Introduction
Chronic obstructive pulmonary disease (COPD) is a
common, preventable and treatable disease characterized by
persistent respiratory symptoms and progressive airflow obstruction
documented on spirometry; it is associated with an abnormal
inflammatory response of the lungs to noxious particles or gases
(1). COPD affects ~400 million
individuals and is already the third leading cause of death
worldwide, which the World Health Organization predicted would not
occur until 2030(1). Pulmonary
emphysema is one of the most important pathological manifestations
of COPD, and the pathogenesis of pulmonary emphysema includes
non-specific inflammation propagated by cigarette smoke injury,
increased oxidative stress, imbalanced proteases/anti-proteases and
subsequent alveolar cell apoptosis, which results in the
histological hallmark of enlarged alveolar spaces (2).
Necroptosis is a mechanism of genetically programmed
lytic cell death that is considered to serve a role in the
destruction of pathogen-infected cells and/or dysfunctional cells
during certain degenerative or inflammatory disorders. Necroptosis
can be triggered by multiple innate immune signaling pathways that
lead to the phosphorylation and activation of the necroptotic
kinase, receptor-interacting serine/threonine-protein kinase
(RIPK3). RIPK3 activates the mixed lineage kinase domain-like
protein (MLKL) via phosphorylation, which causes substantial
conformational changes that enables the trafficking of MLKL to the
plasma membrane, where it induces membrane permeabilization
(3). It has previously been
reported that that necroptosis may contribute to the development of
pulmonary emphysema (4). However,
the exact molecular mechanism remains largely unknown. Therefore,
studying the role of necroptosis in the pathogenesis of pulmonary
emphysema is of vital importance.
Black tea, one of the most common types of tea, is
made of fresh green tea leaves fermented and oxidized by polyphenol
oxidase. This process converts a large proportion of catechins into
theaflavins and thearubigins (5).
The theaflavins of black tea include theaflavin (TF-1),
theaflavin-3-gallate (TF-2a), theaflavin-3'-gallate (TF-2b) and
theaflavin-3,3'-digallate (TF-3). A previous study has demonstrated
that theaflavins, especially TF-3, attenuate oxidative stress and
inflammation (6). TF-3 also has
been indicated to decrease the incidence of coronary heart disease,
exhibit an advantageous effect on the bone mineral density and the
potential to prevent cancer (7-9).
However, the effect of TF-3 in protecting against cigarette smoke
extract (CSE)-induced pulmonary emphysema and the mechanism of
action remains unclear. It can therefore be hypothesized that
drinking black tea may prevent CSE-induced pulmonary emphysema. The
aim of the present study was to investigate the protective effect
of TF-3 on pulmonary emphysema in CSE-treated mice via the
necroptotic signaling pathway.
Materials and methods
Ethics statement
All animal procedures and experiments were approved
by the Institutional Animal Care and Use Committee of Shanghai Jiao
Tong University (Shanghai, China). The experiments were performed
following the Guide for the Care and Use of Laboratory Animals
published by the National Institutes of Health, 8th edition
(10).
Drugs and antibodies
TF-3,
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (z-VAD),
necrostatin-1 and SB203580 were purchased from MedChemExpress. All
antibodies were purchased from Cell Signaling Technology, Inc. The
following primary antibodies were used: phosphorylated (p)-RIPK3
(1:1,000; cat. no. 93654), RIPK3 (1:1,000; cat. no. 13526), p-MLKL
(1:1,000; cat. no. 91689), MLKL (1:1,000; cat. no. 14993), p-p38
MAPK (1:1,000; cat. no. 9211), p38 MAPK (1:1,000; cat. no. 9212),
caspase-3 (1:1,000; cat. no. 9662) and GAPDH (1:1,000; cat. no.
2118). The secondary antibody used was HRP-conjugated goat
anti-rabbit IgG (1:10,000; cat. no. 7074). Lipopolysaccharide
(Escherichia coli 055:B5) was purchased from Sigma-Aldrich
(Merck KGaA).
CSE preparation
The CSE was prepared using a previously described
technique (11). Briefly, a
cigarette (Marlboro; Philip Morris Products S.A.) was burned and
the smoke was passed through a 0.22 µm filter (MilliporeSigma) to
remove particles and bacteria before being added to a vessel
containing PBS (2 ml per cigarette for mouse experiments) or DMEM
(Gibco; Thermo Fisher Scientific, Inc.; 10 ml per cigarette as 100%
CSE used for cell experiments) using a vacuum pump. The
reproducibility of the extract was assessed using an AB Sciex 3200
Qtrap mass spectrometer (SCIEX) connected to an Agilent 1200 Series
high-performance liquid chromatography (Agilent Technologies,
Inc.). The following conditions were used: Chromatographic column,
Diamonsil-C18 (length, 150 mm; inner diameter, 4.6 mm; particle
size, 5 µm; Dikma Technologies, Inc.); flow rate, 0.6 ml/min;
mobile phase, phosphoric acid-citric acid buffer containing 30%
(v/v) acetonitrile [30 mM K2HPO4, 30 mM
citric acid, 0.5% (v/v) triethylamine, adjusted to pH 6.7 with 10 M
NaOH]; column temperature, 25˚C; sample quantity, 20 µl; internal
standard, the nicotine standard (cat. no. N0267; Sigma-Aldrich;
Merck KGaA) was dissolved in methanol to prepare the experimental
standard (10 mg/ml). The ionization mode was positive ion mode, the
source temperature was 700˚C and the nebulizer pressure was 50 psi.
The pH of the CSE-PBS/DMEM solution was 7.2-7.4 and the solution
was freshly prepared for each experiment.
Cell culture and treatment
The human normal lung epithelial BEAS-2B cell line
was purchased from the American Type Culture Collection (cat. no.
