Introduction
Cytochrome P450 (CYP) enzymes serve a pivotal role in the metabolism of numerous xenobiotics, including drugs and environmental chemicals (1). These enzymes are constitutively expressed in a tissue-specific manner, which can be increased further following exposure to xenobiotics (2). This induction system enhances detoxification capacity and protects organisms from the harmful effects of xenobiotics (3). In particular, transcription of CYP1A1, CYP1A2 and CYP1B1 is induced by the specific binding of polycyclic aromatic hydrocarbons, such as 2,3,7,8-tetrachlorodibenzo-p dioxin (TCDD) and β-naphthoflavone (βNF), to the aryl hydrocarbon receptor (AhR) (4,5). After translocating into the nucleus and recruiting the AhR nuclear translocator (Arnt), the AhR/Arnt complex then binds to the target xenobiotic responsive element (XRE) sequence within the regulatory enhancer region of the CYP1 gene (6).
It has been frequently reported that transcriptional regulation is at least in part mediated by cytosine methylation in the CpG sites of promoter regions (7). In humans, CYP1A1 and CYP1B1 genes comprise clusters of 103 and 307 CpG sites, respectively, termed CpG islands, in their promoter and enhancer regions (8). These CYP genes, as with common tumor suppressor genes, can be re-activated in cancer cell lines upon treatment with certain DNA methyltransferase inhibitors, including 5-aza-2'-deoxycytidine (DAC) (9,10). Therefore, DNA methylation at CpG sites within the promoter can cause the transcriptional silencing of CYP genes. Furthermore, exposure to TCDD in different cell lines, such as MCF-7 and HeLa cells has been found to induce CYP1B1 expression at various levels, which was enhanced further by co-treatment with DAC (11,12). These findings suggest that DNA methylation in CYP1B1 can alter the AhR-mediated transcription activity of these cells. However, the detailed mechanism underlying this process remains poorly understood.
The XRE core sequence CACGC (reverse complement, GCGTG) contains a CpG site (13,14). Therefore, this suggests that DNA methylation at XRE sequences can suppress its transcriptional activation by AhR by interfering with its interaction with AhR/Arnt complexes. To test this hypothesis, the potential interaction between βNF-induced AhR/Arnt complexes and unmethylated or methylated XRE sequences of the CYP1B1 gene was examined. In the present study, AhR-XRE complexes were isolated from βNF-induced liver cancer cells using the method of cleavage under targets and release using nuclease (CUT&RUN) (15), which is a modified form of the chromatin immunoprecipitation (ChIP) assay. The precipitate from the CUT&RUN assay was then used to examine the methylation status of the XRE sequences that were bound to the AhR/Arnt complexes. This novel approach, unlike the reporter and gel shift assays, does not depend on artificial conditions and can provide direct evidence for the binding of AhR/Arnt complexes to its target sequences under physiological intracellular conditions following exposure to ligands.
Materials and methods
Cells and reagents
Human liver cancer cell lines HepG2 and HuH7 were obtained from the Japanese Cancer Research Resources Bank. DMEM and 100X antibiotic-antimycotic solution were purchased from Thermo Fisher Scientific, Inc. FBS, PBS and DAC were purchased from Sigma-Aldrich; Merck KGaA. DMSO, βNF and proteinase K were purchased from Fujifilm Wako Pure Chemical Corporation. DAC and βNF were dissolved in PBS and DMSO, respectively.
Cell culture and treatment
HepG2 and HuH7 cells were cultured in DMEM containing 10% FBS and 1X antibiotic-antimycotic solution at 37˚C in a humidified atmosphere under 5% CO2. Thereafter, CYP1B1 expression was induced by treatment with 10 µM βNF at 37˚C for 4 h. To assess DNA demethylation, cells were exposed to 0.5 µM DAC at 37˚C for 72 h and the medium was changed every 24 h, prior to βNF treatment. PBS and 0.1% DMSO were used as solvent controls. After treatment, cells were further assessed using various assays.
From the results of preliminary experiments using reverse transcription-quantitative PCR analysis and bisulfite direct sequencing, it was revealed that 10 µM (between 0.01 and 100 µM) βNF and 0.5 µM (between 0.5 and 5 µM) DAC were the minimum concentration levels that caused substantial CYP1B1 gene induction and gene demethylation, respectively (data not shown). In addition, these concentration levels fell within the range of those reported by previous studies (5,16). Therefore, this dose combination was selected for the detection of DNA demethylation.
Reverse transcription-quantitative (q)PCR
Total RNA was isolated from cells and purified using the RNeasy Mini kit and DNase Set (Qiagen GmbH). First-strand cDNA was prepared from total RNA (3 µg) using the PrimeScript™ II 1st strand cDNA Synthesis kit (Takara Bio, Inc.). The following temperature protocol was used for reverse transcription: 30˚C for 10 min followed by 42˚C for 60 min and the reaction was terminated by 95˚C for 5 min. Thereafter, the levels of CYP1B1, AHR, ARNT and ACTB (β-actin) transcripts were measured using qPCR and the FastStart™ Universal SYBR®-Green Master (ROX) kit (Roche Diagnostics) in the ABI 7500 System (Thermo Fisher Scientific, Inc.). The following thermocycling conditions were used for qPCR: Initial denaturation for 10 min at 95˚C; followed by 45 cycles for 15 sec at 95˚C and 60 sec at 60˚C. The primer sequences used for qPCR are listed in Table I. Relative mRNA expression levels were determined using the standard curve method and a 10-fold dilution series, followed by normalization to ACTB mRNA expression levels (17). In all qPCR experiments, three independent replicates were analyzed for each treatment and the data were expressed as the mean ± standard deviation. P<0.05 was considered to indicate a statistically significant difference. This was determined using Shaffer's multiple comparison test using R statistical language and RStudio (ver. 1.2.5042; RStudio, Inc.).
 |
Table I
Primers used for reverse transcription-quantitative PCR and methylation analysis (29).
|
Table I
Primers used for reverse transcription-quantitative PCR and methylation analysis (29).
