was
removed, and H9c2 cells were subsequently incubated with 1 ml of
serum-free DMEM supplemented with 1 µl of fluorescent
dichloro-dihydro-fluorescein diacetate (DCFH-DA; Beyotime Institute
of Biotechnology) at 37˚C for 30 min in the dark. Cells were washed
twice with PBS and fluorescence was observed under an Olympus
LCX100 imaging system (Olympus Corporation). The average
fluorescence intensity was measured using ImageJ v1.8.0.112
software (National Institutes of Health).
Cell viability assay
Following treatment with LPS, Cell Counting Kit-8
(CCK-8) reagent (APExBIO Technology LLC) was added to a 96-well
plate and H9c2 cells were incubated at 37˚C for 4 h in the dark.
Absorbance was measured using an enzyme-labeled instrument (BioTek
Instruments, Inc.), at a wavelength of 450 nm. The average optical
density (OD) was used to calculate cell viability using the
following equation: Cell viability = (experimental group OD - blank
control group OD)/(normal control group OD - blank control group
OD) x100%.
Lactate dehydrogenase (LDH)
cytotoxicity assay
Following treatment with LPS, the cell culture
medium was collected and centrifuged at 11,588 x g for 15 min at
4˚C, and the supernatant was collected. LDH activity was measured
to evaluate the damage status of H9c2 cells using Lactate
dehydrogenase assay kit (Nanjing Jiancheng Bioengineering
Institute), according to the manufacturer's instructions.
Absorbance was measured at a wavelength of 440 nm using an
enzyme-labeled instrument (BioTek Instruments, Inc.). LDH activity
was calculated according to the manufacturer's instructions.
Propidium iodide (PI) staining
Following treatment with LPS, the culture medium was
removed and cells were washed twice with PBS. PI (Beyotime
Institute of Biotechnology) was subsequently added to each culture
dish, according to the manufacturer's instructions. H9c2 cells were
stained with PI at 37˚C for 30 min. Fluorescent images of the cells
were captured using an Olympus LCX100 imaging system. The average
fluorescence intensity was measured using ImageJ v1.8.0.112
software (National Institutes of Health).
Statistical analysis
Statistical analysis was performed using GraphPad
Pro Prism 8.0 software (GraphPad Software, Inc.). All experiments
were performed in triplicate and data are presented as the mean ±
SEM. Unpaired Student's t-test was used to compare differences
between two groups, while one-way ANOVA followed by Tukey's post
hoc test were used to compare differences between multiple groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
DDX3X expression in LPS-treated H9c2
cardiomyocytes
RT-qPCR and western blot analyses were performed to
detect DDX3X expression in H9c2 cells treated with different
concentrations of LPS (0, 100, 500 and 1,000 ng/ml). The results
demonstrated that the mRNA and protein expression levels of DDX3X
increased following treatment with LPS for 24 h, with DDX3X mRNA
