Introduction
Ovarian cancer is the most common lethal
gynecological malignancy. More than two-thirds of patients are
diagnosed at a late stage [Federation Internationale of Gynecologie
and Obstetrigue (FIGO), stages III or IV], and the five-year
survival rate is <30% (1,2).
Carcinoma diagnosed in situ could elevate the five-year survival
rate to 92% (1,3), but a diverse group of tumors in
epithelial ovarian cancer impedes the early detection and new
therapeutic approaches. Accumulating evidence has indicated that
epithelial-to-mesenchymal transition (EMT) plays a crucial role in
the development of ovarian cancer, which is a major driver of
mortality in patients with cancer, whereas EMT is upregulated by
the mitogen-activated protein kinase (MEK)-extracellular
signal-regulated kinase (ERK) pathway (4,5). The
ERK/MAPK cascade mediates mitogenic signaling and is essential for
the control of cell fate, differentiation and proliferation, which
is frequently hyperactivated in human cancer (6).
MEK/ERK pathways are regulated by various mechanisms
(6,7). Protein phosphorylation is one of the
most important formats: Activated Raf phosphorylation and activates
MEK1 and MEK2, which then phosphorylates ERK1 and ERK2,
respectively (8,9). The protein Small ubiquitin-like
modifier (SUMO) is an important pathway for modulating cellular
function (10). SUMOs are small
ubiquitin-like modifiers containing 92-97-amino-acid polypeptides
(11). Similar to phosphorylation,
SUMOylation is a reversible and dynamic process involving the
activation of enzyme and cysteine proteases, which in turn modulate
the function, subcellular localization, complex formation and/or
stability of substrate proteins.
Monensin (Rumensin™) is a polyether ionophore
antibiotic secreted by the bacterium Streptomyces
cinnamonensis (12,13). Previous studies have demonstrated
that monensin exerts cytotoxic effects against several types of
cancer cells (14-16),
but the underlying mechanism has not yet been thoroughly
explored.
In the present study, the anticancer activity of
monensin against human ovarian cancer cells was investigated by
using a series of human ovarian cancer lines. Further studies
indicated that monensin effectively abrogated MEK SUMOylation,
resulting in the enhanced activation of the ERK pathway, whereas
SUMOylation faded with medium change and monensin withdrawal.
Overall, the present study results revealed a previously unknown
mechanism of monensin against ovarian carcinogenesis.
Materials and methods
Cell culture
Human ovarian cancer cell lines SK-OV-3, A2780,
OVCAR3 and CAOV-3 were purchased from the American Type Culture
Collection. The aforementioned cells were maintained in complete
Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum (FBS;
Invitrogen; Thermo Fisher Scientific, Inc.) at 37˚C in 5%
CO2.
Chemicals
Monensin (cat. no. S2324; Selleck Chemicals) were
added to the cells following the indicated concentration. Monensin
was dissolved in ethanol at 50 mM for storage in -80˚C. When used,
monensin was diluted 1:1,000 with DMEM, and added to cellular
medium based on the total volume for working solutions, such as
0.2, 1 and 5 µM.
Cell proliferation assay
Cell proliferation in response to monensin in
vitro was assessed by Cell Counting Kit-8 (CCK-8; Dojindo
Molecular Technologies, Inc.) assay according to the manufacturer's
instructions. Ovarian cancer cells were cultured at a density of
5x103 per well in FBS 10% (v/v) DMEM medium on 96-well
plate at 37˚C. After 24 h of incubation, the culture medium were
treated with monensin for 24, 48 and 72 h respectively. Then, 10 µl
CCK-8 reagent was added to each well followed by incubation at 37˚C
for 2 h. Absorbance was measured using a microplate reader
(Molecular Devices, LLC) at 450 nm with a reference wavelength of
650 nm.
The CellTiter-Glo (CTG) Luminescent Cell Viability
assay is a homogenous method of determining the number of viable
cells in culture based on the quantitation of the ATP present,
which signals the presence of metabolically active cells. The cells
were cultured in 96-well plates and treated with monensin for 24,
48 and 72 h respectively. Before luminescence recording, 100 µl CTG
reagent (Promega Corporation) was added to each well, and the
contents were mixed for 2 min on a shaker to induce cell lysis. The
plate was equilibrated to room temperature for 10 min, and the
luminescence signal was recorded on an Ascent luminometer (Thermo
Labsystems) according to the manufacturer's protocols. Data was
recorded as the mean ± SD of three independent experiments.
Cell wounding assay
Exponentially growing ovarian cancer cells were
seeded onto 6-well plates until 80% confluence was reached. A
sterile tip was used to create a wound in a cell monolayer, which
was then washed 3 times to remove non-adherent cells. Fresh medium
with 10% FBS was then added to the cultures. Images were captured
at 0, 24, and 48 h after scratching. Photoshop software (Adobe
Photoshop CS6; Adobe Systems, Inc.) was used to measure the
distance of the wound at each time point, and the wound healing
rate was calculated as follows: Wound healing rate = (control group
wound width - sample group wound width)/control group wound width
x100%. Data was recorded as the mean ± SD of three independent
experiments.
Transwell and invasion assay
Ovarian cancer cells were seeded onto the upper
chamber with 80% confluence. Ovarian cancer cells were starved by
incubating in serum-free medium for 24 h. Culture inserts of 8-µm
pore size (Transwell, Costar; Corning, Inc.) were placed onto the
wells of 24-well culture plates. The solution containing the test
agent was placed below the permeable cell membrane, and
1x105 cells were seeded onto the upper chamber. After 48
h of incubation at 37˚C with 5% CO2, the number of cells
that had migrated through the pores were fixed with 4%
paraformaldehyde at room temperature (RT) for 20 min, and stained
with 0.5% crystal violet for 3 min at RT. The photos were imaged at
x200 magnification under an inverted microscope (NIB-100F; Ningbo
Yongxin Optics Co., Ltd.), and the number of invading cells was
determined with ImageJ software 1.42 (National Institutes of
Health).
For the cell migration assay, culture inserts of
8-µm pore size were coated with Matrigel (BD Biosciences; 100
µg/well) and placed onto the wells of 24-well culture plates. The
subsequent steps were as aforementioned for the Transwell
experiments. The number of migrated cells were imaged at x200
magnification under an inverted microscope (NIB-100F; Ningbo
Yongxin Optics Co., Ltd.), and determined with ImageJ, and data was
recorded as the mean ± SD of three independent experiments.