| Genes/region |
Primer sequence (5'-3') |
| ACTB |
|
| Forward |
TCATGAAGTGTGACGTGGACATC |
| Reverse |
CAGGAGGAGCAATGATCTTGATCT |
| AHR |
|
| Forward |
ACATCACCTACGCCAGTCGC |
| Reverse |
TCTATGCCGCTTGGAAGGAT |
| ARNT |
|
| Forward |
GCTGCTGCCTACCCTAGTCTCA |
| Reverse |
GCTGTCCGTGTCTGGAATTGT |
| CYP1B1 |
|
| Forward |
CGGCTGGATTTGGAGAACGTA |
| Reverse |
TGATCCAATTCTGCCTGCACT |
| XRE1 |
|
| Forward |
CTTTCCRAAAAACAAACTCAAATC |
| Reverse |
TTATTAGTAGGTTTTTATGGGAG |
| XRE2/XRE3 |
|
| Forward |
GYGTGTTAGGTGTYGTGAGAA |
| Reverse |
AAAAACRAATCTCCRCRCTCC |
| XRE4/XRE5/XRE6 |
|
| Forward |
GATYGTAAGYGYGTTTAGGAAGATT |
| Reverse |
CCAATCATATCCCTAAACRCTACCT |
| XRE7/XRE8 |
|
| Forward |
GTTATGTTGGTTAGGTTGGTTTYGA |
| Reverse |
AAACAACCCTACACTTTAAACTCCA |
| XRE2/XRE3 (for |
|
| CUT&RUN assay) |
|
| Forward |
GGCAGCGCCCAGGGATATGACTGGA |
| Reverse |
CGGAGAGTGGCAGGAGGAGGCGAAT |
DNA methylation analysis
In total, eight potential XRE sites are located within 2.3 kb of the 5'-flanking region of the human CYP1B1 gene (18). To assess the original DNA methylation status of these XREs in HepG2 and HuH7 cells without any treatment, genomic DNA samples were isolated from cells using the standard proteinase K/phenol-chloroform extraction method (19) and subjected to bisulfite sequencing.
Bisulfite modification was performed using the EpiTect Plus DNA Bisulfite kit (Qiagen GmbH) according to the manufacturer's protocol. A total of four genomic DNA segments covering the XRE1, XRE2/XRE3, XRE4/XRE5/XRE6 or XRE7/XRE8 regions were amplified by PCR using the Takara EpiTaq HS kit (Takara Bio, Inc.) and the primer sets listed in Table I. Thermocycling conditions were as follows: Initial denaturation at 94˚C for 5 min; followed by 35 cycles of denaturation at 94˚C for 30 sec, annealing at 55˚C (XRE1 and XRE4/XRE5/XRE6) or 60˚C (XRE2/XRE3 and XRE7/XRE8) for 30 sec and extension at 72˚C for 30 sec; and final extension at 72˚C for 7 min. These primers were originally designed based on bisulfite-modified sequences covering the XRE1-8 sites as described previously (18). After amplification and purification using the NucleoSpin Gel and PCR Clean-up kit (Takara Bio, Inc.), the methylation status of these fragments was detected by Sanger direct sequencing, which was performed using the BigDye™ Terminator v3.1 Cycle Sequencing kit (Thermo Fisher Scientific, Inc.) and 3130xl Genetic Analyzer (Thermo Fisher Scientific, Inc.). DNA methylation status of each XRE site was visibly determined according to the peak area ratio of C (methylated) and T (unmethylated) in the electropherogram and categorized as follows: Fully methylated; highly methylated (C>T); lowly methylated (C<T); and rarely methylated (fully unmethylated). The categorization was confirmed to be identical between two independent experiments.
CUT&RUN assay
To assess potential AhR binding to the XRE2/XRE3 sequence of the CYP1B1 gene, the CUT&RUN method was applied to HepG2 and HuH7 cells treated with βNF at 37˚C for 4 h using the antibody-targeted digestion of chromatin, which causes lower background signals than those obtained using the ChIP method (15). AhR-DNA complexes were precipitated from cells using an anti-AhR rabbit monoclonal antibody (cat. no. 83200; Cell Signaling Technology, Inc.) and a CUT&RUN Assay kit (cat. no. 86652S; Cell Signaling Technology, Inc.), according to the manufacturer's protocol.
Briefly, 10 µl concanavalin A magnetic beads suspended in 100 µl concanavalin A bead activation buffer were added to 1x105 cell solution (suspended in 100 µl 1X Wash Buffer) and were mixed on a rotator for 5 min at room temperature. The cell pellets separated on magnetic rack for 2 min at room temperature were mixed with 100 µl antibody binding buffer and 3 µl 1:33 diluted anti-AhR rabbit monoclonal antibody on the rotator for 12 h at 4˚C. The cell pellets were then bound to pAG-MNase enzyme by gentle mixing on the rotator for 1 h at 4˚C with 50 µl digitonin buffer (including 5 µl 10X Wash Buffer, 0.5 µl 100X Spermidine, 0.25 µl 200X Protease Inhibitor Cocktail, 1.25 µl Digitonin Solution, and 43 µl water) and 1.5 µl pAG-MNase Enzyme. After cooling for 5 min on ice with 150 µl digitonin buffer, the cell pellets were subjected to MNase activation by adding 3 µl cold CaCl2. DNA digestion was stopped by adding 150 µl of 1X Stop Buffer (including 3.75 µl Digitonin Solution and 0.75 µl RNaseA) and incubating at 37˚C for 10 min. DNA purification was performed using DNA Purification Buffers and Spin columns (cat. no. 14209; Cell Signaling Technology, Inc.). Briefly, digested DNA was mixed with 1.5 ml DNA Binding Buffer and was transferred to a DNA spin column in a collection tube. After centrifugation at 18,500 x g for 30 sec at 20˚C, DNA was washed with 750 µl DNA wash buffer. Finally, DNA was eluted from the column using 50 µl of DNA Elution Buffer. The eluate was subjected to PCR using the Takara EpiTaq HS kit (Takara Bio, Inc.). To confirm that this assay was optimized, positive control using 3 µl 1:33-diluted Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit monoclonal antibody (cat. no. 9751; Cell Signaling Technology, Inc.) and negative control using 5 µl 1:20-diluted Rabbit (DA1E) monoclonal antibody IgG XP Isotype Control (CUT&RUN; cat. no. 66362; Cell Signaling Technology, Inc.) were tested instead of using the anti-AhR rabbit monoclonal antibody. Input DNA without antibody precipitation (genomic DNA prepared for DNA methylation analysis as aforementioned) was used as an internal control. The primers used for amplifying the CYP1B1 enhancer region covering the XRE2/XRE3 (-872 to -723) sequences are listed in Table I. Thermocycling conditions were as follows: Initial denaturation at 94˚C for 5 min; followed by 35 cycles of denaturation at 94˚C for 30 sec, annealing at 62˚C for 30 sec and extension at 72˚C for 30 sec; and final extension at 72˚C for 7 min. The sizes of the PCR products were confirmed by electrophoresis on a 2% agarose gel with ethidium bromide staining at room temperature for 20 min.