levels significantly increased at LPS 500 (P<0.01) and 1,000
ng/ml (P<0.001; Fig. 1A-C).
Taken together, upregulated DDX3X expression in LPS-treated
cardiomyocytes suggests that DDX3X participates in the development
of LPS-induced cardiomyocyte injury.
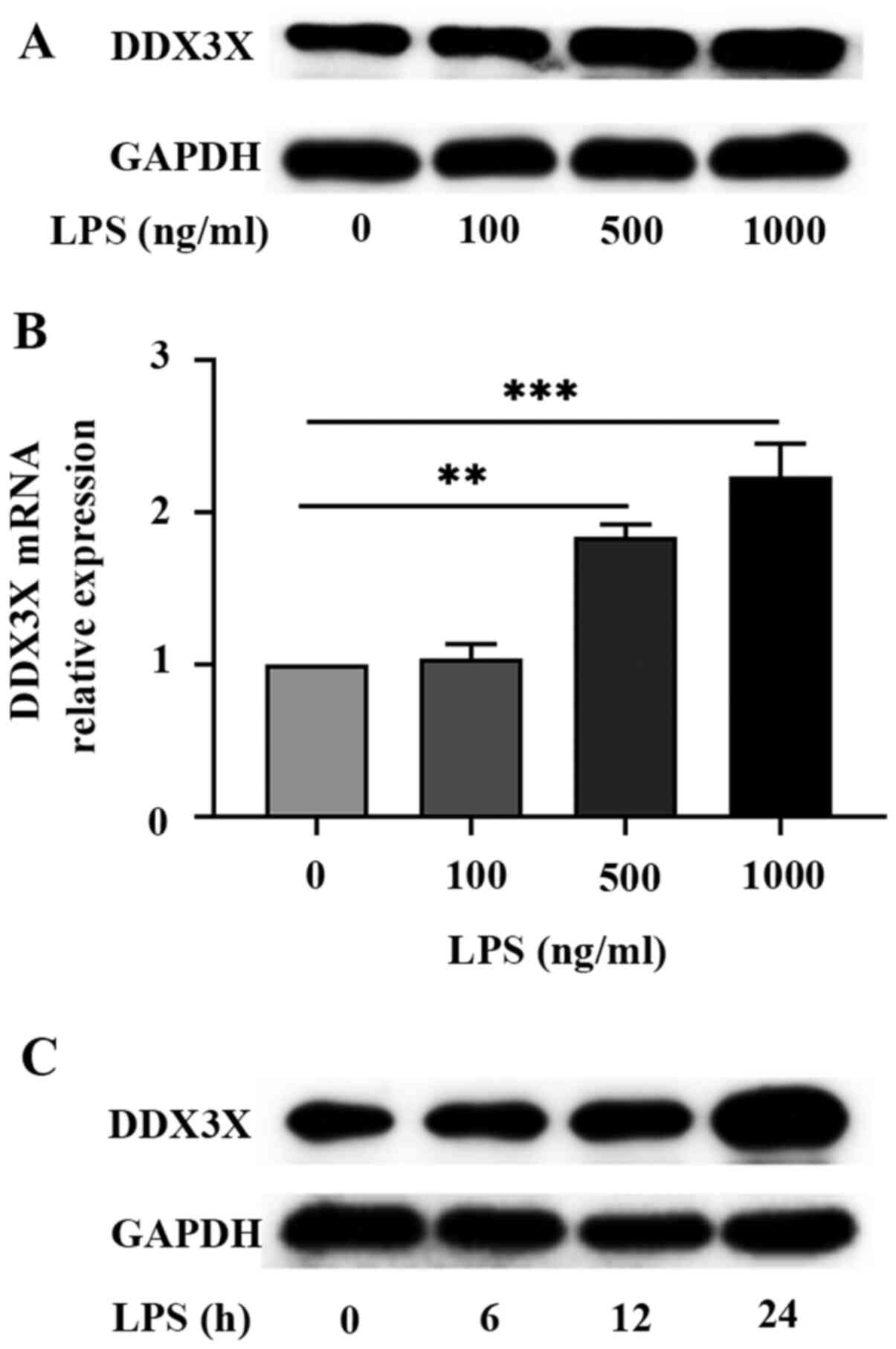 | Figure 1DDX3X expression in LPS-treated
cardiomyocytes. (A) Western blot analysis was performed to detect
DDX3X protein expression in H9C2 cardiomyocytes treated with
different concentrations of LPS (0, 100, 500 and 1,000 ng/ml) for
24 h. Representative blots (n=3). (B) Reverse
transcription-quantitative PCR analysis was performed to detect
DDX3X mRNA expression in H9C2 cardiomyocytes treated with different
concentrations of LPS (0, 100, 500 and 1,000 ng/ml) for 24 h. Data
are presented as the mean ± SEM (n=3). (C) Western blot analysis
was performed to detect DDX3X protein expression in H9C2
cardiomyocytes stimulated with LPS (1,000 ng/ml) for 0, 6, 12 and
24 h. Representative blots (n=3). **P<0.01 and
***P<0.001. LPS, lipopolysaccharide. |
LPS induces pyroptosis by increasing
intracellular ROS levels and activating the NLRP3 inflammasome
To determine whether LPS provides a priming signal
to activate the NLRP3 inflammasome in H9c2 cardiomyocytes, the
present study detected the expression levels of NLRP3 and
pro-IL-1β. The results demonstrated that the protein and mRNA
expression levels of NLRP3 and pro-IL-1β increased following
treatment with LPS for 24 h, with NLRP3 mRNA levels significantly
increased at LPS 100, 500 and 1,000 ng/ml (all P<0.001), and
pro-IL-1β mRNA levels significantly increased at LPS 500 ng/ml
(P<0.05) and 1,000 ng/ml (P<0.001; Fig. 2A-D). The present results suggested
that LPS acted as a priming signal to promote the expression levels
of NLRP3 and pro-IL-1β during NLRP3 inflammasome activation in H9c2
cardiomyocytes. It has been reported that ROS can supply activating
signals for the activation of the NLRP3 inflammasome (22). Thus, the present study measured ROS
levels in LPS-treated cardiomyocytes. As expected, LPS treatment
induced the production of ROS, as ROS levels were positively
correlated with increasing LPS concentrations (Fig. 2E). The protein levels of
pro-caspase-1 and caspase-1 p20, as well as caspase-1 activity,
were assessed using a caspase-1 activity assay kit to determine
whether LPS treatment can activate a functional NLRP3 inflammasome.
As presented in Fig. 2F, the
protein expression levels of intracellular pro-caspase-1 and
caspase-1 p20 increased in H9c2 cells following treatment with LPS.
Cleaved IL-1β was also detected in the supernatant of the cell
culture medium. Consistently, the level of cleaved IL-1β notably
increased at LPS 1,000 ng/ml (Fig.
2F). As presented in Fig. 2G,
caspase-1 activity significantly increased at LPS 100 (P<0.05),
500 (P<0.01) and 1,000 ng/ml (P<0.001). Collectively, the
present results confirm that LPS stimulation activated the NLRP3
inflammasome in H9c2 cells.