Cell cycle analysis
Exponentially growing ovarian cancer cells were
seeded onto 6-well plates at a density of 5x104 and
allowed to culture for 24 h. The cells were later treated with
various concentrations of monensin and stored for 48 h. After
treatment, cells were harvested with trypsin, pelleted by
centrifugation, and washed with PBS. The cells were resuspended in
PBS and fixed in ice-cold 70% ethanol for 24 h at 4˚C. After
fixation, cells were centrifuged at 2,500 x g for 30 sec and washed
twice with PBS at RT, and then 1 ml propidium iodide master mix
solution (40 µl PI, 10 µl RNase and 950 µl PBS) were added and
incubated at 37˚C for 30 min in darkness. The cell cycle arrest was
analyzed by flow cytometry (Accuri C6, BD Biosciences). The
percentage of cell populations in the G1, S, and
G2/M phases was analyzed with Modfit LT 4.0 software
(Verity Software House, Inc.). Measurements were repeated
independently three times.
Western blot analysis
Ovarian cancer cells were lysed in RIPA buffer plus
protease inhibitors (XinShengyuan). Protein concentrations of the
cell lysates were determined by bicinchoninic acid kit (BCA; cat.
no. P0012, Beyotime Institute of Biotechnology). The lysates were
loaded 20-60 µg per lane, separated on 10% SDS-PAGE. Then the
samples were transferred onto a polyvinylidene fluoride membrane
(cat. no. ISEQ00010; EMD Millpore), and blocked by incubation with
5% fat-free milk in TBST buffer (150 mM NaCl, 50 mM Tris-HCl and
0.5% Tween-20, pH 7.6) at RT for 1 h. The membranes were incubated
with primary antibodies at room temperature for 1 h and then with
horseradish peroxide (HRP) conjugated secondary antibodies (OriGene
Technologies, Inc.), such as HRP conjugated goat anti-mouse IgG
(cat. no. ZB-2305) and HRP conjugated goat anti-rabbit IgG (cat.
no. ZB-2301), at RT for 1 h at dilution of 1:5,000. The bands were
visualized using ECL reagent (cat. no. 32106; Thermo Fisher
Scientific, Inc.), and calculated using ImageJ (1.42; National
Institutes of Health). The antibodies used for immunoblotting were
as follows (all from Abcam): E-cadherin rabbit polyclonal
antibodies (Ab; cat. no. ab92486; 1:2,000), claudin rabbit
polyclonal Ab (cat. no. ab124512; 1:3,000), vimentin rabbit
polyclonal Ab (cat. no. ab24525; 1:3,000), MEK1 rabbit polyclonal
Ab (cat. no. ab32091; 1:1,500), phosphor-MEK1/2-S298 rabbit
polyclonal Ab (cat. no. ab96379; 1:2,000), ERK1/2 rabbit polyclonal
Ab (cat. no. ab54230; 1:2,500), phosphor-MEK1/2-T202/T204 rabbit
polyclonal Ab (cat. no. ab214362; 1:2,000), rabbit tubulin
polyclonal Ab (cat. no. ab15568; 1:4,000) and SUMO rabbit
polyclonal Ab (cat. no. ab219724; 1:1,000). The expression of
tubulin was set as the control. All assay conditions were performed
in triplicate.
Immunoprecipitation (IP)
IP analyses were carried out as previously described
(17). The cells were suspended and
lysed in 50mM Tris-HCl buffer (pH 7.5) with 0.5% Triton X-100 and
protease inhibitors (XinShengyuan). Then, anti-MEK1 antibodies
(cat. no. ab32091; Abcam) were added to induce cellular lysis and
placed on a rotating stirrer at 4˚C for 2 h. Then, protein-G
Sepharose 4 Fast Flow beads (30 µl; Sigma-Aldrich; Merck KGaA) were
added according to the manufacturer's protocol. The solutions were
rotated overnight at 4˚C, and the beads were washed 3 times with
Tris-HCl buffer (50 mM; pH 7.5) with 0.5% Triton X-100. In one
experiment, the immune complexes were solubilized in SDS-PAGE
loading buffer and separated on SDS-PAGE gels (12%) following
western blotting to detect sumo-MEK1 with anti-SUMO antibodies
(cat. no. ab219724; Abcam).
Xenograft tumors of human ovarian
cancer cells
The use and care of animals were approved by ‘Animal
Ethics Committee of Affiliated Hospital of Shandong University of
Traditional Chinese Medicine’ (permit no. 2018-010). All
experimental procedures were carried out in accordance with the
approved guidelines, and every effort was made to minimize the
suffering of animals. Animals were kept in a specific pathogen-free
environment, in positive pressure rooms with filtered and
humidified air. The animal housing included a temperature of
23±2˚C, a controlled light and dark cycle (12 h: 12 h), ad libitum
food and ultra-filtered water, 50-55% humidity, ventilated caging
systems and standardized environmental enrichment.
Human ovarian cells SK-OV-3 were cultured in
Dulbecco's modified Eagle's medium supplemented with 10% FBS at
37˚C with 5% CO2. Specific pathogen-free female athymic
nude mice (5-6 week old; 16-18 g; female) were purchased from
Beijing HFK Bioscience and were housed three per cage under
specific pathogen-free conditions. Approximately 5x106
cells were injected subcutaneously into the right flank of 5- to 6-
week-old female athymic nude mice. The mice were divided into three
groups (n=3 per group) and treated with various doses of monensin
(0, 8 or 16 mg/kg body weight) at 3, 6, 9 and 12 days. Monensin was
dissolved in 100% ethanol to prepare 12 mg/ml and 24 mg/ml drug
storage solution; 100% ethanol was used as the control group.
Before injection, 20 µl drug preservation solution or 100% ethanol
was added to ~180 µl sterile PBS (plus 1% penicillin and
streptomycin) and diluted into 200 µl working solution. Each dose
was calculated according to the body weight of nude mice as
follows: 8 mg/kg in the low-concentration group, 16 mg/kg in the
high-concentration group, and 0 mg/kg in the control group
(containing only 10% ethanol). It is necessary to mix the drug
solutions before injection. The weight and palpable tumors were
typically observed within 2 weeks after injection of cells, and
tumor sizes were measured with a caliper 15 days after cell
injection. Tumor volumes were calculated using the following
formula: volume (mm3) = length x width2/2.