Combined bisulfite restriction analysis (COBRA) and subcloning
After the CUT&RUN assay, DNA methylation at the individual CpG sites within the XRE2/XRE3 sequences was examined for anti-AhR antibody-precipitated complexes using COBRA (17,20). The template DNA was purified from the AhR-DNA complexes using proteinase K digestion at 55˚C for 2 h, phenol/chloroform extraction and ethanol precipitation to achieve the complete conversion of unmethylated cytosines to uracils by bisulfite treatment (21). After bisulfite modification, the XRE2/XRE3 sequence was amplified by PCR using the same kit and primers as that used for DNA methylation analysis. For COBRA, the amplified products were digested with the restriction endonuclease HpyCH4IV (New England Biolabs, Inc.) at 37˚C for 3 h, which cleaves only methylated CpG sites within both XRE sequences. Subsequently, the digested DNA was electrophoresed on a 3% agarose gel and visualized by ethidium bromide staining.
For in-depth analysis, the CpG methylation status of a single copy of the CYP1B1 gene was determined through dideoxy sequencing after subcloning the amplified products into the pCR4-TOPO vector using a TOPO™ TA Cloning™ kit for Sequencing (Thermo Fisher Scientific, Inc.) (22). The sequencing reaction was performed using the M13 primer set: Forward, 5'-GTAAAACGACGGCCAG-3' and reverse, 5'-CAGGAAACAGCTATGAC-3' (reverse). In total, ≥10 clones of each amplified product were sequenced. In addition, the input genomic DNA was subjected to COBRA and subcloning as a control to indicate the original DNA methylation status of HepG2 and HuH7 cells.
Results
βNF-induced upregulation of CYP1B1 expression is enhanced by DAC treatment
Fig. 1 demonstrates the levels of CYP1B1 expression in the two liver cancer cell lines. βNF significantly increased CYP1B1 expression in both HepG2 and HuH7 cells compared with the PBS-treated group (Fig. 1A and B). By contrast, cell treatment with DAC alone conferred no significant effects on CYP1B1 expression compared with those in the PBS group. The βNF-induced upregulation of CYP1B1 expression was significantly potentiated by pre-treating the HepG2 cells with DAC compared with that in the βNF group (βNF vs. D + β; Fig. 1A). However, no such potentiation could be observed in HuH7 cells (βNF vs. D + β; Fig. 1B). Treatment with DAC alone had no significant effects on the AHR and ARNT expression levels in HepG2 cells (Fig. 1C and E). However, AHR and ARNT expression was slightly but significantly upregulated in HuH7 cells by DAC treatment alone compared with that in cells treated with PBS (Fig. 1D and F). These results suggested that DNA demethylation induced by DAC treatment enhanced the response to the βNF-activated AhR pathway in HepG2 cells, which increased CYP1B1 expression without increasing AHR or ARNT expression. In HuH7 cells, DNA demethylation upregulated AHR and ARNT expression, but this had little effect on CYP1B1 gene expression.