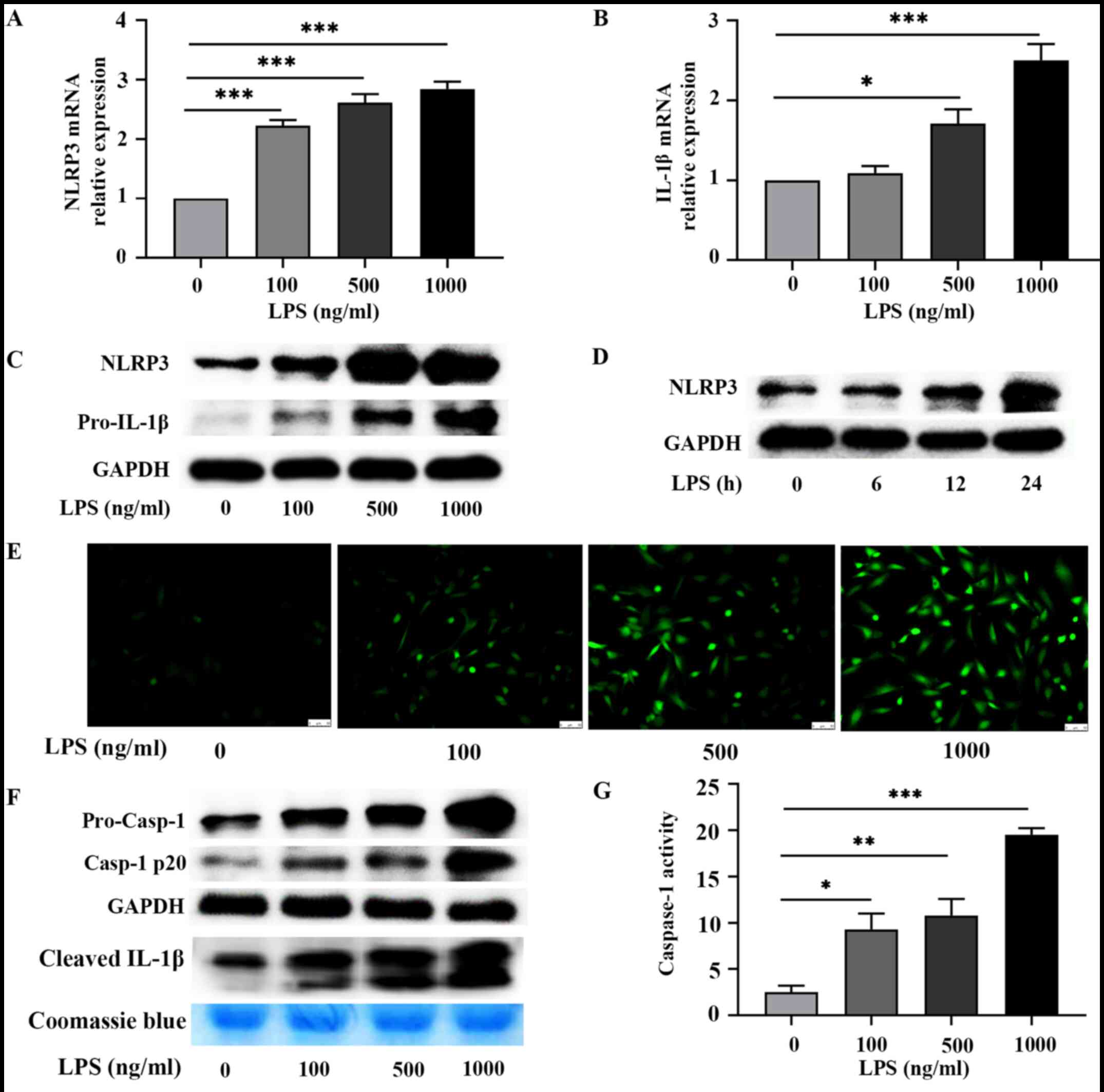 | Figure 2LPS induces pyroptosis by increasing
intracellular reactive oxygen species levels and activating NLRP3
inflammasome. Reverse transcription-quantitative PCR analysis was
performed to detect the mRNA expression levels of (A) NLRP3 and (B)
IL-1β in H9C2 cardiomyocytes treated with different concentrations
of LPS (0, 100, 500 and 1,000 ng/ml) for 24 h. Data are presented
as the mean ± SEM (n=4). (C) Western blot analysis was performed to
detect the protein expression levels of NLRP3 and pro-IL-1β in H9C2
cardiomyocytes treated with different concentrations of LPS (0,
100, 500 and 1,000 ng/ml) for 24 h. Representative blots (n=3). (D)
Western blot analysis was performed to detect NLRP3 protein
expression in H9C2 cardiomyocytes stimulated with LPS (1,000 ng/ml)
for 0, 6, 12 and 24 h. Representative blots (n=3). (E) The DCFH-DA
assay was performed to detect ROS production in H9C2 cardiomyocytes
treated with LPS (1,000 ng/ml) for 6 h. Bar, 50 µm (n=3). (F)
Western blot analysis was performed to detect intracellular protein
levels of pro-caspase-1 and caspase-1 p20, and the level of the
cleaved IL-1β in the supernatant of culture medium in H9C2
cardiomyocytes treated with different concentration of LPS (0, 100,
500 and 1,000 ng/ml) for 24 h. Representative blots (n=3). (G)
Caspase-1 activity was detected in H9C2 cardiomyocytes treated with
different concentrations of LPS (0, 100, 500 and 1,000 ng/ml) for
24 h. Data are presented as the mean ± SEM (n=3).
*P<0.05, **P<0.01 and
***P<0.001. LPS, lipopolysaccharide; NLRP3, NOD-like
receptor protein3. |
Ddx3x knockdown blocks NLRP3
inflammasome activation in LPS-stimulated H9c2 cardiomyocytes
To understand the regulatory effect of DDX3X in
LPS-induced cardiomyocyte pyroptosis and cell injury, H9c2
cardiomyocytes were transfected with Ddx3x siRNA. Western
blot and RT-qPCR analyses confirmed the efficiency of Ddx3x
knockdown (Fig. 3A and B). The present study also compared the
levels of intracellular DDX3X, NLRP3 and IL-1β, as well as the
levels of caspase-1 cleavage and cleaved IL-1β in the supernatant
of the culture medium, in H9c2 cells treated with or without LPS or
in the absence or presence of DDX3X. As presented in Fig. 3C and D, in the absence of LPS stimulation, no
significant differences in the expression levels of NLRP3 and
pro-IL-1β, the level of caspase-1 cleavage and the accumulation of
cleaved IL-1β were observed between the Ddx3x-silenced and
control groups. These results suggest that Ddx3x knockdown
has no influence on the signaling pathway regulating NLRP3
inflammasome activation in the absence of LPS treatment. When H9c2
cells were stimulated with LPS 1,000 ng/ml, the levels of caspase-1
(P<0.01) cleavage and cleaved IL-1β (P<0.001) significantly
decreased in DDX3X-deficient cells, while no significant difference
in NLRP3 expression was observed between LPS-stimulated cells with
normal or altered DDX3X expression (Fig. 3C). Caspase-1 activity also decreased
(P<0.01) following Ddx3x knockdown in H9c2 cells treated
with LPS 1,000 ng/ml (Fig. 3E).