The maximum diameter of a single subcutaneous tumor was <1 cm.
All mice were sacrificed at 3 weeks by inhalation of CO2
gas, with a flow rate 20% replacement of home cage volume/min.
Death was verified by cessation of the heartbeat and lack of
movement, and the CO2 flow was maintained for at least 3
min after cardiac arrest. The tumors were retrieved for western
blotting assay. Multiple tumors were not observed in any of the
mice.
Statistical analysis
Data are expressed as the mean ± standard deviation
and analysis performed using SPSS v19.0 (IBM Corp.). All assays
were performed in triplicate and/or repeated three times.
Statistical significances were determined by one-way ANOVA test and
Tukey's test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Monensin inhibits the cell
proliferation of human ovarian cancer cells
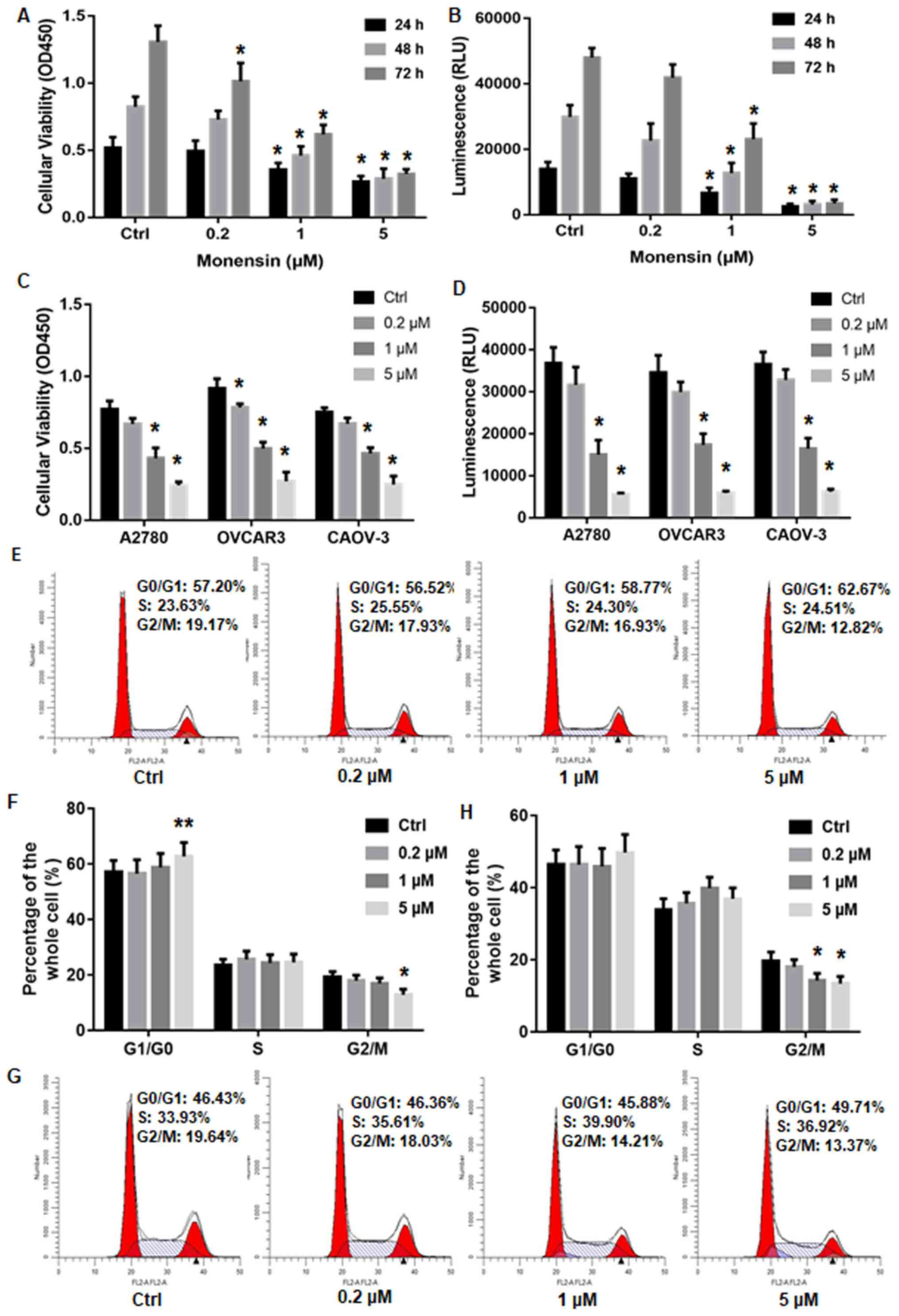
The inhibitory effects of antibiotic monensin on
human ovarian cancer were initially tested on SK-OV-3 cells by
CCK-8 assay. SK-OV-3 is commonly used as a model for ovarian cancer
to detect the effects and potency of various chemicals.
Sub-confluent SK-OV-3 cells were treated with 0, 0.2, 1 and 5 µM
monensin for 24, 48 or 72 h, respectively, and then detected. CCK-8
cell viability assay suggested that cell proliferation was
suppressed with increased monensin concentration and incubation
duration. As low as 0.2 µM monensin showed an inhibitory effect,
whereas completely inhibited cell proliferation was observed in the
first 24 h as high as 5 µM monensin (Fig. 1A). CTG luminescent cell viability
assay determined metabolically active cells according to the
quantification of the ATP present. Based on the luminescence
signal, CTG assay results indicated a similar inhibitory effect of
monensin on cell proliferation with CCK-8 assay (Fig. 1B).
In order to validate the inhibitory effects of
monensin on the proliferation of ovarian cancer cell SK-OV-3,
monensin was applied on other ovarian cancer cell lines including
A2780, OVCAR3 and CAOV-3. Cell viability was detected by CCK-8 and
CTG assays. When increased concentration of monensin (0.2, 1, and 5
µM) were added to the culture, all ovarian cancer cells showed
similar results at 48 h post-treatment; their proliferative
abilities were all effectively restrained by monensin (Fig. 1C and D).