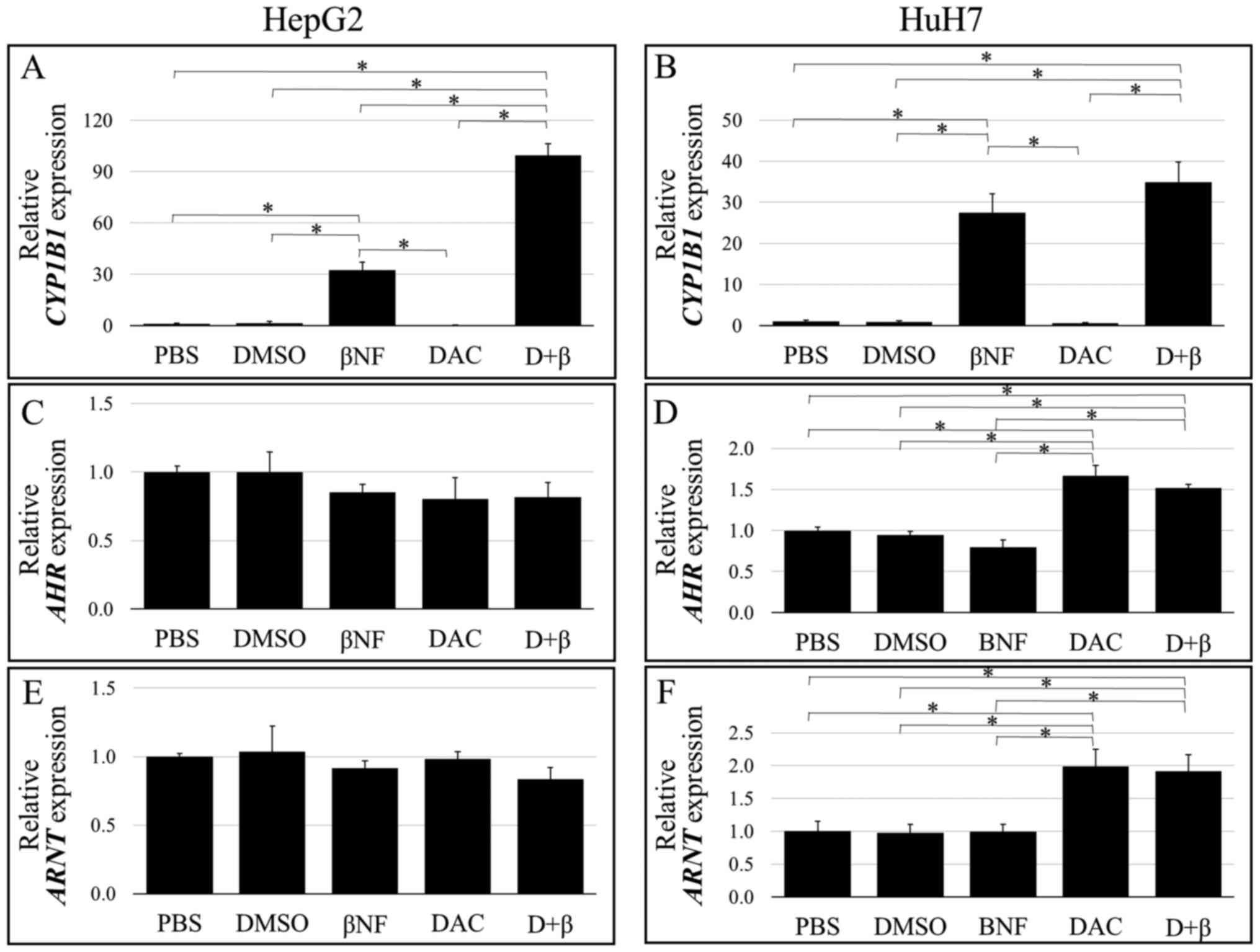 |
Figure 1
Basal and induced mRNA expression levels of CYP1B1, AhR and Arnt as determined using quantitative reverse transcription-quantitative PCR. (A-F) HepG2 and HuH7 cells were treated with either PBS, DMSO, βNF, DAC alone or D+β. CYPB1 expression in (A) HepG2 and (B) HuH7 cells. AhR expresion in (C) HepG2 and (D) HuH7 cells. Arnt expression in (E) HepG2 and (F) HuH7 cells. The vertical axes indicate relative transcription levels normalized to those of ACTB (plotted as a ratio to levels in PBS-treated cells). Transcript levels are expressed as means ± SD. Statistically significant differences between the two groups are depicted as *P<0.05. ACTB, β-actin; XRE, xenobiotic response element; AHR, aryl hydrocarbon receptor; Arnt, AhR nuclear translocator; CYP1B1, cytochrome P450 family 1 subfamily B member 1; βNF, β-naphthoflavone; DAC, 5-aza-2'-deoxycytidine; D + β, DAC + βNF.
|
DNA methylation status of the XRE sequences
Fig. 2 illustrates the DNA methylation status of the eight XRE sites detected in the two liver cancer cell lines without any treatment. XRE4, XRE5 and XRE6 were found to be rarely methylated (mostly unmethylated) in both cell lines. This finding indicates that DAC treatment-enhanced the βNF-induced CYP1B1 expression in the HepG2 cells in a manner independent of the methylation status of these XRE sequences. By contrast, highly methylated XRE7 and fully methylated XRE8 sequences were observed in both cell lines. Since DAC treatment (inhibition of methylation) did not markedly enhance βNF-induced CYP1B1 expression in HuH7 cells, demethylation of the XRE7/8 is unlikely to exert any effects on the enhanced inducibility in HepG2 cells.
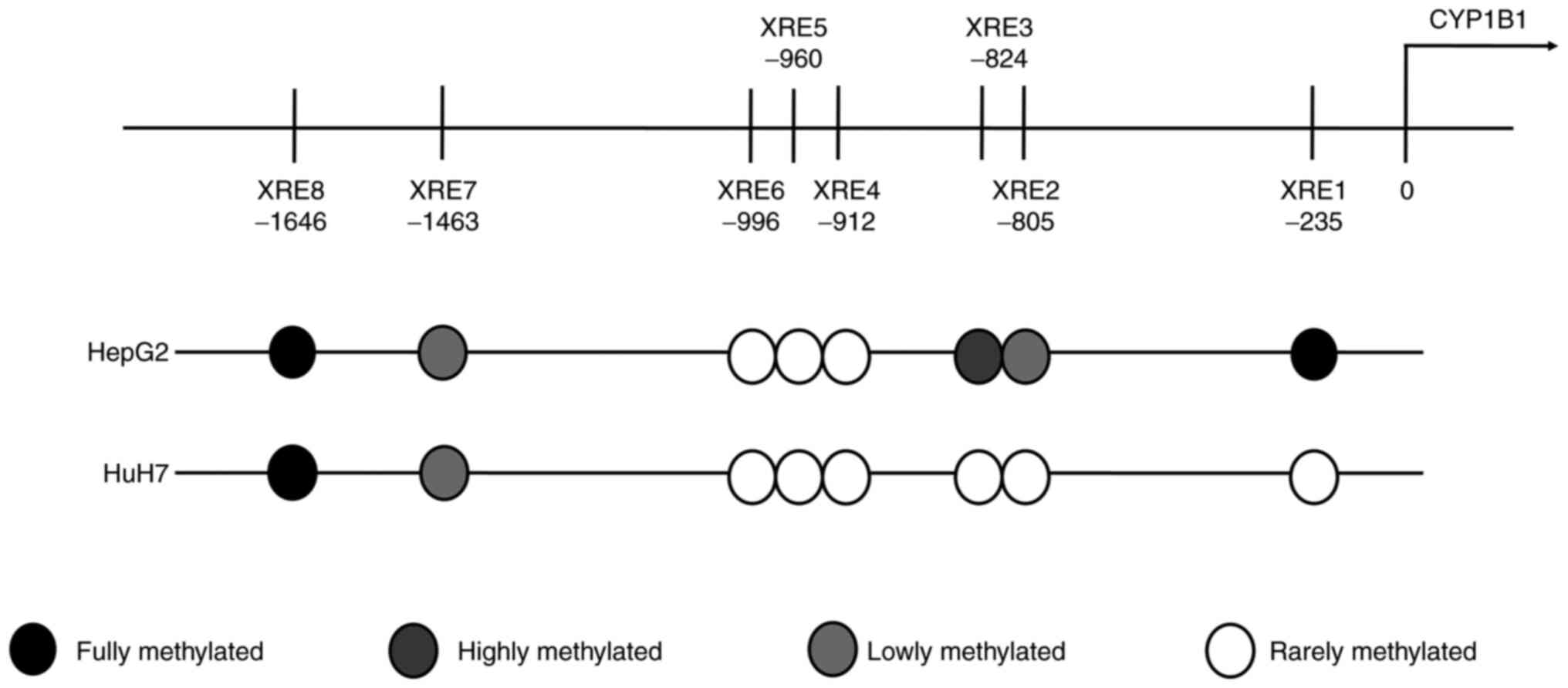 |
Figure 2
DNA methylation status of the eight XRE sites detected in the two liver cancer cell lines. XRE sites and their proposed nucleotide positions in the CYP1B1 gene as depicted in the map. DNA methylation status of each XRE site is presented as one of the following four categories: Fully methylated; highly methylated; lowly methylated; and rarely methylated (mostly unmethylated). XRE, xenobiotic response element; CYP1B1, cytochrome P450 family 1 subfamily B member 1.
|
Subsequently, the potential binding of AhR to the XRE2 and XRE3 sites was next analyzed for two reasons. These two XREs have been reported to serve a pivotal role in TCDD-mediated induction of CYP1B1 expression (18,23). In addition, lowly methylated XRE2 and highly methylated XRE3 were observed in HepG2 cells (Fig. 2), where methylated and unmethylated target sequences coexist. Therefore, XRE2 and XRE3 were selected for evaluating whether AhR preferentially binds to unmethylated or methylated XREs.