CRL-9609™) and maintained in DMEM supplemented with 10% FBS (cat.
no. 10270-106; Gibco; Thermo Fisher Scientific, Inc.) and 1%
vol/vol streptomycin/penicillin (Gibco; Thermo Fisher Scientific,
Inc.) in an incubator at 37˚C with 5% CO2. BEAS-2B cells
were pretreated with TF-3 (5-20 µM) for 24 h at 37˚C before the CSE
challenge. The pan-caspase inhibitor z-VAD (20 µM), the necroptosis
inhibitor necrostatin-1 (20 µM) or the p38 MAPK inhibitor SB203580
(0.5 µM) were preincubated with the cells for 1 h at 37˚C before
the CSE challenge to stimulate necroptosis, inhibit necroptosis or
inhibit p38 MAPK, respectively. For all drug treatments, cells were
maintained in DMEM supplemented with 0.2% FBS at 37˚C. The dose and
treatment duration of all drugs were selected according to previous
studies (7,12-14).
Cell viability
Cell viability was determined using the Cell
Counting Kit-8 (CCK-8; Dojindo Laboratories, Inc.) assay. In brief,
BEAS-2B cells (2.5x104 cells/ml) were seeded into
96-well plates and incubated overnight at 37˚C. The cells were
treated with CSE (1, 5, 10, 20 or 50%) alone, TF-3 (5, 10 or 20 µM)
alone or CSE (10%) in combination with TF-3 (5, 10 or 20 µM).
Following stimulation, 10 µl of CCK-8 reagent was added to each
well and then incubated for 2 h at 37˚C. The absorbance of each
well was measured at 450 nm using a microplate reader (Molecular
Devices, LLC). The viability of the treated cells was calculated as
follows: (absorbance450 nm of the therapeutic
group/absorbance450 nm of the control group) x100%.
LDH assay
Lactate dehydrogenase was determined using an LDH
Assay Kit (Beyotime Institute of Biotechnology). In brief, BEAS-2B
cells (~80% cell confluence) treated with CSE (10%) and/or TF-3 (20
µM) were incubated with 10 µl LDH releasing reagent for 1 h at
37˚C. The samples were centrifuged (400 x g; 5 min; 25˚C) and the
LDH production levels of the supernatant were detected according to
the manufacturer's instructions.
ROS generation assay
ROS generation was determined using a ROS Assay Kit
(Beyotime Institute of Biotechnology). In brief, BEAS-2B cells
(~80% cell confluence) treated with CSE (10%) and/or TF-3 (20 µM)
were incubated with 10 µM 2',7'-dichlorodihydrofluorescein
diacetate for 30 min at 37˚C in the dark. The green fluorescence
intensity was determined using a fluorescence microscope (Leica
Microsystems GmbH; magnification, x200) and analyzed using ImageJ
v1.8.0 software (National Institutes of Health).
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
The mRNA expression levels of proinflammatory
cytokines were determined using RT-qPCR. In brief, BEAS-2B cells
treated with CSE (10%) and/or TF-3 (20 µM) and were lysed with
TRIzol® reagent (Thermo Fisher Scientific, Inc.). mRNA
was quantified using an ultraviolet spectrophotometer and
subsequently, 1 µg of RNA was reverse transcribed into cDNA using a
HiScript® II Q RT SuperMix kit (Vazyme Biotech Co.,
Ltd.) at 50˚C for 15 min and then at 85˚C for 5 sec. qPCR was
performed using ChamQ Universal SYBR qPCR Master Mix (Vazyme
Biotech Co., Ltd.) and a deep-well qPCR detection system
(LightCycler 480; Roche Diagnostics) using the following
thermocycling conditions: Initial denaturation at 95˚C for 5 min;
followed by 35 cycles of denaturation at 95˚C for 60 sec, annealing
at 58˚C for 60 sec and extension at 72˚C for 50 sec. The primers
used for qPCR were as follows: IL-6 forward (F),
5'-CACTGGTCTTTTGGAGTTTGAG-3' and reverse (R),
5'-GGACTTTTGTACTCATCTGCAC-3'; IL-1β F, 5'-AGCTACGAATCTCCGACCAC-3'
and R, 5'CGTTATCCCATGTGTCGAAGAA-3'; TNF-α F,
5'-GAGGCCAAGCCCTGGTATG-3' and R, 5'-CGGGCCGATTGATCTCAGC-3'; and
GAPDH F, 5'-ACAACTTTGGTATCGTGGAAGG-3' and R,
5'-GCCATCACGCCACAGTTTC-3'. GAPDH was used as the internal reference
gene. Quantification of mRNA expression levels was performed using
the 2-IICq method (15).
These data are presented as the relative mRNA expression level
compared to that in control group, which was arbitrarily set to
1.
Immunofluorescence microscopy for
PI/DAPI staining
Necroptotic cells were identified using
immunofluorescence. In brief, BEAS-2B cells were treated with CSE
(10%)/z-VAD (20 µM) with either TF-3 (20 µM) or necrostatin-1 (20
µM), stained with PI for 30 min at 25˚C in the dark (5 µg/ml; BD
Biosciences), fixed in 3.7% paraformaldehyde for 10 min at 25˚C and
stained with DAPI (Sigma-Aldrich; Merck KGaA) for 5 min at 25˚C in
the dark. Immunofluorescence images were captured using a
fluorescence microscope (Leica Microsystems GmbH). For each
experiment, 100 cells were monitored with a fluorescence microscope
and analyzed using ImageJ v1.8.0 software (National Institutes of
Health).
Apoptosis assay
The necroptotic rate was determined using the
Annexin V FITC Apoptosis Detection Kit I (BD Biosciences). In
brief, BEAS-2B cells were treated with CSE (10%)/z-VAD (20 µM) with
either TF-3 (20 µM) or necrostatin-1 (20 µM), washed twice with
cold PBS and then resuspended in 1X binding buffer at a
concentration of 1x106 cells/ml. The solution was
subsequently transferred to a 5 ml culture tube. A total of 5 µl
FITC Annexin V and 5 µl PI were added, the cells were gently
vortexed, and the samples were incubated for 15 min at room
temperature (25˚C) in the dark. Cells were then diluted in 1X
binding buffer (400 ml) and analyzed using flow cytometry (BD
FACSCelesta; BD Biosciences) and FlowJo software (v10.8.0; FlowJo
LLC).