Meanwhile, cell cycle distribution was analyzed by
flow cytometry to detect the effect of monensin. The SK-OV-3 cells
at the G2/M phase was significantly decreased from 19.17
to 12.82% when treated with 5 µM monensin (Fig. 1E and F). The A2780 cells showed similar results,
and the ratio of G2/M phase was decreased from 19.64 to
13.37% when 5 µM monensin was applied (Fig. 1G and H). The aforementioned results indicated
that monensin inhibited ovarian cancer cell proliferation by
blocking DNA replication.
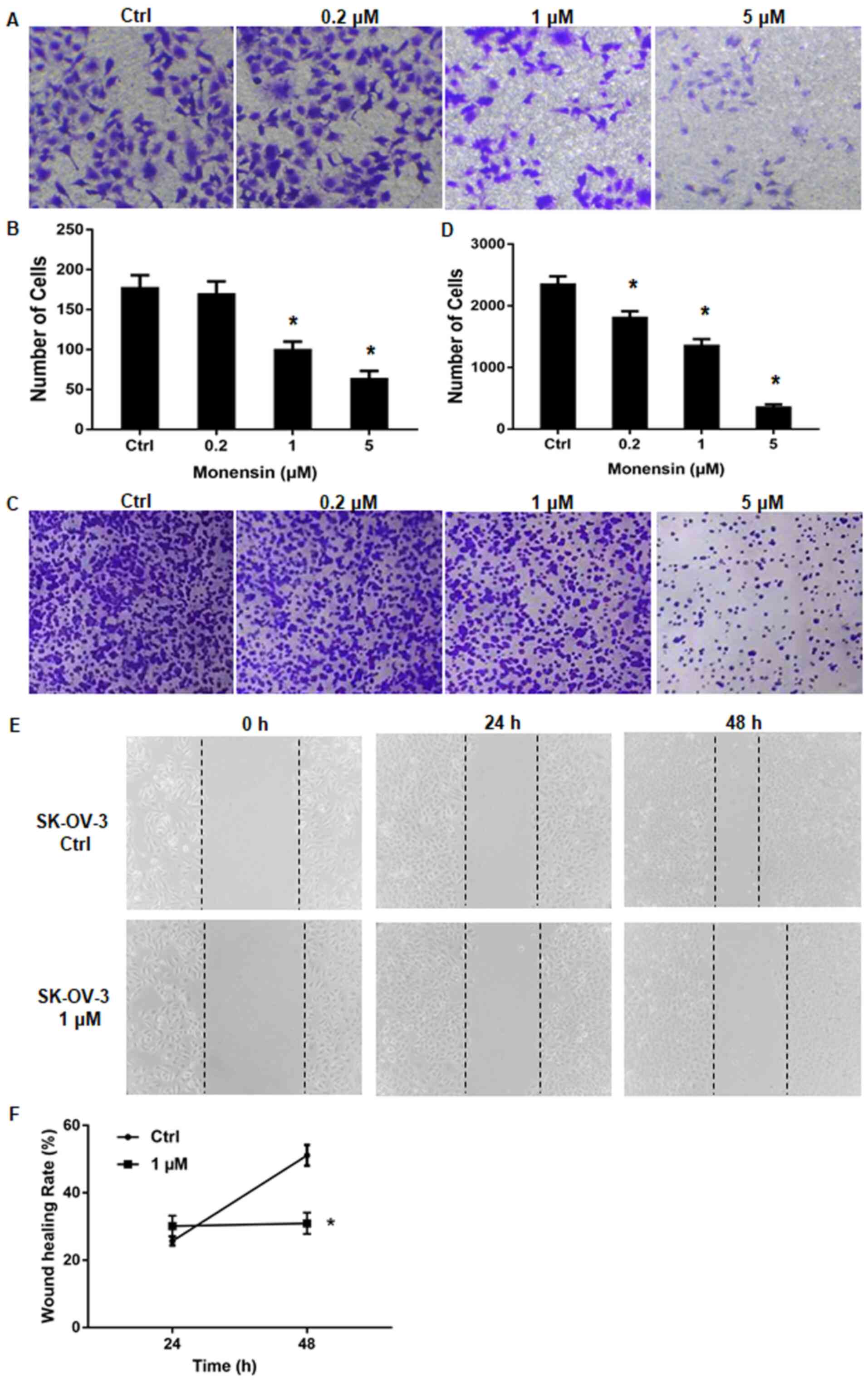
Monensin inhibits cell invasion and
malignant transformation of human ovarian cancer cells
Migration, invasion and transformation are key
procedures and important features of malignant cancer. In the
Transwell assay, cells treated with 0.2 µM monensin had similar
colon number and passing-through-membrane percentage compared with
the control group. However, the number of Transwell cells dropped
to about 100, whereas the percentage was decreased to 30% when 1 µM
monensin was applied to cells. The number and percentage dropped
dramatically to 50 and 10% when more monensin was added (5 µM)
(Fig. 2A and B). Overall, a series of concentrations (0,
0.2, 1 and 5 µM) of monensin was applied in the Transwell assay,
and the ability of malignant transformation of human ovarian cancer
showed significant impairment and does-dependent effect (Fig. 2C and D). Moreover, when 1 µM monensin was
applied to cells for 48 h, the gap closure showed time-dependent
decrease (Fig. 2E), and the wound
healing rate of cells treated with 1 µM monensin was significantly
decreased compared with the control group (Fig. 2F).
Monensin restrains the EMT-associated
signaling pathway
Next, the EMT-associated signaling molecules were
investigated in ovarian cancer cell lines. E-cadherin and claudin
are EMT promoters, and vimentin enables dynamic cell elongation. In
SK-OV-3 cells treated with monensin, the expression level of
E-cadherin and claudin increased to 30% of the control, whereas the
expression level of vimentin decreased to half of the control since
monensin accumulated to 5 µM. The aforementioned findings showed a
concentration-dependent response (Fig.
3A and B).