AhR preferentially binds to unmethylated XRE sequences
COBRA using input DNA revealed that amplified DNA covering the XRE2/XRE3 sequences was partially digested by the restriction enzyme HpyCH4IV in βNF-treated HepG2 cells but not in HuH7 cells (Fig. 3). Therefore, the CpG sites within the XRE2/XRE3 sequence were partially methylated in the HepG2 cells but not in the HuH7 cells. These results are consistent with those from the direct sequencing for the assessment of methylation status without any treatment (Fig. 2). In addition, this partial methylation in the HepG2 cells was mostly eliminated after combined treatment with DAC and βNF (Fig. 3), indicating successful DNA demethylation. To determine if AhR preferentially bounds to unmethylated XRE sequences, AhR-DNA complexes were precipitated from cells using the CUT&RUN method with the anti-AhR antibody. Specific DNA amplification of the CYP1B1 XRE2/XRE3 sequence was confirmed by electrophoresis and direct sequencing. The AhR-DNA complexes were then subjected to bisulfite modification and methylation analysis. Unlike input DNA, the precipitated AhR-bound DNA from these cell lines exhibited no detectable methylation in either cell line (Fig. 3).
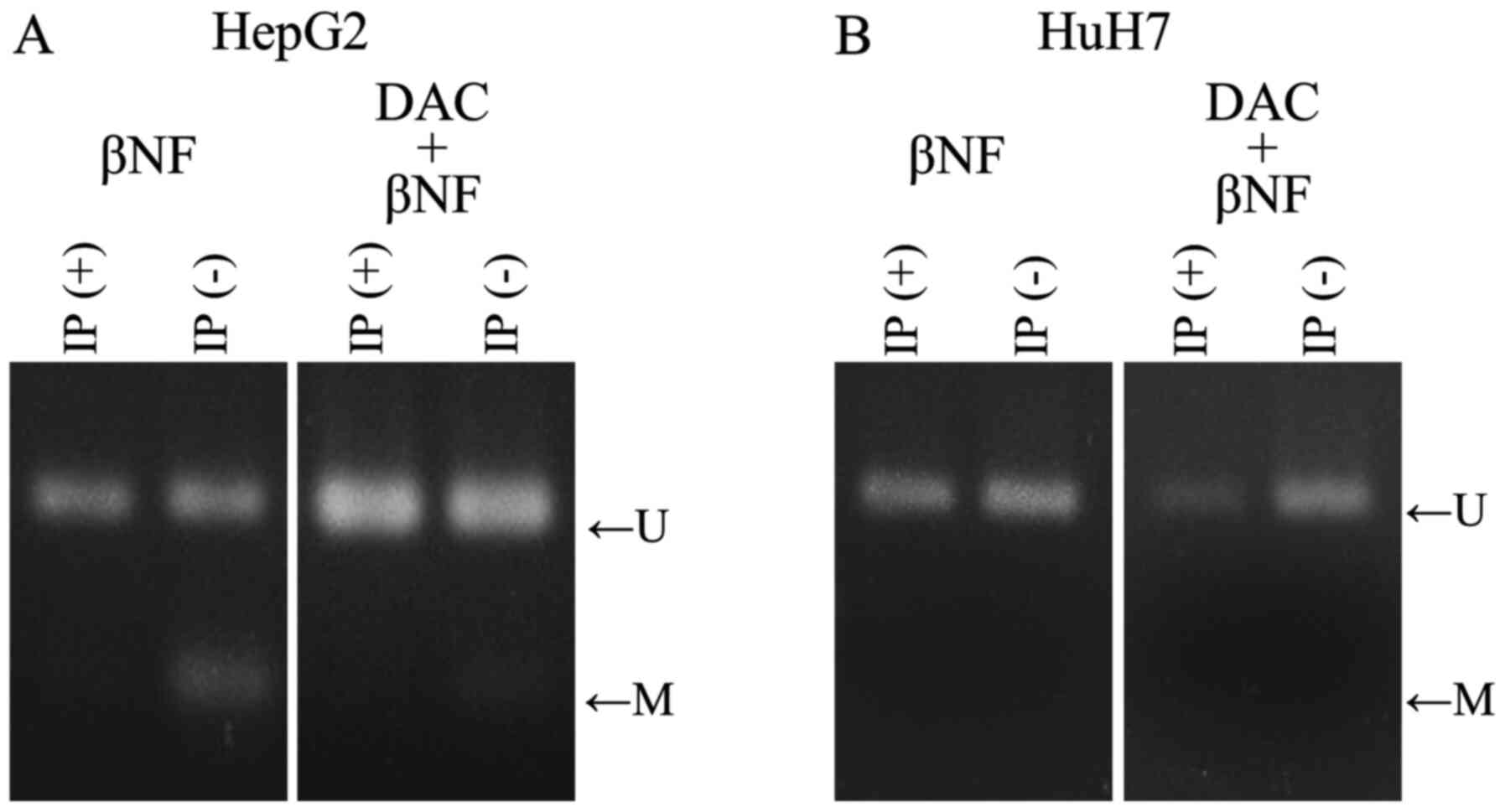 |
Figure 3
DNA methylation status of the XRE2/XRE3 sequences as examined using the COBRA method in the (A) HepG2 and (B) HuH7 liver cancer cell lines. Methylated XRE2/XRE3 fragments were digested using the restriction endonuclease HpyCH4IV, whereas unmethylated XRE2/XRE3 fragments were not. IP(-) indicates the result of input DNA obtained from HepG2 or HuH7 cells after treatment with βNF alone and/or DAC. IP(+) indicates the results of precipitated AhR-bound XRE2/XRE3 sequences obtained from HepG2 or HuH7 cells after the aforementioned treatments. XRE, xenobiotic response element; COBRA, combined bisulfite restriction analysis; IP, immunoprecipitation; U, unmethylated; M, methylated; CYP1B1, cytochrome P450 family 1 subfamily B member 1; βNF, β-naphthoflavone; DAC, 5-aza-2'-deoxycytidine.