Western blotting
Protein expression levels of the relevant proteins
were determined via western blotting. In brief, BEAS-2B cells
treated with CSE (10%)/z-VAD (20 µM) with either TF-3 (5-20 µM) or
SB203580 (0.5 µM), were lysed in RIPA lysis buffer (Epizyme, Inc.)
supplemented with a protease inhibitor and phosphatase inhibitor
cocktail (Epizyme, Inc.). After the protein concentration was
measured using a BCA protein assay kit (Epizyme, Inc.) and
verified, the sample was mixed with 5X SDS loading buffer. Total
protein was separated using SDS-PAGE (20 µg protein/well) on an
8-12% gel. Separated proteins were subsequently transferred to
polyvinylidene difluoride membrane (MilliporeSigma), which were
blocked with 5% BSA (Sigma-Aldrich; Merck KGaA) in TBS with 0.1%
Tween-20 (TBST) for 1 h at room temperature (25˚C). The membranes
were immunoblotted with primary antibodies overnight at 4˚C. After
washing with TBST three times, the membranes were incubated with
secondary antibody for 1 h at room temperature (25˚C) and were then
washed again with TBST three times. The membranes were scanned
using Immobilon Western Chemiluminescent HRP Substrate
(MilliporeSigma) and an imaging system (Amersham Imager 600;
Cytiva). ImageJ v1.8.0 software (National Institutes of Health) was
used for densitometry. GAPDH was used as the loading control. The
data are presented as the relative protein expression level
compared to the control group, which was arbitrarily set to 1.
Experimental animal protocols
All studies involving animals were performed in
compliance with the ARRIVE guidelines (16). All mice were housed at a constant
room temperature of 22-23˚C and humidity of 50-60% with an
alternating 12 h light/dark cycle and free access to water and
standard food. Twenty male C57BL/6J mice maintained in the animal
facility (20-25 g body weight; specific pathogen free) of Shanghai
Jiao Tong University were randomly selected and divided into the
following four groups at 7 weeks of age: i) Control; ii) TF-3; iii)
CSE; and iv) CSE + TF-3 (n=5 each group). The total experimental
period was four weeks. The control group was intraperitoneally
(i.p.) injected with vehicle (normal saline), the TF-3 group was
given TF-3 (5 mg/kg body weight) by oral gavage daily for 28 days
and the CSE group was injected with 0.3 ml of CSE/PBS on day 0, 11
and 22. The CSE + TF-3 group was i.p. injected with 0.3 ml of CSE
on day 0, 11 and 22 along with TF-3 (5 mg/kg body weight). The
doses and administration routes of CSE and TF-3 were based on
previous studies (6,11,17).
ELISA assays
TNF-α (cat. no. DY410; Mouse TNF-alpha DuoSet ELISA)
and IL-1β (cat. no. DY401; Mouse IL-1 beta/IL-1F2 DuoSet ELISA)
levels in the lung tissue were measured with commercially available
ELISA kits (Novus Biologicals, LLC) according to the manufacturer's
instructions.
Malondialdehyde (MDA), superoxide
dismutase (SOD) and glutathione (GSH) assays
The lung homogenate was dissolved in extraction
buffer (cell lysis buffer; Beyotime Institute of Biotechnology) to
detect MDA (cat. no. A003-1-2; MDA Assay Kit), SOD (cat. no.
A001-3-2; SOD Assay Kit) and GSH (cat. no. A006-2-1; GSH Assay Kit)
levels using commercially available assay kits (Nanjing Jiancheng
Bioengineering Institute), according to the manufacturer's
instructions.