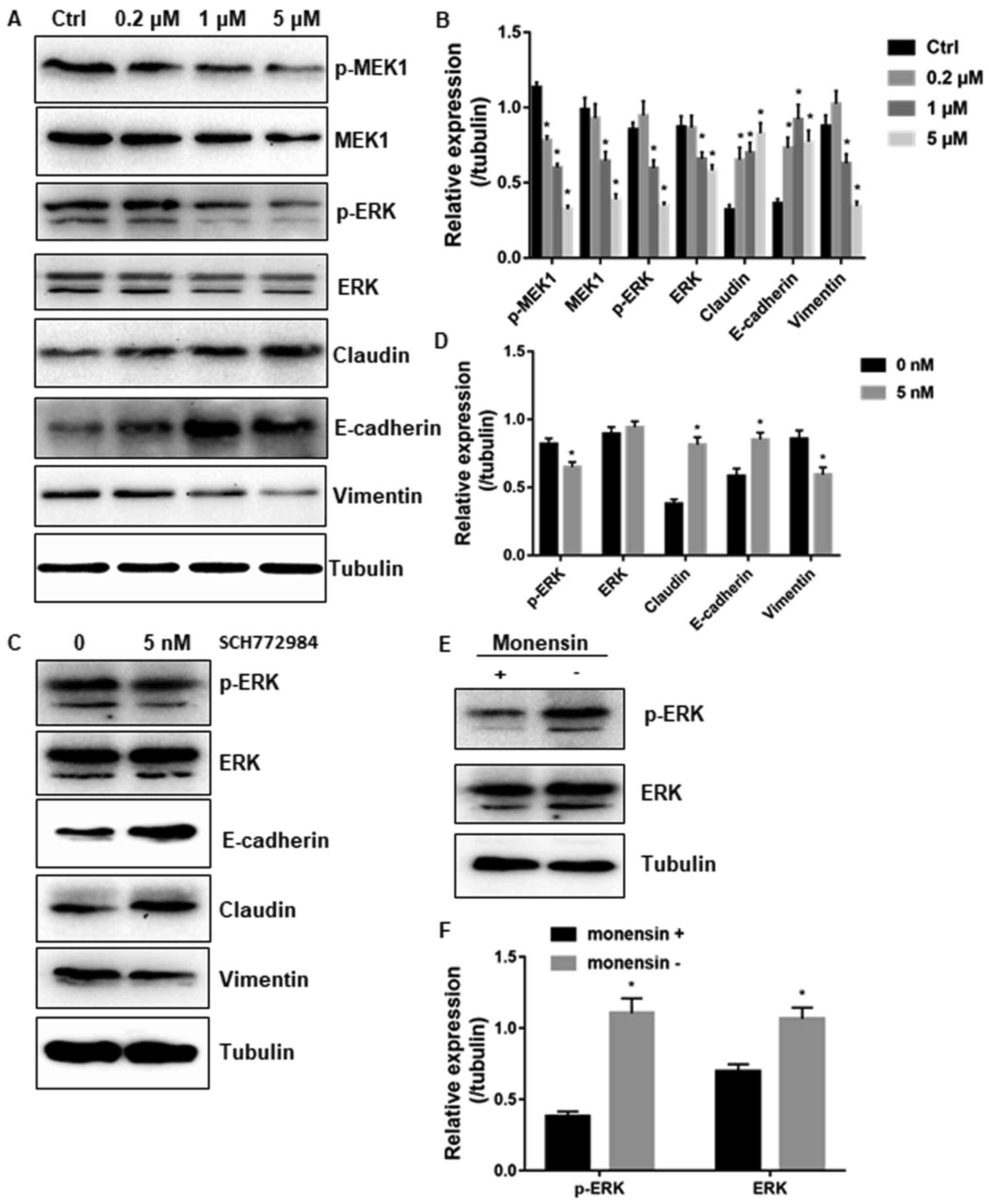 | Figure 3Monensin inhibits the MEK/ERK pathway
and epithelial-mesenchymal transition. (A) p-MEK, ERK, total MEK,
ERK, E-cadherin, vimentin and claudin expression levels were
measured by western blotting at indicated concentrations of
monensin. The densitometry analysis is shown in (B). (C) The
inhibitor of ERK, SCH772984, was used to detect the E-cadherin,
vimentin and claudin expression levels by western blotting, and
gray scale scanning analysis is shown in (D). (E) Monensin was
removed after incubation for 24 h, and the p-ERK level in cells 12
h after monensin removal was detected by western blotting. The
densitometry analysis is shown in (F). All assay conditions were
conducted in triplicate, and the representative results are
presented. *P<0.01 vs. control. p-,
phosphorylated. |
Moreover, E-cadherin and claudin can be activated by
the RAS-RAF-MEK-ERK cascade, and the potential attenuation of this
pathway by monensin was evaluated by western blotting. The results
revealed a specific decrease in the levels of phosphorylated ERK
and MEK protein in SK-OV-3 cells treated with monensin compared
with the control cells. The total MEK and ERK levels were also
decreased when treated with monensin. The regulation of the MEK-ERK
pathway was highly associated with cell proliferation (18), which explained the monensin-induced
growth inhibition. The specific inhibitor of ERK, SCH772984, served
as a positive control, which showed similar results (Fig. 3C and D). Recovery assay further showed that
monensin inhibited ERK phosphorylation. Monensin was removed after
24 h of incubation, and the phospho-ERK level in cells after 12 h
of monensin removal became much higher compared with that of cells
incubated with monensin (Fig. 3E
and F).