|
To determine the CpG methylation status in a single copy of the gene, the input and precipitated DNA used for COBRA in Fig. 3 were subcloned and sequenced. Fig. 4A shows the nucleotide sequence around the CYP1B1 XRE2/XRE3 sequence whereas Fig. 4B shows the representative electropherograms of methylated and unmethylated XRE sequences. Fig. 4C demonstrates the methylation status of the individual clones. In HepG2 cells, of the 10 clones derived from input DNA, eight exhibited XRE3 methylation and three revealed simultaneous XRE2 methylation (Fig. 4C). However, such methylation could not be detected in the clones isolated from the precipitated AhR-bound DNA, whereas methylation outside the XRE2/XRE3 sequences was frequently retained even after precipitation (Fig. 4C). This finding that AhR-bound DNA clones always revealed the unmethylated XRE2/3 status was consistent with the COBRA results (Fig. 3). In HuH7 cells, clones with methylated XRE2 and XRE3 could not be obtained from input or AhR-precipitated DNA. The results suggested that βNF-induced AhR recognizes and selectively bind to unmethylated XRE2 and XRE3 sequences.
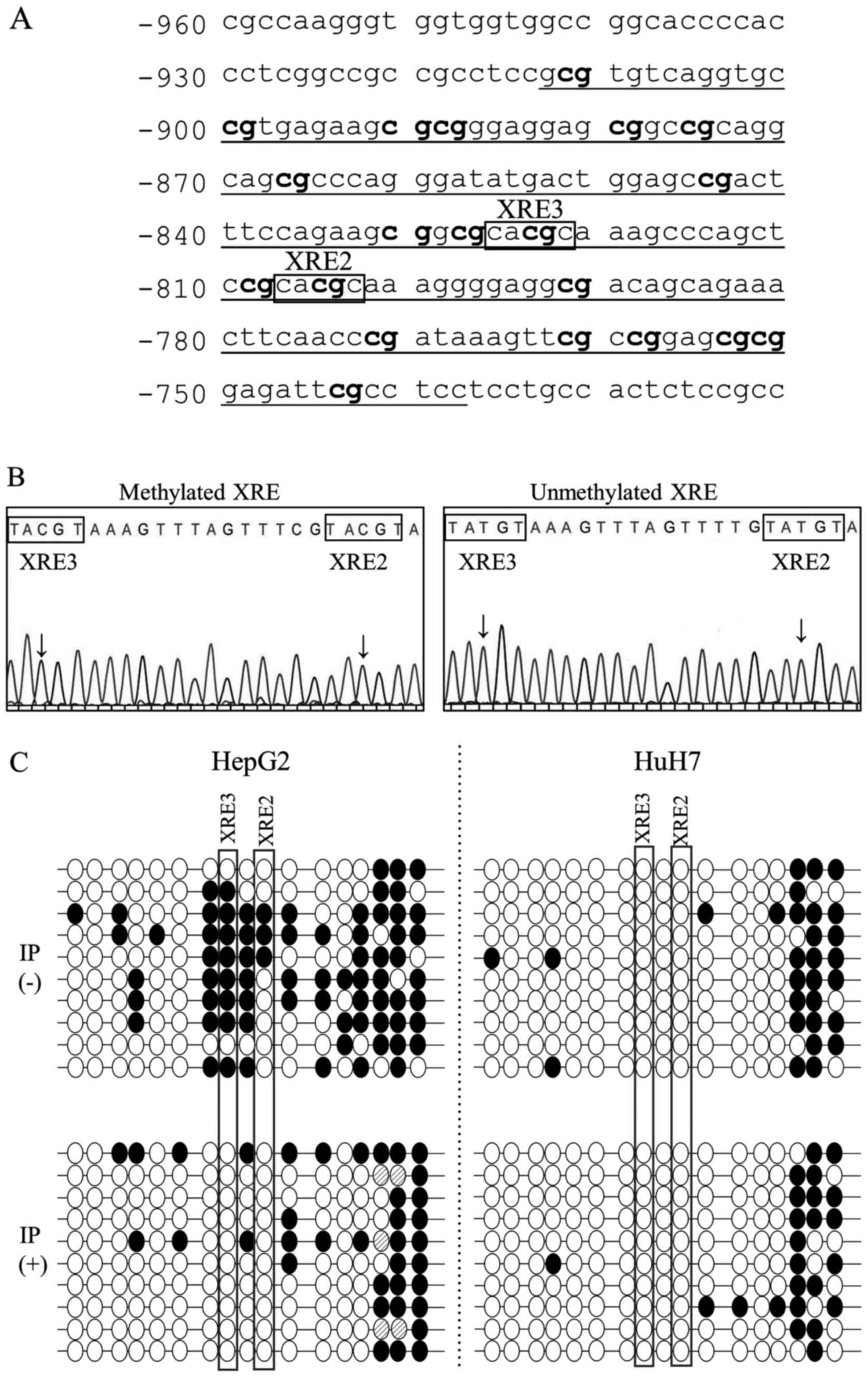 |
Figure 4
DNA methylation status around the XRE2/XRE3 sequences as determined by sequencing the individual copies of the gene in the two liver cancer cell lines. (A) Nucleotide sequence of XRE2/XRE3 in the CYP1B1 enhancer region (DDBJ/EMBL/GenBank, NG_0088386). The numbers on the left of each line represent the distance from the putative transcription start site (bp). The amplified region and individual CpG sites are indicated by underlined text and bold text, respectively. (B) Sequencing electropherograms from representative clones comprising either methylated or unmethylated XRE2/XRE3 sequences. Arrows indicate CpG dinucleotides. (C) Methylation profiles around the XRE2/XRE3 sequences in HepG2 and HuH7 cells after treatment with βNF. Each line represents a DNA sequence of an individual clone. IP(-) indicates the methylation status of individual clones derived from input DNA. IP(+) shows the methylation status of clones from precipitated AhR-bound DNA. CpG dinucleotides in the XRE2/XRE3 region are depicted as circles horizontally aligned in the 5'-3' direction. White and black circles represent unmethylated and methylated CpG sites, respectively. Shaded circles represent the CpG sites where the methylation status could not be determined in this study. Two CpG dinucleotides within the XRE2 and XRE3 sequences are enclosed by square boxes. XRE, xenobiotic response element; CYP1B1, cytochrome P450 family 1 subfamily B member 1; IP, immunoprecipitate; βNF, β-naphthoflavone.