H&E staining
Mice were anesthetized by inhalation of isoflurane
(4%) and euthanized by exsanguination via right ventricle
aspiration, followed by cervical dislocation. The left lungs of the
mice were harvested and fixed with 4% paraformaldehyde at 25˚C for
24 h. After fixation, lung tissue was embedded in paraffin and
sectioned at 5-µm thickness. The sections were stained with
hematoxylin at 25˚C for 10 min and eosin at 25˚C for 3 min and
visualized under a light microscope (magnification, x400).
Emphysema was quantified by measuring the mean linear intercept and
determining the destructive index (11).
Statistical analysis
All the data were normally distributed and are
presented as the mean ± SEM of three independent cell experiments
or five independent animal experiments. Statistical analyses were
performed using GraphPad Prism v8 software (GraphPad Software,
Inc.). One-way ANOVA was performed followed by Tukey's post hoc
test for comparisons among multiple groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
TF-3 prevents CSE-induced cytotoxicity
in BEAS-2B cells
In order to explore the cytotoxic effect of CSE and
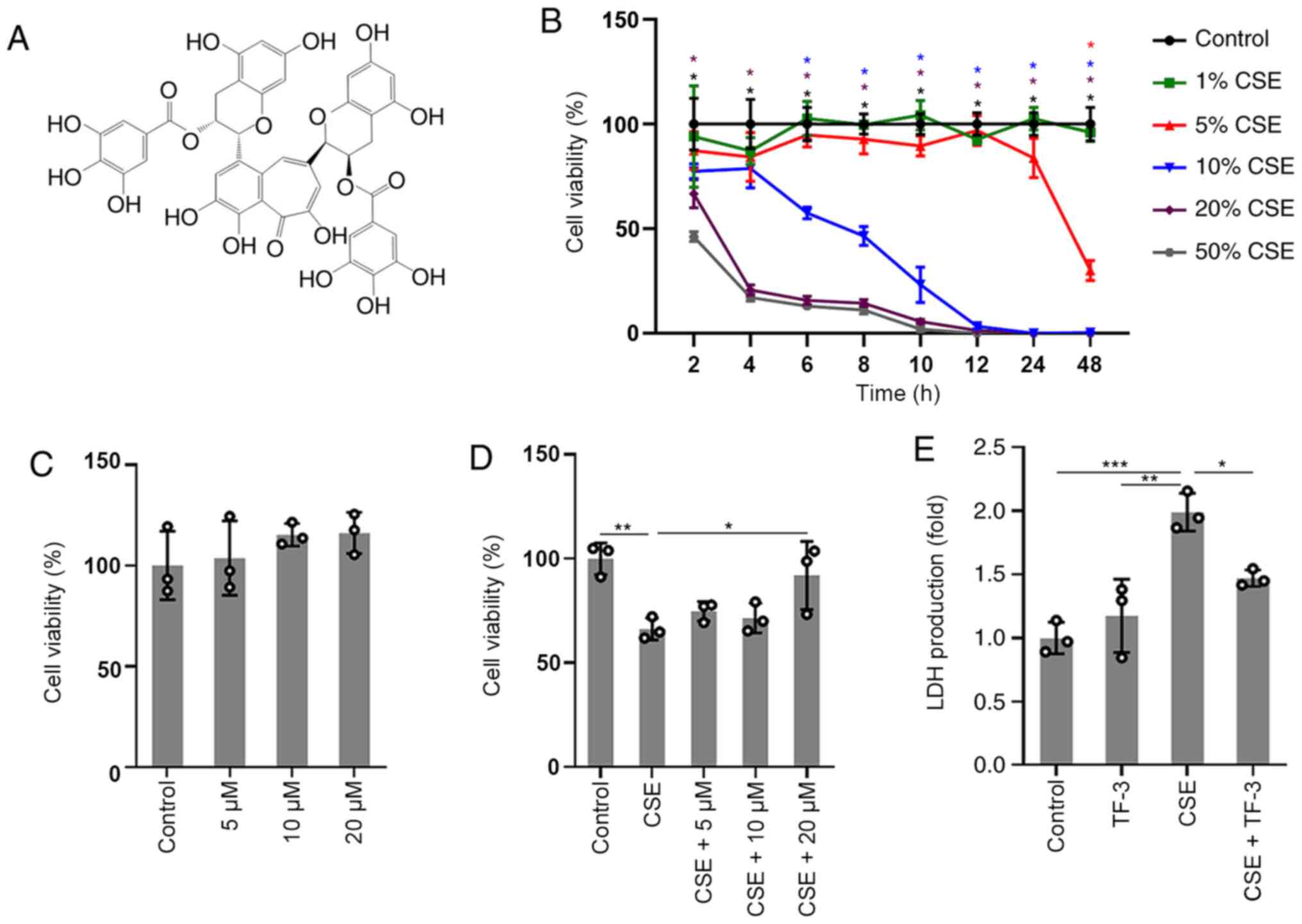
the protective effect of TF-3 (Fig.
1A) on BEAS-2B cells, CCK-8 assay was used to detect cell
viability. The results of the concentration/time-response study, in
which BEAS-2B cells were subjected to different concentrations of
CSE (1-50%) for up to 48 h, are displayed in (Fig. 1B). The viability of BEAS-2B cells
treated with 1 or 5% CSE was not significantly different compared
with the control cells before 24 h. However, significant decreases
in cell viability were observed at 20 or 50% CSE exposure compared
with the control. Gradual decreases in cell viability were observed
after exposure to 10% CSE and cell viability was close to 58±3% at
6 h. Therefore, cells were treated with 10% CSE for 6 h or vehicle
as a control. No significant cytotoxicity of TF-3 (5-20 µM) was
observed, as displayed in (Fig.
1C). The results in (Fig. 1D)
demonstrated that TF-3 significantly reversed the decrease in
viability of the CSE-induced BEAS-2B cells compared with the CSE
group. Moreover, the membrane integrity of the cells was evaluated
via the LDH assay. TF-3 significantly reduced the production of LDH
released into the medium (Fig. 1E)
in CSE-induced BEAS-2B cells compared with the CSE group.
TF-3 reduces ROS generation and mRNA
expression levels of TNF-α, IL-6 and IL-1β in CSE-treated BEAS-2B
cells
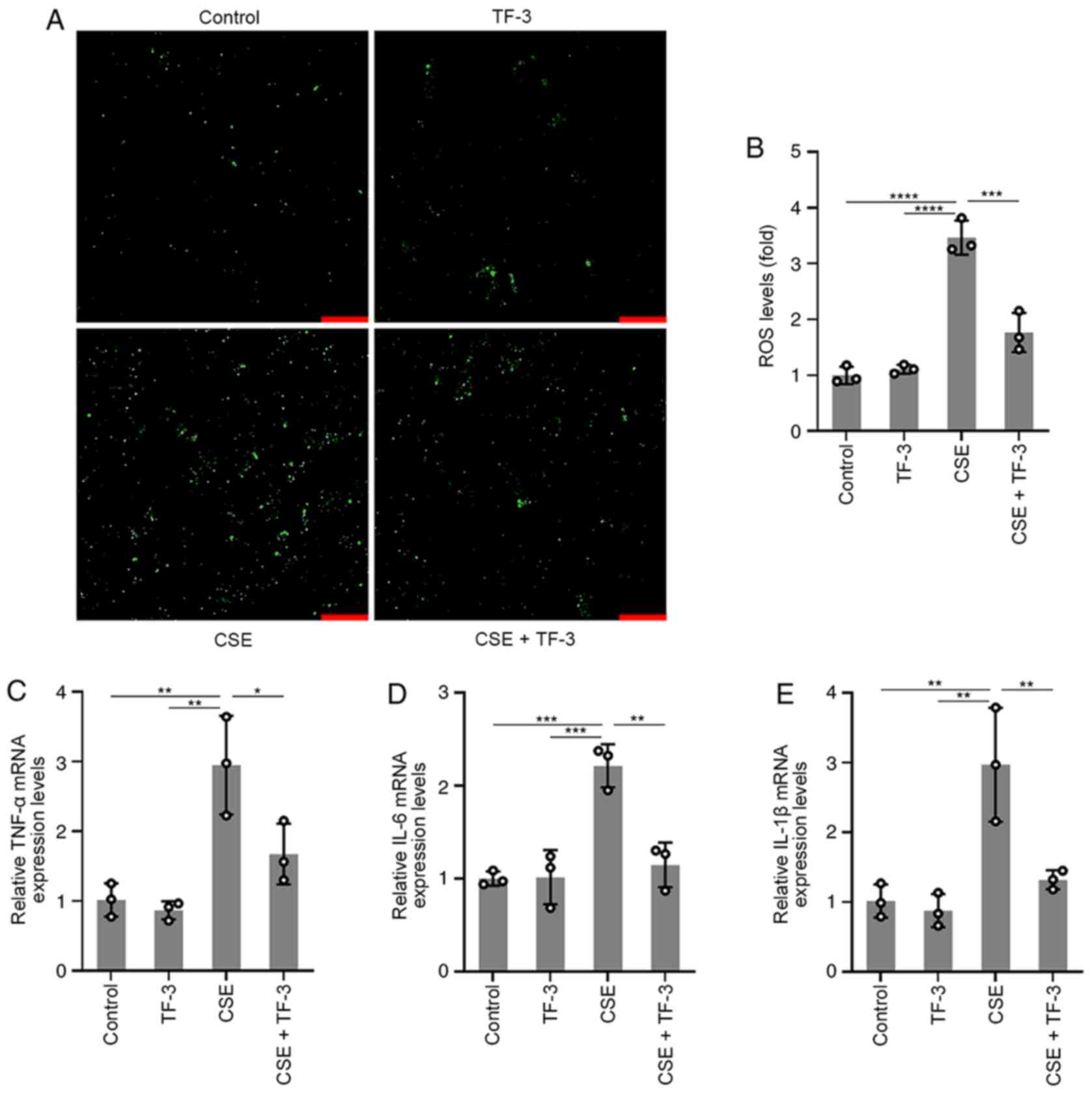
To further understand the role of TF-3 in ROS and
inflammatory cytokine generation, BEAS-2B cells were pretreated
with TF-3 (20 µM) for 24 h prior to CSE challenge. The results
displayed in (Fig. 2A and B) demonstrated that CSE-induced ROS
generation was significantly suppressed by TF-3 compared with the
CSE group, whereas TF-3 treatment on healthy cells did not affect
ROS generation compared with the control. The mRNA expression
levels of TNF-α, IL-6 and IL-1β were detected using RT-qPCR. As
displayed in (Fig. 2C-E), the mRNA
expression levels of TNF-α, IL-1β and IL-6 in CSE-induced BEAS-2B
cells were significantly diminished by TF-3 treatment compared with
the CSE only group. These results indicated that TF-3 may serve a
crucial role in antioxidation and anti-inflammation.