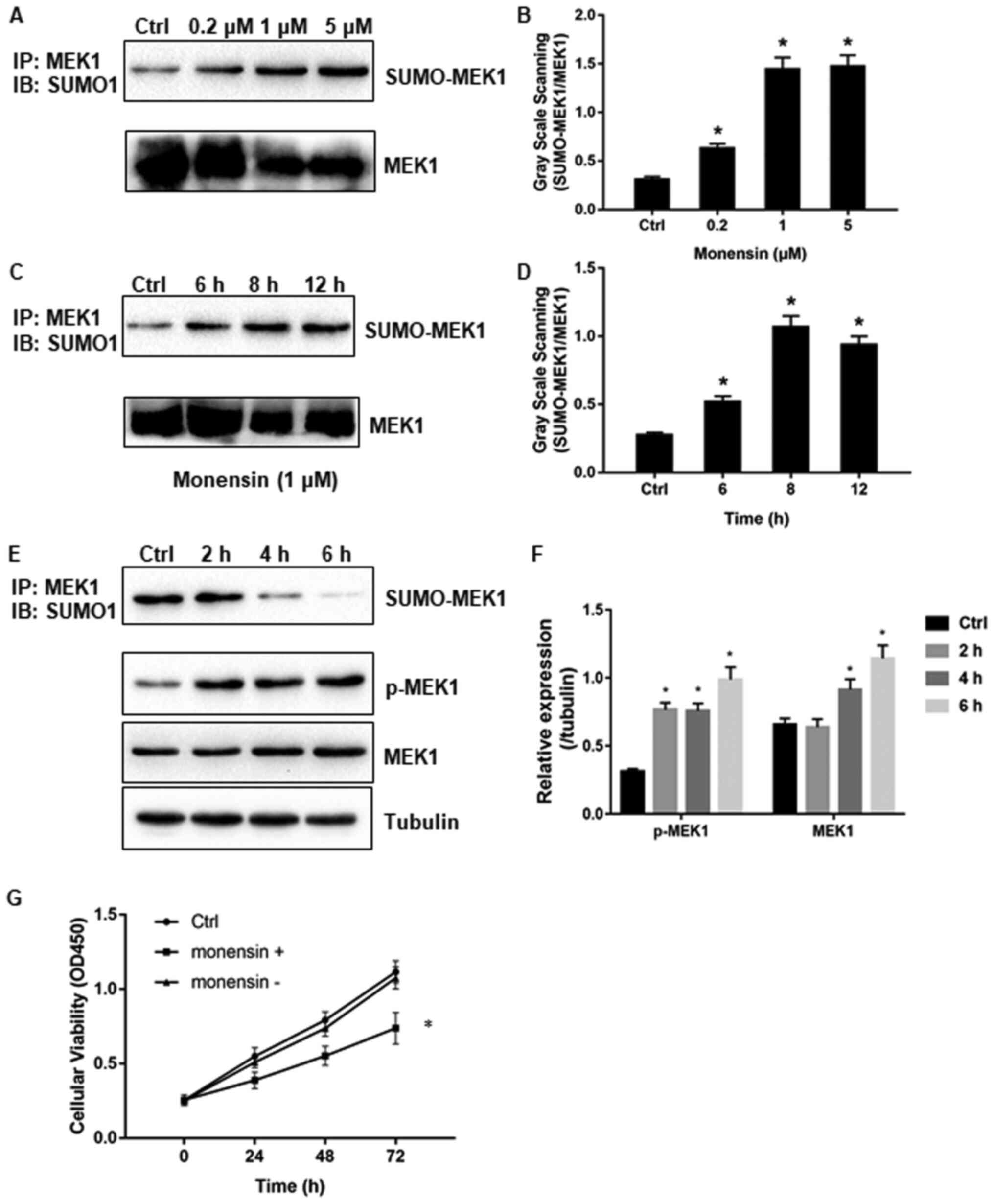
Monensin enhances the SUMOylation of
MEK1
Protein SUMOylation is associated with various
cellular functions, such as cell proliferation and invasion. MEK1
SUMOylation is an important way to regulate the MEK-ERK pathway. To
determine whether MEK underwent SUMOylation in ovarian cancer cell
line, MEK1 was expressed together with SUMO1 in HEK293 cells. MEK1
was immunoprecipitated with anti-MEK1 monoclonal antibodies and
protein G, separated by SDS-PAGE, and detected with anti-SUMO
monoclonal antibodies (Fig. 4A and
B). The results indicated that
monensin enhanced the SUMOylation modification of MEK1 with
increased drug concentration and growth of drug action time
(Fig. 4C and D).
To identify the function of monensin on MEK
SUMOylation and MEK-ERK phosphorylation cascade, cells transfected
with MEK1 and SUMO1 were incubated with the drug for 24 h and the
fresh medium was changed to a fresh one. At the time points of 2, 4
and 6 h, SUMOylated MEK1, phosphorylated MEK1 and total MEK1 were
detected by IP analysis. The results indicated that SUMO
modification was detached upon drug removal, thereby confirming the
effects of monensin on MEK1 SUMOylation. Moreover, with the
withdrawal of SUMO modification, the phosphorylation of MEK1 was
increased (Fig. 4E and F), whereas cell viability and
proliferation were recovered (Fig.
4G). It can be concluded that SUMOylation significantly
attenuated MEK1 activity in ovarian cancer cells by downregulating
ERK signaling, and the process was reversible.