|
Discussion
In the present study, it was found that βNF-induced CYP1B1 expression in HepG2 cells was upregulated by DAC pre-treatment whilst AHR and ARNT expression was not upregulated. Although basal CYP1B1 expression levels in HepG2 cells was lower compared with that in HuH7 cells (data not shown), these levels did not change by treatment with DAC alone. Therefore, basal CYP1B1 promoter activity may be retained in both cells before DAC treatment and this promoter activity could not be further increased by DNA demethylation alone. This suggest that DAC pre-treatment upregulated βNF-induced CYP1B1 expression in HepG2 cells by increasing AhR-mediated enhancer activity due to the demethylation of XRE sequences.
In the current study, eight putative XRE sequences (XRE1-8) were present in the human CYP1B1 enhancer region. Among these, XRE2-6 were previously found to serve an important role in the TCDD-induced upregulation of CYP1B1 expression in human cancer cell lines, including HepG2, MCF-7 (breast), LS-180 (colon), OMC-3 (ovarian) and SCC12 (squamous carcinoma) cells (18,24). However, analysis in the present study revealed that this enhanced inducibility could not be explained by the methylation status of the XRE4, XRE5 or XRE6 sequences, because these XREs were mostly unmethylated prior to DAC treatment. By contrast, DAC treatment did not enhance CYP1B1 expression in HuH7 cells, where the XRE7 and XRE8 sequences were either highly or fully methylated. Therefore, AhR binding to XRE7 or XRE8 likely exerted minimal contributions to the βNF-induced CYP1B1 expression in both cell lines. In the present study, the XRE1 sequence was found to be fully methylated in the HepG2 cells but not in HuH7 cells. The binding of XRE1 is likely to be less important for the βNF-activated AhR pathway because the XRE1 sequence could not be amplified in the chromatin immunoprecipitates isolated from both cell lines (data not shown). Therefore, the XRE2 and XRE3 sequences were focused on for the evaluation of their binding to AhR. In addition, the partially methylated status of the XRE2/XRE3 sequences in the HepG2 cells rendered them suitable for determining whether AhR preferentially binds to unmethylated or methylated XREs.
Previous studies have reported the possibility of AhR preferentially binding to unmethylated CYP1B1 XRE sequences instead of their methylated counterparts. For example, TCDD-induced upregulation of CYP1B1 expression was found to be markedly enhanced after DAC treatment in HepG2 cells compared with that in MCF-7 cells, due to the presence of higher levels of methylated XRE sequences in HepG2 cells than those in MCF-7 cells (11). In addition, other studies have demonstrated selective AhR binding to unmethylated XRE sequences using reporter and gel shift assays (25,26). However, these previous findings were obtained under artificial conditions, which were generally not influenced by other factors and may not truly reflect the function of AhR in the intracellular physiological chromatin context of the cells. To date, no direct evidence has been provided for the selective binding of activated AhR to unmethylated XRE sequences under physiological intracellular conditions after the cells were exposed to ligands.
In the present study, AhR-bound XRE fragments were isolated from βNF-induced cells by immunoprecipitation using an anti-AhR antibody before the methylation status of these XRE sequences were assessed. Through COBRA, no detectable methylation could be observed in the AhR-bound XRE2/XRE3 sequences, whereas a partial methylation pattern was observed in the corresponding input DNA derived from HepG2 cells. This result was consistent with that found by a more detailed sequencing analysis evaluating individual copies of the CYP1B1 gene in the present study. The CpG sites within the XRE2 and XRE3 sequences of HepG2 cells were always unmethylated in the clones derived from AhR-bound XRE fragments whereas the surrounding CpG sites were frequently methylated. By contrast, DAC treatment showed no effect on βNF-induced CYP1B1 expression in HuH7 cells, where the XRE2 and XRE3 sequences were fully unmethylated. These results suggest that methylation of the two CpG sites in XRE2 and XRE3 prevents binding of the AhR/Arnt heterodimer. XRE3 has been previously reported to be the most important site for enhancing TCDD-induced CYP1B1 expression (18,23). In the present study, both XRE2 and XRE3 sites were unmethylated in all clones examined following AhR immunoprecipitation. However, binding of AhR to the two XRE sequences could not be assessed separately due to the proximity of XRE2 and XRE3 to each other on the sequence. Therefore, the dominant XREs in terms of AhR binding and βNF-mediated AhR activation remain unclear at present.
To conclude, data from the present study suggested that methylation at the XRE2 and XRE3 sequences prevented the binding of AhR, which affect the efficacy of the XRE sequence in mediating the induction of CYP1B1 expression following βNF exposure. To the best of our knowledge, this is the first study to provide direct evidence that ligand-activated AhR complexes can preferentially bind unmethylated XRE sequences in a physiological chromatin context. In addition, the COBRA method applied in the present study was easily reproducible in determining the methylation status of XRE sequences. Therefore, the combination of ChIP and COBRA is advised for the future assessment of other XRE sequences located outside the CYP1B1 gene.
Similar to the AhR/XRE pathway, the activity of other transcription factors can also be regulated by the methylation of target DNA sequences. The CCCTC-binding factor exclusively binds to unmethylated target sequences and functions as an insulator to control the imprinting of insulin-like growth factor 2 and H19 genes (27). By contrast, methyl-CpG-binding protein 2 specifically binds to methylated CpG sites and negatively regulates gene transcription (7,28). The novel approach in the present study can also be applied to assess the effects of DNA methylation on the binding of transcription factors other than AhR. Therefore, this technique can be a powerful tool for identifying the unknown functions of DNA-binding proteins that are strictly regulated by the methylation status of their target genes.
Acknowledgements
Not applicable.