Taken together, these results suggest that Ddx3x knockdown
blocks NLRP3 inflammasome activation but has no effect on NLRP3
expression in LPS-treated H9c2 cardiomyocytes.
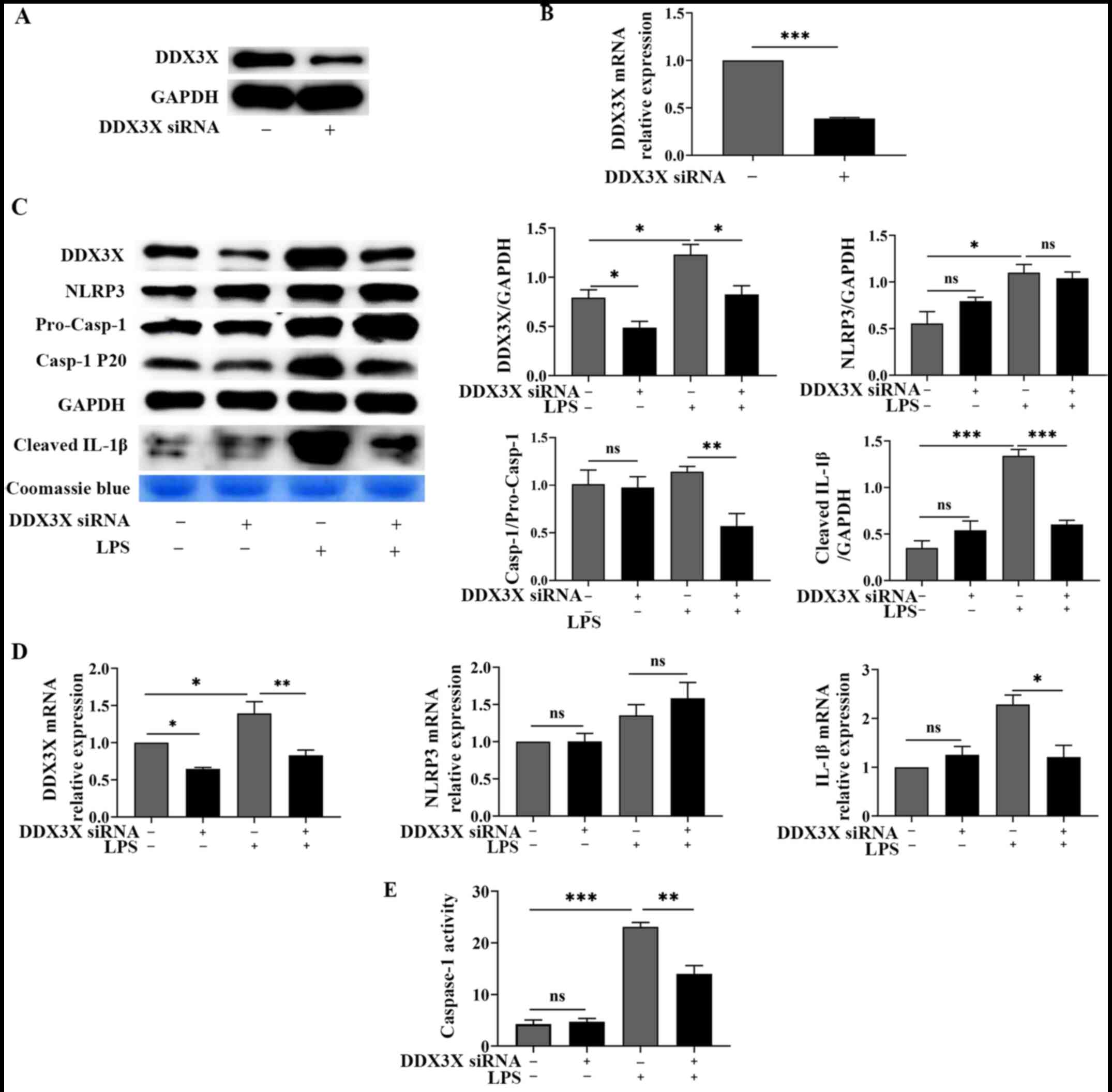 | Figure 3Ddx3x knockdown blocks NLRP3
inflammasome activation in LPS-stimulated H9C2 cardiomyocytes. (A)
Western blotting confirmed the efficiency of Ddx3x
knockdown. Representative blots (n=3). (B) RT-qPCR analysis
confirmed the efficiency of Ddx3x knockdown. Data are
presented as the mean ± SEM (n=3). (C) Western blot analysis was
performed to detect the protein expression levels of DDX3X, NLRP3,
caspase-1 cleavage and cleaved IL-1β in the supernatant of culture
medium in H9C2 cardiomyocytes treated with or without LPS (1,000
ng/ml) for 24 h following siRNA-mediated knockdown of Ddx3x.
Data are presented as the mean ± SEM (n=3). (D) RT-qPCR analysis
was performed to detect the mRNA expression levels of DDX3X, NLRP3
and IL-1β in H9C2 cardiomyocytes treated with or without LPS (1,000
ng/ml) for 24 h following siRNA-mediated knockdown of Ddx3x.
Data are presented as the mean ± SEM (n=3). (E) Caspase-1 activity
was detected in H9C2 cardiomyocytes treated with or without LPS
(1,000 ng/ml) for 24 h following siRNA-mediated knockdown of
Ddx3x. Data are presented as the mean ± SEM (n=3).