TF-3 inhibits nonapoptotic cell death
in CSE/z-VAD-treated BEAS-2B cells
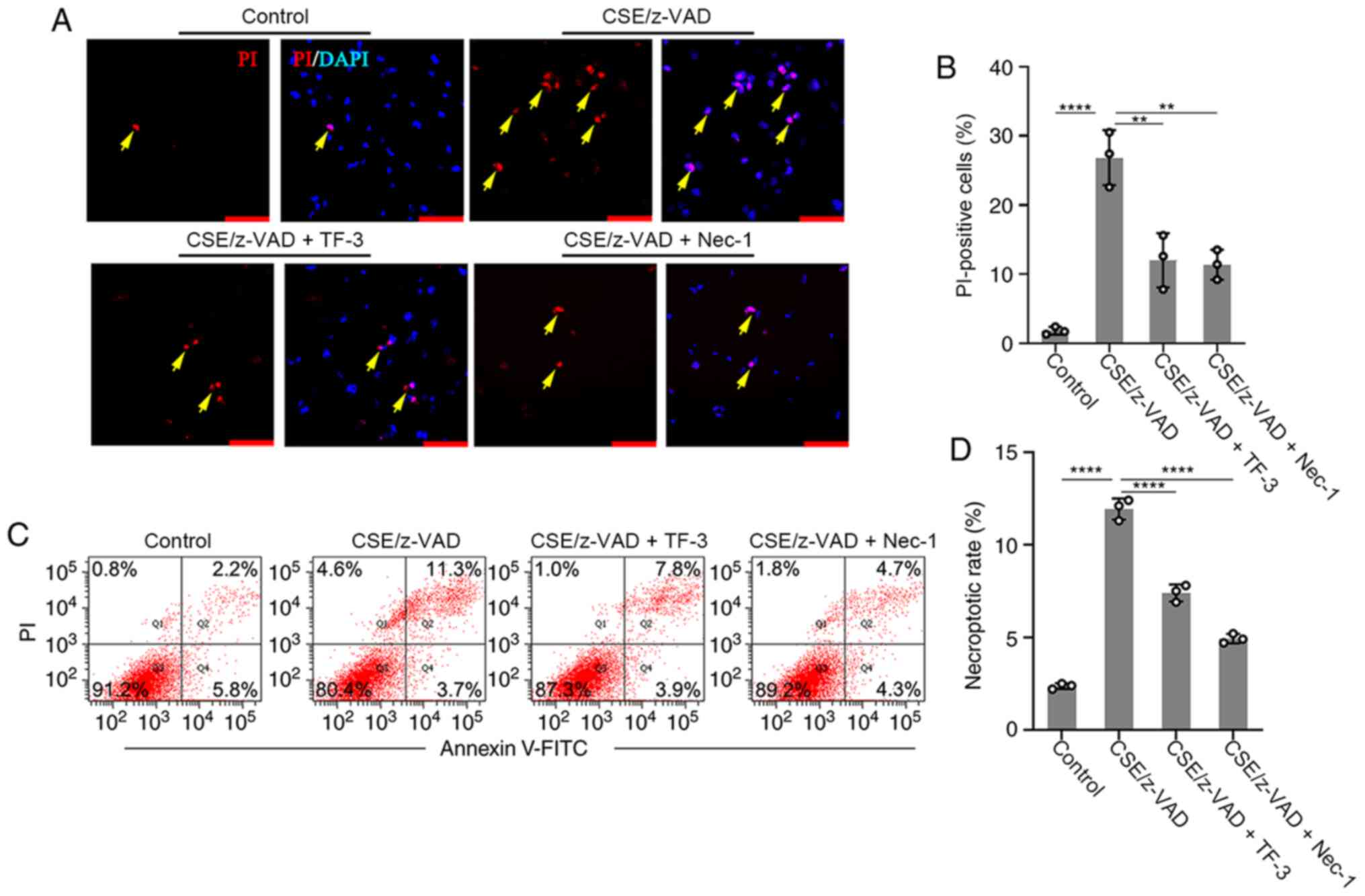
The effect of TF-3 on necroptosis was examined. The
necroptotic rate was determined using immunofluorescence and flow
cytometry. Necrostatin-1, an inhibitor of necroptosis, was used to
examine whether CSE/z-VAD induced apoptosis. The positive rate of
PI was significantly increased following necroptotic stimulation in
the immunofluorescence assay compared with the control group,
whereas PI-positive cells were significantly decreased by
Necrostatin-1 compared with the CSE/z-VAD only group, which
indicated that CSE/z-VAD induced necroptosis in BEAS-2B cells.