Funding
The present study was supported by a Grant-in-Aid for Scientific Research type (C) from the Japan Society for the Promotion of Science (JSPS KAKENHI; grant no. 19K07200).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
TM and WH contributed to the study conception and design. TM and RO performed the experiments. TM, JT, SO and WH contributed to the data analysis and interpretation. TM and WH confirm the authenticity of all the raw data. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
1
|
Zanger UM, Turpeinen M, Klein K and Schwab M: Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal Bioanal Chem. 392:1093–1108. 2008.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Pelkonen O, Turpeinen M, Hakkola J, Honkakoski P, Hukkanen J and Raunio H: Inhibition and induction of human cytochrome P450 enzymes: Current status. Arch Toxicol. 82:667–715. 2008.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wolf CR, Mahmood A, Henderson CJ, McLeod R, Manson MM, Neal GE and Hayes JD: Modulation of the cytochrome P450 system as a mechanism of chemoprotection. IARC Sci Publ. 139:165–173. 1996.PubMed/NCBI
|
|
4
|
Kress S and Greenlee WF: Cell-specific regulation of human CYP1A1 and CYP1B1 genes. Cancer Res. 57:1264–1269. 1997.PubMed/NCBI
|
|
5
|
Krusekopf S, Roots I, Hildebrandt AG and Kleeberg U: Time-dependent transcriptional induction of CYP1A1, CYP1A2 and CYP1B1 mRNAs by H+/K+ -ATPase inhibitors and other xenobiotics. Xenobiotica. 33:107–118. 2003.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Fujii-Kuriyama Y and Mimura J: Molecular mechanisms of AhR functions in the regulation of cytochrome P450 genes. Biochem Biophys Res Commun. 338:311–317. 2005.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Lund AH and van Lohuizen M: Epigenetics and cancer. Genes Dev. 18:2315–2335. 2004.PubMed/NCBI View Article : Google Scholar
|
|
8
|
University of California: Santa Cruz Genomics Institute. UCSC Genome Browser. Available from: http://genome.ucsc.edu/index.html.
|
|
9
|
Ivanov M, Kacevska M and Ingelman-Sundberg M: Epigenomics and interindividual differences in drug response. Clin Pharmacol Ther. 92:727–736. 2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Fisel P, Schaeffeler E and Schwab M: DNA methylation of ADME genes. Clin Pharmacol Ther. 99:512–527. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Beedanagari SR, Taylor RT, Bui P, Wang F, Nickerson DW and Hankinson O: Role of epigenetic mechanisms in differential regulation of the dioxin-inducible human CYP1A1 and CYP1B1 genes. Mol Pharmacol. 78:608–616. 2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Nakajima M, Iwanari M and Yokoi T: Effects of histone deacetylation and DNA methylation on the constitutive and TCDD-inducible expressions of the human CYP1 family in MCF-7 and HeLa cells. Toxicol Lett. 144:247–256. 2003.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yao EF and Denison MS: DNA sequence determinants for binding of transformed Ah receptor to a dioxin-responsive enhancer. Biochemistry. 31:5060–5067. 1992.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Swanson HI, Tullis K and Denison MS: Binding of transformed Ah receptor complex to a dioxin responsive transcriptional enhancer: Evidence for two distinct heteromeric DNA-binding forms. Biochemistry. 32:12841–12849. 1993.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Skene PJ and Henikoff S: An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife. 6(e21856)2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Nishimura M, Ueda N and Naito S: Effects of dimethyl sulfoxide on the gene induction of cytochrome P450 isoforms, UGT-dependent glucuronosyl transferase isoforms, and ABCB1 in primary culture of human hepatocytes. Biol Pharm Bull. 26:1052–1056. 2003.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Habano W, Gamo T, Sugai T, Otsuka K, Wakabayashi G and Ozawa S: CYP1B1, but not CYP1A1, is downregulated by promoter methylation in colorectal cancers. Int J Oncol. 34:1085–1091. 2009.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Tsuchiya Y, Nakajima M and Yokoi T: Critical enhancer region to which AhR/ARNT and Sp1 bind in the human CYP1B1 gene. J Biochem. 133:583–592. 2003.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Sambrook J and Russell DW: Isolation of high-molecular-weight DNA from mammalian cells using proteinase K and phenol. In: Molecular Cloning: A Laboratory Manual. 3rd edition. Cold Spring Harbor Laboratory Press, New York, NY, pp6.4-6.12, 2001.
|
|
20
|
Xiong Z and Laird PW: COBRA: A sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 25:2532–2534. 1997.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Warnecke PM, Stirzaker C, Song J, Grunau C, Melki JR and Clark SJ: Identification and resolution of artifacts in bisulfite sequencing. Methods. 27:101–107. 2002.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Habano W, Gamo T, Terashima J, Sugai T, Otsuka K, Wakabayashi G and Ozawa S: Involvement of promoter methylation in the regulation of Pregnane X receptor in colon cancer cells. BMC Cancer. 11(81)2011.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Shehin SE, Stephenson RO and Greenlee WF: Transcriptional regulation of the human CYP1B1 gene. Evidence for involvement of an aryl hydrocarbon receptor response element in constitutive expression. J Biol Chem. 275:6770–6776. 2000.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Tang YM, Wo YY, Stewart J, Hawkins AL, Griffin CA, Sutter TR and Greenlee WF: Isolation and characterization of the human cytochrome P450 CYP1B1 gene. J Biol Chem. 271:28324–28330. 1996.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Shen ES and Whitlock JP Jr: The potential role of DNA methylation in the response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Biol Chem. 264:17754–17758. 1989.PubMed/NCBI
|
|
26
|
Takahashi Y, Suzuki C and Kamataki T: Silencing of CYP1A1 expression in rabbits by DNA methylation. Biochem Biophys Res Commun. 247:383–386. 1998.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wolffe AP: Transcriptional control: Imprinting insulation. Curr Biol. 10:R463–R465. 2000.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Siegfried Z and Cedar H: DNA methylation: A molecular lock. Curr Biol. 7:R305–R307. 1997.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Chang H, Chang LW, Cheng YH, Tsai WT, Tsai MX and Lin P: Preferential induction of CYP1A1 and CYP1B1 in CCSP-positive cells. Toxicol Sci. 89:205–213. 2006.PubMed/NCBI View Article : Google Scholar
|