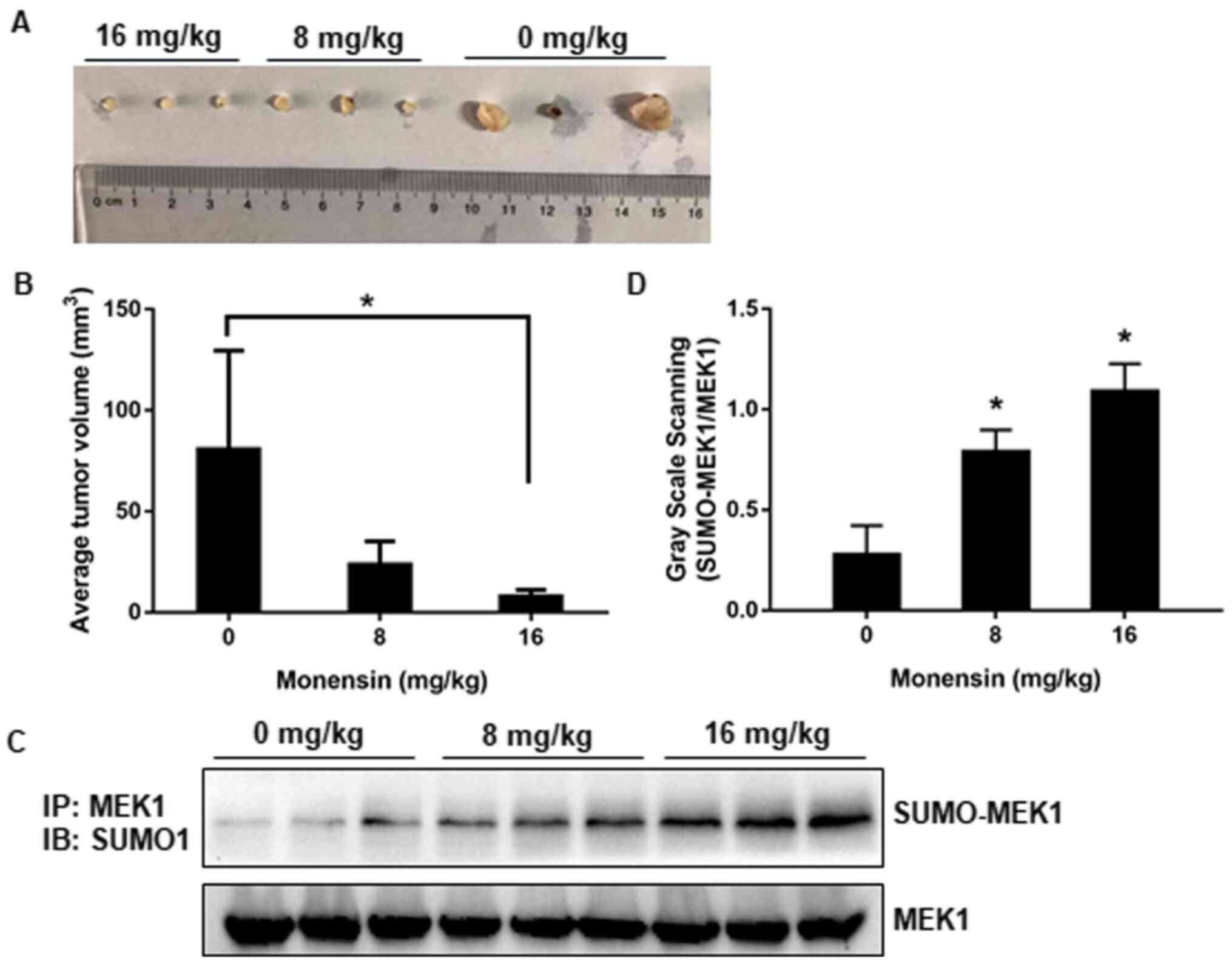
Monensin suppresses xenograft tumor
growth and enhanced MEK1 SUMOylation in vivo
Monensin exerted significant effects on the
proliferation, migration and invasion of ovarian cancer cells in
vitro. Accordingly, the anticancer effects of monensin on the
establishment and progression of ovarian cancer was explored in
vivo. SK-OV-3 ovarian cancer cells were injected subcutaneously
into the flanks of nude mice. At 3 days post-injection, the animals
were treated with three doses (0, 8 and 16 mg/kg) of monensin. The
tumor masses were retrieved on day 20, and the control group had
significantly larger individual tumors and higher average tumor
volume compared with monensin-treated groups (Fig. 5A and B). Besides, MEK1 SUMOylation was
strengthened with increased monensin dosage in vivo,
consistent with the data in vitro (Figs. 5C and 4D). Together, monensin effectively
inhibited tumor growth in ovarian cancer xenograft model by MEK1
SUMOylation reinforcement.
Discussion
Ovarian cancer is the most lethal tumor type among
malignancies in females with high incidence and mortality rate.
Most patients are diagnosed at a late stage (III and IV) and
undergo different responses to chemotherapy (1). A gap currently exists between
efficient treatment and clinical agents for ovarian cancer. Thus,
the demand for developing effective and novel therapies to ovarian
cancer is urgent.
Monensin has been approved by the Food and Drug
Administration as a safety medicine in veterinary medicine. Several
studies have also indicated its inhibitory effects on different
types of cancer, such as prostate, melanoma and acute myeloid
leukemia (15,17,18).
Monensin can work by freely passing through the lipid bilayer of
the cytoplasmic membrane along with passive diffusion (19), but the mechanism on anticancer
activity is unclear. The present study explored the possible
mechanism underlying the effects of monensin on ovarian cancer.
Firstly, monensin was found to suppress the
proliferation of different type of ovarian cells to minor varying
degrees. Cell-cycle distribution analysis verified the inhibitory
effect of monensin at the G1/S phase. Meanwhile,
monensin was detected to inhibit the invasion and migration of
ovarian cells, as well as to be highly associated with the
expression of EMT-associated signaling molecules (such as
E-cadherin, claudin and vimentin) and the central kinase of MEK and
ERK (20,21). E-cadherin is required for
progression through EMT, which contributes to the loss of adherent
junctions (22), whereas vimentin
is an intermediate filament composition that changes cytoskeletal
and polarity complex proteins and in turn contributes to EMT
(23). Claudin-linked protein
complexes exist between adjacent epithelial or endothelial cells,
and the dissolution of tight junctions during EMT is accompanied by
decreased Claudin (24). Overall,
E-cadherin, claudin and vimentin were all associated with
accelerated tumor growth, invasion and poor prognosis.
Furthermore, it was found that monensin intensified
the SUMO1 modification of MEK1, which attenuated MEK1 activity by
inhibiting MEK1 activation and negatively regulated the ERK
pathway, finally suppressing cellular overgrowth. The
aforementioned finding suggested that SUMO modification was a
reversible and highly dynamic process, which changed with drug
concentration and duration time. Results of in vivo
xenograft studies further confirmed that monensin effectively
inhibited xenograft tumor growth by inhibiting cell proliferation
and enhancing the SUMOylation of MEK. SUMOylation is an important
modulation of protein function and cell fate, and is highly
responsive to various stimuli and cellular microenvironment
(25).
Together, the results of the present study strongly
suggested that monensin functioned as an anti-ovarian cancer agent
and provided first evidence that the SUMO1 modification of MEK
played crucial roles in ovarian carcinogenesis. The present study
can guide future studies on the anticancer efficacy of monensin in
preclinical and clinical studies. In the present study, monensin
was shown to inhibit the MEK-ERK pathway and EMT, through in
vitro and in vivo experiments, by enhancing MEK1
SUMOylation.
Acknowledgements
Not applicable.