*P<0.05, **P<0.01 and
***P<0.001. NLRP3, NOD-like receptor protein 3; LPS,
lipopolysaccharide; RT-qPCR, reverse transcription-quantitative
PCR; si, small interfering; +, presence; -, absence; ns, not
significant. |
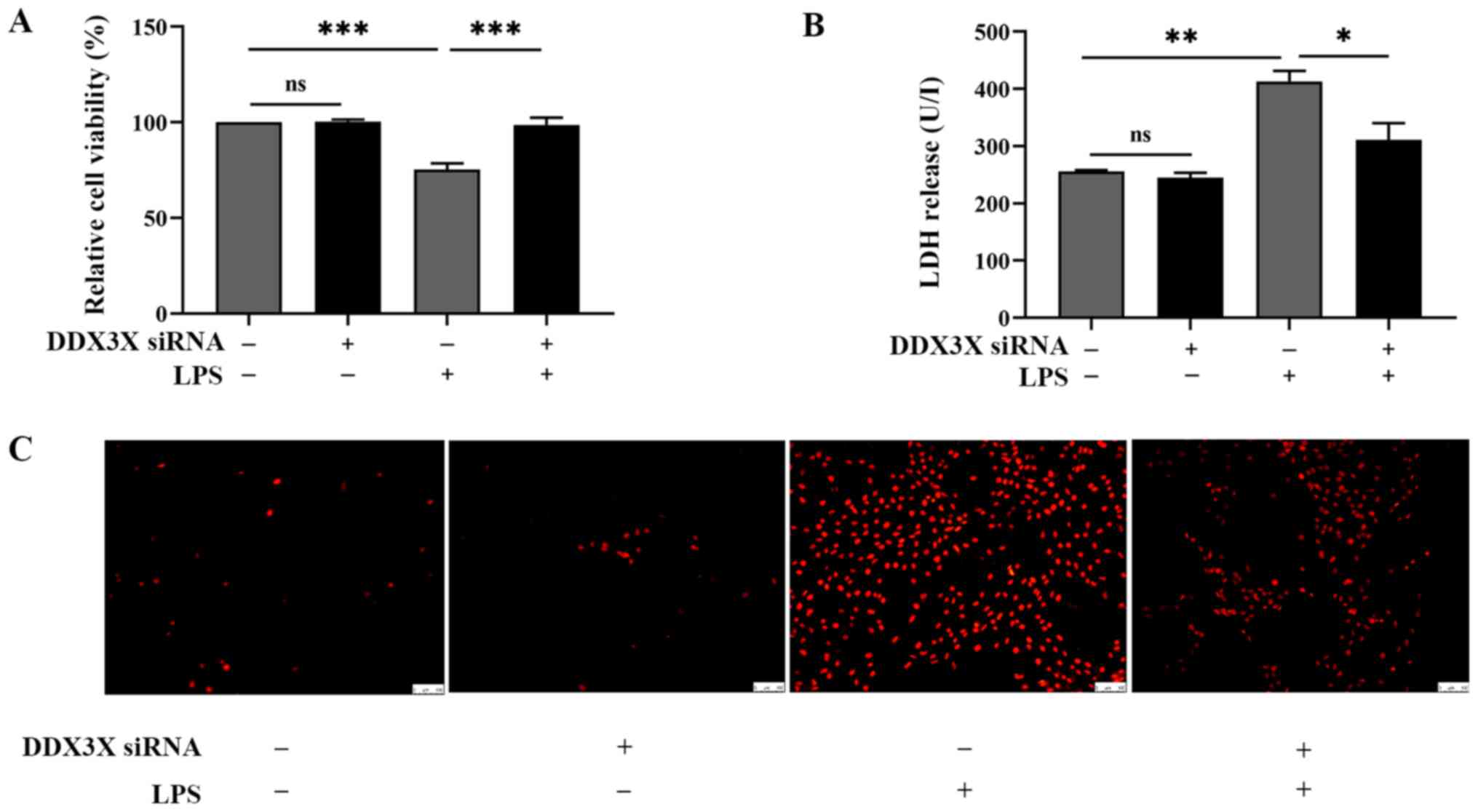
Ddx3x knockdown attenuates pyroptotic
cell death and cell injury in LPS-treated H9c2 cardiomyocytes
Cell cytotoxicity was assessed by detecting the
levels of released LDH, and cell viability was detected via the
CCK-8 assay. LDH release is a sensitive biomarker of cardiac injury
(34); thus, the present study
detected the levels of LDH in the supernatant of the culture
medium. As presented in Fig. 4A,
DDX3X deficiency reversed the increase in LDH level in cells
treated with LPS. In addition, cell viability increased following
Ddx3x knockdown in H9c2 cells treated with LPS (Fig. 4B). However, in H9c2 cells not
treated with LPS, knockdown of Ddx3x had no significant
effect on cell viability or LDH release. PI staining was performed
to detect pyroptotic cell death in H9c2 cells. As expected,
Ddx3x knockdown attenuated pyroptotic cell death in
LPS-treated cardiomyocytes (Fig.