Furthermore, TF-3 decreased the number of PI-positive cells in
CSE/z-VAD-treated cells compared with the CSE/z-VAD only group
(Fig. 3A and B). Following necroptotic stimulation, the
necrotic or late apoptotic (Annexin V+/PI+ in Q2) rates were
increased. By contrast, no obvious elevation of early apoptotic
(Annexin V+/PI− in Q4) rates was observed. Similar effects of
necrostatin-1 and TF-3 were observed in the flow cytometry assay,
and they both reduced the rates of cells in the area Q2 compared
with the CSE/z-VAD only group, which indicated that TF-3 decreased
the necroptotic rates of CSE/z-VAD-treated cells (Fig. 3C and D). To further exclude the role of TF-3 in
necroptosis induced by CSE/z-VAD, caspase-3 protein expression
levels were examined. CSE/z-VAD treatment did not significantly
affect caspase-3 activities in BEAS-2B cells (Fig. 4A and E). Furthermore, no cleavage of caspase-3
was detected via western blotting analysis (Fig. 4A) indicating that apoptosis was not
responsible for CSE/z-VAD-induced cell death in BEAS-2B cells.
These results suggested that TF-3 may inhibit CSE/z-VAD-induced
nonapoptotic cell death in BEAS-2B cells.
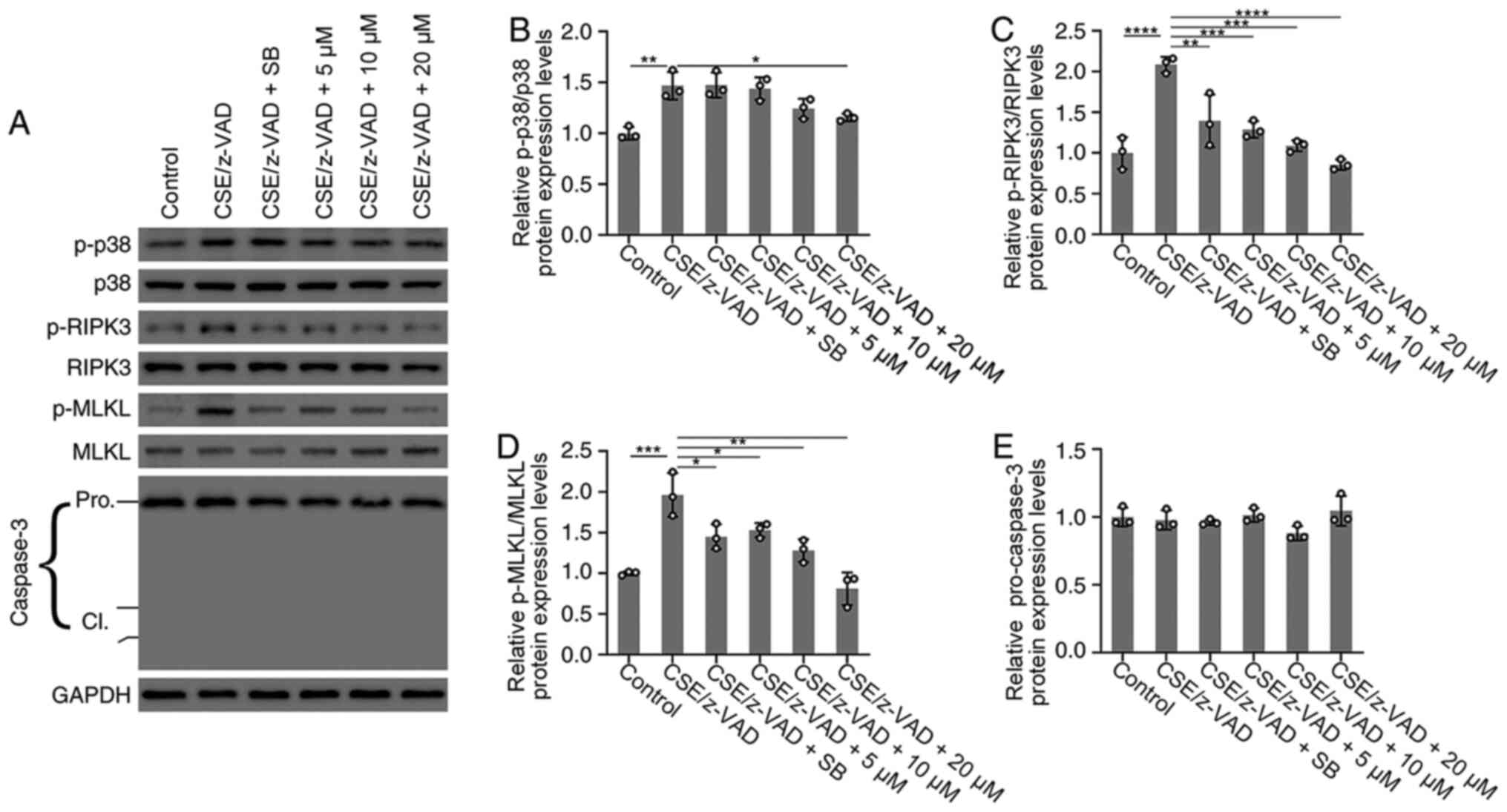 | Figure 4Effects of TF-3 on the protein
expression levels of p38 MAPK, RIPK3, MLKL and caspase-3. BEAS-2B
cells were treated with CSE (10%)/z-VAD (20 µM) alone or in
combination with either TF-3 (5-20 µM) or SB203580 (0.5 µM). (A)
Protein expression levels of p-p38 MAPK/p38 MAPK, p-RIPK3/RIPK3,
p-MLKL/MLKL and caspase-3 were determined via western blotting with
GAPDH as an internal control. Semi-quantification of the protein
expression levels of (B) p-p38 MAPK, (C) p-RIPK3, (D) p-MLKL and
(E) caspase-3. Data are presented as the mean ± SEM of three
independent experiments indicated by dots. One-way ANOVA was
performed followed by Tukey's post hoc test for statistical
comparisons among more than two groups. *P<0.05,
**P<0.01, ***P<0.001 and
****P<0.0001. CSE, cigarette smoke extract; TF-3,
theaflavin-3,3'-digallate; RIPK3, receptor-interacting
serine/threonine-protein kinase 3; MLKL, mixed lineage kinase
domain-like; p, phosphorylated; cl, cleaved. |
TF-3 suppresses necroptosis via the
p38 MAPK/RIPK3/MLKL signaling pathway in CSE/z-VAD-induced BEAS-2B
cells
To investigate the effect of TF-3 on necroptosis at
the protein expression level of RIPK3 and MLKL, these key mediators
of the necroptosis pathway were investigated via western blotting.