Funding
This study was supported by Funds for Teachers' research of
Jining Medical University (grant no. JYFC2019FKJ136).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SZ conceived and supervised the study; SY designed
experiments and wrote the original manuscript; WW established the
xenograft model; BZ and XC cultured cells and performed cell-based
assay; HY analysed the data. SY and SZ confirm the authenticity of
all the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The use and care of animals were approved by ‘Animal
Ethics Committee of Affiliated Hospital of Shandong University of
Traditional Chinese Medicine’ (permit no. 2018-010).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Phillips-Chavez C, Watson M, Coward J and
Schloss J: A systematic literature review assessing if genetic
biomarkers are predictors for platinum-based chemotherapy response
in ovarian cancer patients. Eur J Clin Pharmacol. 76:1059–1074.
2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Zhang R, Shi H, Ren F, Feng W, Cao Y, Li
G, Liu Z, Ji P and Zhang M: MicroRNA-338-3p suppresses ovarian
cancer cells growth and metastasis: Implication of Wnt/catenin beta
and MEK/ERK signaling pathways. J Exp Clin Cancer Res.
38(494)2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wen J, Zhao Z, Huang L, Wang L, Miao Y and
Wu J: IL-8 promotes cell migration through regulating EMT by
activating the Wnt/β-catenin pathway in ovarian cancer. J Cell Mol
Med. 24:1588–1598. 2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kan T, Wang W, Ip PP, Zhou S, Wong AS,
Wang X and Yang M: Single-cell EMT-related transcriptional analysis
revealed intra-cluster heterogeneity of tumor cell clusters in
epithelial ovarian cancer ascites. Oncogene. 39:4227–4240.
2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Wang S, Wang C, Li X, Hu Y, Gou R, Guo Q,
Nie X, Liu J, Zhu L and Lin B: Down-regulation of TRIB3 inhibits
the progression of ovarian cancer via MEK/ERK signaling pathway.
Cancer Cell Int. 20(418)2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Liu J, Hu HB, Liu YM, Li FX, Zhang LP and
Liao ZM: LncRNA HOTTIP promotes the proliferation and invasion of
ovarian cancer cells by activating the MEK/ERK pathway. Mol Med
Rep. 22:3667–3676. 2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Wu PK, Becker A and Park JI: Growth
inhibitory signaling of the Raf/MEK/ERK pathway. Int J Mol Sci.
21(21)2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Parker MI, Nikonova AS, Sun D and Golemis
EA: Proliferative signaling by ERBB proteins and RAF/MEK/ERK
effectors in polycystic kidney disease. Cell Signal.
67(109497)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Nayak A and Amrute-Nayak M: SUMO system -
a key regulator in sarcomere organization. FEBS J. 287:2176–2190.
2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Varejão N, Lascorz J, Li Y and Reverter D:
Molecular mechanisms in SUMO conjugation. Biochem Soc Trans.
48:123–135. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wan W, Zhang X, Huang C, Chen L, Yang X,
Bao K and Peng T: Monensin inhibits glioblastoma angiogenesis via
targeting multiple growth factor receptor signaling. Biochem
Biophys Res Commun. 530:479–484. 2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Gu J, Huang L and Zhang Y: Monensin
inhibits proliferation, migration, and promotes apoptosis of breast
cancer cells via downregulating UBA2. Drug Dev Res. 81:745–753.
2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Dayekh K, Johnson-Obaseki S, Corsten M,
Villeneuve PJ, Sekhon HS, Weberpals JI and Dimitroulakos J:
Monensin inhibits epidermal growth factor receptor trafficking and
activation: Synergistic cytotoxicity in combination with EGFR
inhibitors. Mol Cancer Ther. 13:2559–2571. 2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Markowska A, Kaysiewicz J, Markowska J and
Huczyński A: Doxycycline, salinomycin, monensin and ivermectin
repositioned as cancer drugs. Bioorg Med Chem Lett. 29:1549–1554.
2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Rajendran V, Ilamathi HS, Dutt S,
Lakshminarayana TS and Ghosh PC: Chemotherapeutic potential of
monensin as an anti-microbial agent. Curr Top Med Chem.
18:1976–1986. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Xin H, Li J, Zhang H, Li Y, Zeng S, Wang
Z, Zhang Z and Deng F: Monensin may inhibit melanoma by regulating
the selection between differentiation and stemness of melanoma stem
cells. PeerJ. 7(e7354)2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yusenko MV, Trentmann A, Andersson MK,
Ghani LA, Jakobs A, Arteaga Paz MF, Mikesch JH, Peter von Kries J,
Stenman G and Klempnauer KH: Monensin, a novel potent MYB
inhibitor, suppresses proliferation of acute myeloid leukemia and
adenoid cystic carcinoma cells. Cancer Lett. 479:61–70.
2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Vanneste M, Huang Q, Li M, Moose D, Zhao
L, Stamnes MA, Schultz M, Wu M and Henry MD: High content screening
identifies monensin as an EMT-selective cytotoxic compound. Sci
Rep. 9(1200)2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Hu M, Zhang Y, Li X, Cui P, Li J,
Brännström M, Shao LR and Billig H: Alterations of endometrial
epithelial-mesenchymal transition and MAPK signalling components in
women with PCOS are partially modulated by metformin in vitro. Mol
Hum Reprod. 26:312–326. 2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Xin L, Zhao R, Lei J, Song J, Yu L, Gao R,
Ha C, Ren Y, Liu X, Liu Y, et al: SND1 acts upstream of SLUG to
regulate the epithelial-mesenchymal transition (EMT) in SKOV3
cells. FASEB J. 33:3795–3806. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Sommariva M and Gagliano N: E-cadherin in
pancreatic ductal adenocarcinoma: A multifaceted actor during EMT.
Cells. 9(9)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Avtanski D, Garcia A, Caraballo B,
Thangeswaran P, Marin S, Bianco J, Lavi A and Poretsky L: Resistin
induces breast cancer cells epithelial to mesenchymal transition
(EMT) and stemness through both adenylyl cyclase-associated protein
1 (CAP1)-dependent and CAP1-independent mechanisms. Cytokine.
120:155–164. 2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wang K, Li T, Xu C, Ding Y, Li W and Ding
L: Claudin-7 downregulation induces metastasis and invasion in
colorectal cancer via the promotion of epithelial-mesenchymal
transition. Biochem Biophys Res Commun. 508:797–804.
2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Jansen NS and Vertegaal ACO: A chain of
events: Regulating target proteins by SUMO polymers. Trends Biochem
Sci. 46:113–123. 2021.PubMed/NCBI View Article : Google Scholar
|