4C). Collectively, these results suggest that Ddx3x
knockdown improves the viability of H9c2 cardiomyocytes treated
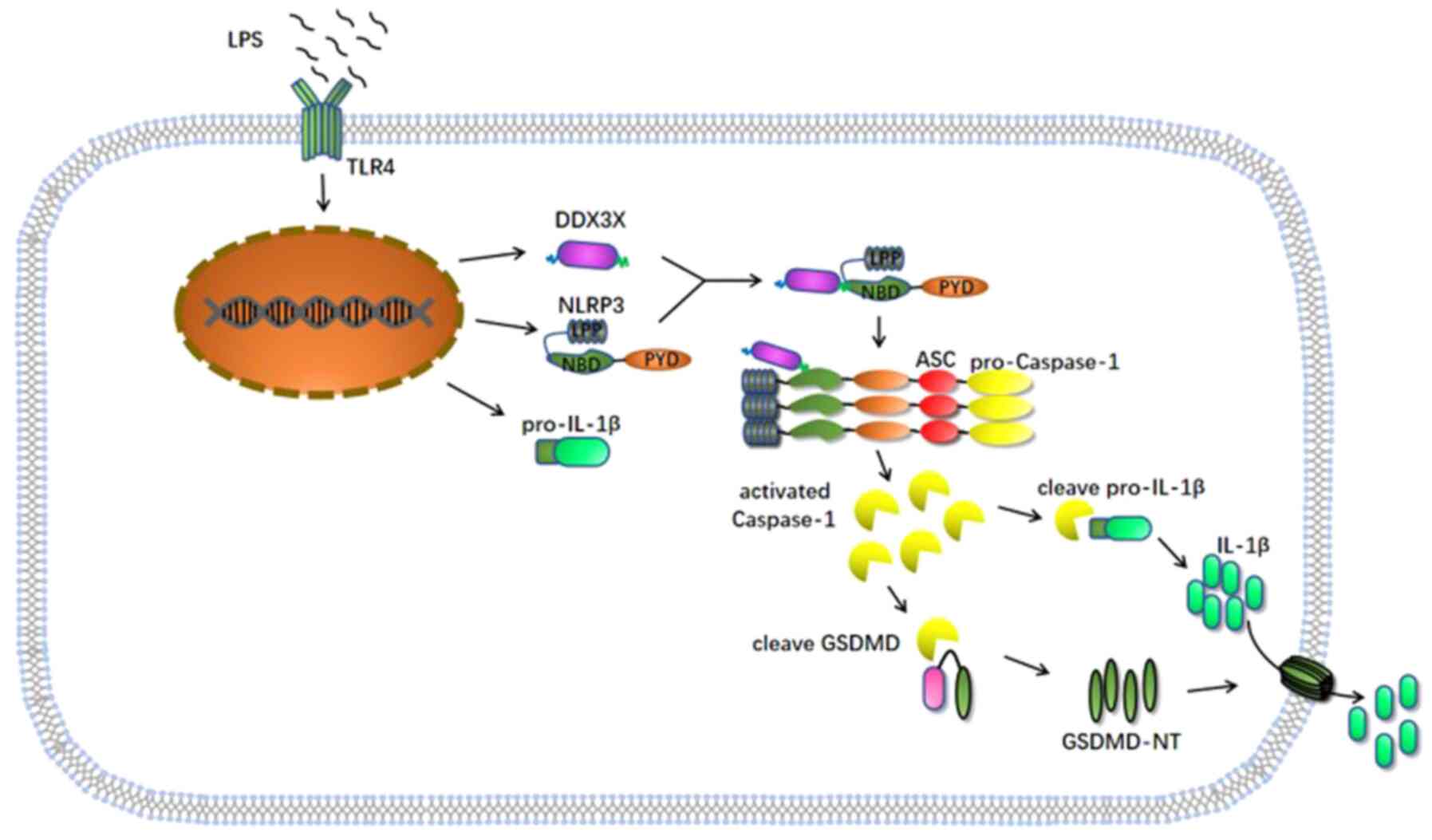
with LPS. The interactions uncovered by the present results are
presented in Fig. 5.
Discussion
To the best of our knowledge, the present study was
the first study to demonstrate that DDX3X is an essential molecular
component of the signaling pathway of LPS-induced cardiomyocyte
pyroptosis and cell injury. The results demonstrated that DDX3X
expression was significantly increased in LPS-treated
cardiomyocytes in vitro. Notably, Ddx3x knockdown
attenuated LPS-induced cardiomyocyte pyroptosis and cell
injury.
Pyroptosis is a major pathophysiological mechanism
in several cardiovascular diseases, such as atherosclerosis,
ischemic heart disease, diabetic cardiomyopathy, and cardiac
hypertrophy (35). During sepsis,
pyroptosis protects the host by eliminating infected cells;
however, overactivated pyroptosis can lead to systemic
inflammation, resulting in septic shock, MODS and an increased risk
of secondary infection (36). The
present study detected the upregulation of NLRP3 and pro-IL-1β, and
the activation of caspase-1 in H9c2 cells treated with LPS,
demonstrating that LPS stimulation can directly trigger pyroptosis
by activating the NLRP3 inflammasome in H9c2 cardiomyocytes and
lead to cardiomyocyte damage.
Increasing evidence suggest that the NLRP3
inflammasome participates in sepsis-induced cardiomyopathy
(34,37-39).
The NLRP3 inflammasome mediates caspase-1 activation and the
excretion of the proinflammatory cytokines, IL-1β and IL-18,
resulting in pyroptosis and systemic inflammatory responses. NLRP3
inflammasome activation is essential for normal host defense
against microbial infections. However, dysregulated activation of
the NLRP3 inflammasome can give rise to severe auto-inflammatory
states (37). Thus, activation of
the NLRP3 inflammasome must be tightly regulated to provide
adequate immune safeguards rather than damage to the host. Several
mechanisms have been demonstrated to regulate inflammasome
activation, including molecular post-translational modifications of
NLRP3 and its interacting partners (21).
DDX3X is a functionally multifaceted helicase, which
plays various roles in RNA metabolism, cell cycle control, stress
granule formation, apoptosis, innate immunity, viral infection and
cancer (40). A previous study
suggested that the availability of DDX3X molecules regulated
pyroptosis mediated by the NLRP3 inflammasome in macrophages, and
that DDX3X was involved in NLRP3 inflammasome assembly through
direct binding to the NACHT domain of NLRP3(28). Inhibition of the ATPase activity of
DDX3X by RK-33 does not affect NLRP3 inflammasome activation,
suggesting that DDX3X exerts a scaffold function by facilitating
the assembly of ASC specks, rather than a catalytic function
(41). In the present study, DDX3X
expression in cardiomyocytes increased following treatment with
LPS, suggesting that DDX3X is involved in LPS-induced cardiomyocyte
stress response. The results also demonstrated that Ddx3x
knockdown inhibited LPS-induced NLRP3 inflammasome activation but
did not affect NLRP3 expression. The results further confirmed that
DDX3X promotes NLRP3 inflammasome assembly by interacting with
NLRP3. The inhibition of NLRP3 inflammasome activation further
hindered caspase-1 activation, which attenuated pyroptosis and cell
death in LPS-treated cardiomyocytes. Taken together, these results
suggest that DDX3X acts as a regulator of pyroptosis during
cardiomyocyte stress response.