As demonstrated in (Fig. 4A,
C and D), western blotting determined that the
protein expression levels of p-RIPK3 and p-MLKL were significantly
increased following CSE/z-VAD treatment compared with the control.
However, TF-3 treatment significantly decreased the protein
expression levels of p-RIPK3 and p-MLKL compared with the CSE/z-VAD
group. Furthermore, the protein expression levels of p38 MAPK were
examined. Western blotting demonstrated that TF-3 (20 µM)
significantly reduced the protein expression levels of p-p38 MAPK
compared with the CSE/z-VAD group (Fig.
4A and B). Furthermore, the
results demonstrated that SB203580, a specific inhibitor of the p38
MAPK signaling pathway, significantly reduced the level of
p-RIPK3/p-MLKL in CSE/z-VAD-treated BEAS-2B cells, compared with
the CSE/z-VAD only group. These results indicated that TF-3 may
inhibit necroptosis via the p38 MAPK/RIPK3/MLKL signaling pathway
in CSE/z-VAD-induced BEAS-2B cells.
TF-3 attenuates morphological lung
injury in CSE-induced emphysema mice
In vivo, a mouse model of emphysema by i.p.
injection of CSE within 4 weeks was established (17). Compared with the normal alveolar
architecture of the control group, structural damage in the CSE
group was observed, with an increase in alveolar size, broken
alveolar wall and pulmonary emphysema. This indicated that the i.p.
injection of CSE induced emphysema within 4 weeks. However, TF-3
markedly suppressed these emphysematous changes (Fig. 5A-C).
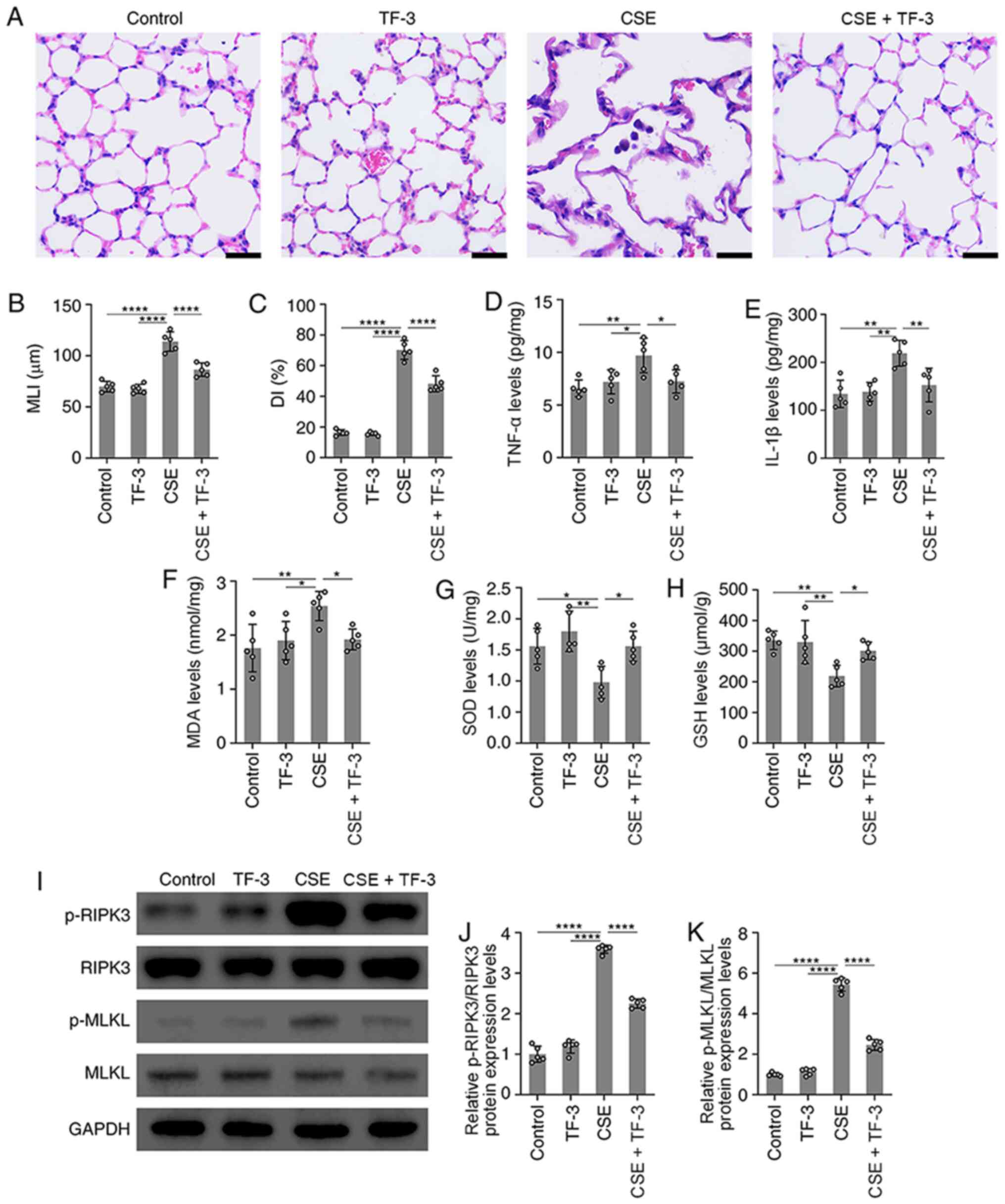 | Figure 5TF-3 attenuates emphysema in
CSE-treated mice. (A) Images of lung tissue stained with H&E.