In conclusion, the results of the present study
demonstrated that LPS stimulation induced DDX3X expression in
cardiomyocytes, and Ddx3x knockdown attenuated LPS-induced
cardiomyocyte pyroptosis and cell injury by suppressing NLRP3
inflammasome activation. However, no in vivo data were
provided in the present study. Thus, further studies are required
to confirm whether DDX3X may be a potential therapeutic target for
sepsis-induced cardiomyopathy.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Nature Science
Foundation of China (grant no. 81873473), the Academic Promotion
Program of Shandong First Medical University (grant no. 2019QL014)
and Shandong Taishan Scholarship (no grant number available).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DF, LG, JiL, HH, JuL and EH were involved in the
study design. DF, YS and XM performed the experiments. DF analyzed
the data and drafted the initial manuscript. All authors have read
and approved the final manuscript. DF and EH confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Deutschman CS and Tracey KJ: Sepsis:
Current dogma and new perspectives. Immunity. 40:463–475.
2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
van Engelen TSR, Wiersinga WJ, Scicluna BP
and van der Poll T: biomarkers in sepsis. Crit Care Clin.
34:139–152. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Sato R and Nasu M: A review of
sepsis-induced cardiomyopathy. J Intensive Care.
3(48)2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Li F, Lang F, Zhang H, Xu L, Wang Y, Zhai
C and Hao E: Apigenin alleviates endotoxin-induced myocardial
toxicity by modulating inflammation, oxidative stress, and
autophagy. Oxid Med Cell Longev. 2017(2302896)2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Hao E, Lang F, Chen Y, Zhang H, Cong X,
Shen X and Su G: Resveratrol alleviates endotoxin-induced
myocardial toxicity via the Nrf2 transcription factor. PLoS One.
8(e69452)2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Peng F, Chang W, Sun Q, Xu X, Xie J, Qiu H
and Yang Y: HGF alleviates septic endothelial injury by inhibiting
pyroptosis via the mTOR signalling pathway. Respir Res.
21(215)2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Dai XG, Li Q, Li T, Huang WB, Zeng ZH,
Yang Y, Duan ZP, Wang YJ and Ai YH: The interaction between C/EBPβ
and TFAM promotes acute kidney injury via regulating NLRP3
inflammasome-mediated pyroptosis. Mol Immunol. 127:136–145.
2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Fu Q, Wu J, Zhou XY, Ji MH, Mao QH, Li Q,
Zong MM, Zhou ZQ and Yang JJ: NLRP3/caspase-1 pathway-induced
pyroptosis mediated cognitive deficits in a mouse model of
sepsis-associated encephalopathy. Inflammation. 42:306–318.
2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wu C, Lu W, Zhang Y, Zhang G, Shi X,
Hisada Y, Grover SP, Zhang X, Li L, Xiang B, et al: Inflammasome
activation triggers blood clotting and host death through
pyroptosis. Immunity. 50:1401–1411.e4. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Bordon Y: Mucosal immunology:
Inflammasomes induce sepsis following community breakdown. Nat Rev
Immunol. 12:400–401. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
11
|
Pu Q, Gan C, Li R, Li Y, Tan S, Li X, Wei
Y, Lan L, Deng X, Liang H, Ma F and Wu M: Atg7 deficiency
intensifies inflammasome activation and pyroptosis in pseudomonas
sepsis. J Immunol. 198:3205–3213. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Li Z, Guo J and Bi L: Role of the NLRP3
inflammasome in autoimmune diseases. Biomed Pharmacother.
130(110542)2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Martinon F, Burns K and Tschopp J: The
inflammasome: A molecular platform triggering activation of
inflammatory caspases and processing of proIL-beta. Mol Cell.
10:417–426. 2002.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kanneganti TD, Ozören N, Body-Malapel M,
Amer A, Park JH, Franchi L, Whitfield J, Barchet W, Colonna M,
Vandenabeele P, et al: Bacterial RNA and small antiviral compounds
activate caspase-1 through cryopyrin/Nalp3. Nature. 440:233–236.
2006.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Mariathasan S, Weiss DS, Newton K, McBride
J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM and
Dixit VM: Cryopyrin activates the inflammasome in response to
toxins and ATP. Nature. 440:228–232. 2006.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Karki R and Kanneganti TD: Diverging
inflammasome signals in tumorigenesis and potential targeting. Nat
Rev Cancer. 19:197–214. 2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Malireddi RKS, Gurung P, Mavuluri J,
Dasari TK, Klco JM, Chi H and Kanneganti TD: TAK1 restricts
spontaneous NLRP3 activation and cell death to control myeloid
proliferation. J Exp Med. 215:1023–1034. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Man SM and Kanneganti TD: Converging roles
of caspases in inflammasome activation, cell death and innate
immunity. Nat Rev Immunol. 16:7–21. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Venegas C, Kumar S, Franklin BS, Dierkes
T, Brinkschulte R, Tejera D, Vieira-Saecker A, Schwartz S,
Santarelli F, Kummer MP, et al: Microglia-derived ASC specks
cross-seed amyloid-β in Alzheimer's disease. Nature. 552:355–361.
2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
He Y, Hara H and Núñez G: Mechanism and
regulation of NLRP3 inflammasome activation. Trends Biochem Sci.
41:1012–1021. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kelley N, Jeltema D, Duan Y and He Y: The
NLRP3 Inflammasome: An overview of mechanisms of activation and
regulation. Int J Mol Sci. 20(3328)2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Tschopp J and Schroder K: NLRP3
inflammasome activation: The convergence of multiple signalling
pathways on ROS production? Nat Rev Immunol. 10:210–215.
2010.PubMed/NCBI View
Article : Google Scholar
|
|
23
|
Zhou R, Tardivel A, Thorens B, Choi I and
Tschopp J: Thioredoxin-interacting protein links oxidative stress
to inflammasome activation. Nat Immunol. 11:136–140.
2010.PubMed/NCBI View
Article : Google Scholar
|
|
24
|
Beckham C, Hilliker A, Cziko AM, Noueiry
A, Ramaswami M and Parker R: The DEAD-box RNA helicase Ded1p
affects and accumulates in Saccharomyces cerevisiae P-bodies. Mol
Biol Cell. 19:984–993. 2008.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Garbelli A, Radi M, Falchi F, Beermann S,
Zanoli S, Manetti F, Dietrich U, Botta M and Maga G: Targeting the
human DEAD-box polypeptide 3 (DDX3) RNA helicase as a novel
strategy to inhibit viral replication. Curr Med Chem. 18:3015–3027.
2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Soulat D, Bürckstümmer T, Westermayer S,
Goncalves A, Bauch A, Stefanovic A, Hantschel O, Bennett KL, Decker
T and Superti-Furga G: The DEAD-box helicase DDX3X is a critical
component of the TANK-binding kinase 1-dependent innate immune
response. EMBO J. 27:2135–2146. 2008.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Botlagunta M, Vesuna F, Mironchik Y, Raman
A, Lisok A, Winnard P Jr, Mukadam S, Van Diest P, Chen JH,
Farabaugh P, et al: Oncogenic role of DDX3 in breast cancer
biogenesis. Oncogene. 27:3912–3922. 2008.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Bol GM, Vesuna F, Xie M, Zeng J, Aziz K,
Gandhi N, Levine A, Irving A, Korz D, Tantravedi S, et al:
Targeting DDX3 with a small molecule inhibitor for lung cancer
therapy. EMBO Mol Med. 7:648–669. 2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Samir P, Kesavardhana S, Patmore DM,
Gingras S, Malireddi RK, Karki R, Guy CS, Briard B, Place DE,
Bhattacharya A, et al: DDX3X acts as a live-or-die checkpoint in
stressed cells by regulating NLRP3 inflammasome. Nature.
573:590–594. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Lu A, Magupalli VG, Ruan J, Yin Q,
Atianand MK, Vos MR, Schröder GF, Fitzgerald KA, Wu H and Egelman
EH: Unified polymerization mechanism for the assembly of
ASC-dependent inflammasomes. Cell. 156:1193–1206. 2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Franklin BS, Bossaller L, De Nardo D,
Ratter JM, Stutz A, Engels G, Brenker C, Nordhoff M, Mirandola SR,
Al-Amoudi A, et al: The adaptor ASC has extracellular and
‘prionoid’ activities that propagate inflammation. Nat Immunol.
15:727–737. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
32
|
Duncan JA, Bergstralh DT, Wang Y,
Willingham SB, Ye Z, Zimmermann AG and Ting JP: Cryopyrin/NALP3
binds ATP/dATP, is an ATPase, and requires ATP binding to mediate
inflammatory signaling. Proc Natl Acad Sci USA. 104:8041–8046.
2007.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Mavridis K, Stravodimos K and Scorilas A:
Downregulation and prognostic performance of microRNA 224
expression in prostate cancer. Clin Chem. 59:261–269.
2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Li N, Zhou H, Wu H, Wu Q, Duan M, Deng W
and Tang Q: STING-IRF3 contributes to lipopolysaccharide-induced
cardiac dysfunction, inflammation, apoptosis and pyroptosis by
activating NLRP3. Redox Biol. 24(101215)2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhaolin Z, Guohua L, Shiyuan W and Zuo W:
Role of pyroptosis in cardiovascular disease. Cell Prolif.
52(e12563)2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Gao YL, Zhai JH and Chai YF: Recent
advances in the molecular mechanisms underlying pyroptosis in
sepsis. Mediators Inflamm. 2018(5823823)2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Kalbitz M, Fattahi F, Grailer JJ, Jajou L,
Malan EA, Zetoune FS, Huber-Lang M, Russell MW and Ward PA:
Complement-induced activation of the cardiac NLRP3 inflammasome in
sepsis. FASEB J. 30:3997–4006. 2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zhang W, Tao A, Lan T, Cepinskas G, Kao R,
Martin CM and Rui T: Carbon monoxide releasing molecule-3 improves
myocardial function in mice with sepsis by inhibiting NLRP3
inflammasome activation in cardiac fibroblasts. Basic Res Cardiol.
112(16)2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Guo T, Jiang ZB, Tong ZY, Zhou Y, Chai XP
and Xiao XZ: Shikonin Ameliorates LPS-induced cardiac dysfunction
by SIRT1-dependent inhibition of NLRP3 inflammasome. Front Physiol.
11(570441)2020.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Mo J, Liang H, Su C, Li P, Chen J and
Zhang B: DDX3X: Structure, physiologic functions and cancer. Mol
Cancer. 20(38)2021.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Samir P and Kanneganti TD: DDX3X Sits at
the crossroads of liquid-liquid and prionoid phase transitions
arbitrating life and death cell fate decisions in stressed cells.
DNA Cell Biol. 39:1091–1095. 2020.PubMed/NCBI View Article : Google Scholar
|