Scale bars, 20 µm. (B and C) Morphometric measurements of the MLI
and DI in the four groups were quantified. (D and E) TNF-α and
IL-1β levels in lung tissue were detected using ELISA and
quantified. (F-H) MDA, SOD and GSH levels in lung tissue were
detected with assay kits and quantified. (I) Protein expression
levels of p-RIPK3/RIPK3 and p-MLKL/MLKL were determined via western
blotting with GAPDH as an internal control. Semi-quantification of
protein expression levels of (J) p-RIPK3 and (K) p-MLKL were
determined. Data are presented as the mean ± SEM of five
independent experiments indicated by dots. One-way ANOVA was
performed followed by Tukey's post hoc test for statistical
comparisons among more than two groups. *P<0.05,
**P<0.01 and ****P<0.0001. MLI, mean
linear intercept; DI, destructive index; MDA, malondialdehyde; SOD,
superoxide dismutase; GSH, glutathione; RIPK3, receptor-interacting
serine/threonine-protein kinase 3; MLKL, mixed lineage kinase
domain-like; p, phosphorylated; CSE, cigarette smoke extract; TF-3,
theaflavin-3,3'-digallate. |
TF-3 suppresses oxidative stress and
inflammatory responses in CSE-induced emphysema mice
Subsequently, the antioxidative and
anti-inflammatory capacity of TF-3 in CSE-treated emphysema mice
was explored. Compared with CSE only group, the results
demonstrated that TF-3 significantly decreased TNF-α, IL-1β and MDA
levels but significantly increased the levels of SOD and GSH in the
lungs (Fig. 5D-H). These results
indicated that TF-3 may prevent CSE-induced emphysema by inhibiting
oxidative stress and inflammatory responses.
TF-3 suppresses necroptosis in
CSE-induced emphysema mice
The effect of TF-3 on necroptosis in CSE-induced
emphysema mice was also detected. The western blotting results
demonstrated that the protein expression levels of p-RIPK3 and
p-MLKL were significantly increased in the lung tissue of
CSE-treated mice compared with the control. However, TF-3
significantly decreased these protein expression levels compared
with the CSE group (Fig. 5I-K).
Discussion
In the present study, the results demonstrated that
TF-3 may exhibit a protective effect against emphysema in
CSE-induced mice via the inhibition of necroptosis.
Black tea is a well-known beverage that is commonly
consumed and contains theaflavins, which are considered to have
anti-inflammatory and antioxidant effects (18). Among theaflavins, TF-3, which has
two gallic acid moieties, exhibits the strongest anti-inflammatory
and antioxidant activity (6).
Currently, all available evidence firmly suggests
that oxidative stress modulated by ROS serves a significant role in
the pathogenesis of pulmonary emphysema (19,20).
Under physiological conditions, ROS can help prevent pathological
injury or noxious stimulation. However, excessive generation of ROS
is thought to lead to cellular injury and oxidative stress
(21). In the present study, the
results demonstrated that antioxidant activity was significantly
decreased in CSE-challenged BEAS-2B cells. However, TF-3 was
capable of significantly blocking this decrease, which suggested
that TF-3 may be involved in the pathophysiological pathway of
oxidative stress in the development of emphysema.
Inflammation is a type of stress protection against
tissue or cell damage induced by adverse external stimulation. A
large number of proinflammatory cytokines are produced in the
process of inflammation, including TNF-α, IL-1β and IL-6, which are
generally considered to be the most relevant proinflammatory
cytokines in the inflammatory response (22). In the present study, TF-3
significantly reduced the mRNA expression levels of TNF-α, IL-1β
and IL-6 in CSE-induced BEAS-2B cells.
Increasing numbers of reports have shown that
necroptosis, a novel nonapoptotic cell death mechanism, may
contribute to the pathogenesis of pulmonary emphysema (4,23-26).
The difference between necrosis and necroptosis is that necrosis is
a non-programmed and passive method of cell death, whereas
necroptosis can be regulated (27).
A previous study has also indicated that inhibition of the
RIPK3/MLKL signaling pathway attenuates necroptosis (28). The results of the present study
demonstrated that TF-3 significantly decreased the protein
expression levels of p-RIPK3 and p-MLKL in CSE/z-VAD-treated
BEAS-2B cells. Moreover, the flow cytometry and immunofluorescence
assay results determined that TF-3 significantly suppressed the
necroptosis stimulated by CSE/z-VAD. Collectively this evidence
demonstrated that TF-3 may ameliorate CSE/z-VAD-induced
inflammation and oxidative stress by inhibiting the
RIPK3/MLKL-mediated necroptosis signaling pathway. Furthermore, it
was confirmed that the p38 MAPK signaling pathway was strongly
associated with necroptosis. SB203580, a specific inhibitor of p38
MAPK, was used to suppress the autophosphorylation and substrate
phosphorylation of p38 MAPK (14).
However, SB203580 does not inhibit the phosphorylation of p38 MAPK
by upstream kinases. In CSE/z-VAD-treated BEAS-2B cells, the
phosphorylation of RIPK3 and MLKL was significantly increased, but
this was significantly ameliorated by SB203580, which indicated
that p38 MAPK may promote RIPK3/MLKL-mediated necroptosis (Fig. 4A-D).
In vivo, it was observed that TF-3
significantly alleviated the emphysematous changes induced by CSE
treatment in the lung tissue of mice, which indicated that TF-3 can
inhibit emphysema morphologically. Furthermore, TF-3 may suppress
inflammatory responses in the pathogenesis of emphysema and
inflammatory cytokines (TNF-α and IL-1β) were significantly reduced
following TF-3 therapy. Moreover, the detection results of
oxidative stress-related indicators (MDA, SOD and GSH) indicated
that the antioxidant capacity of TF-3 may serve an important role
in the inhibition of emphysema. Furthermore, the results of the
present study demonstrated that necroptosis may be involved in the
pathogenesis of emphysema by detecting the protein expression
levels of p-RIPK3 and p-MLKL in the lung tissue of CSE-induced
mice. The results demonstrated that necroptosis was significantly
inhibited by TF-3 in the lung tissue of CSE-induced emphysema mice.
All this aforementioned evidence indicated that TF-3 may attenuate
emphysema by suppressing necroptosis in CSE-induced mice.
The emphysema mouse model used in the present study
has been previously described (17). The mechanism of this new method for
establishing an animal model of emphysema is not well understood,
but it has been reported to be related to the autoimmune mechanism
of alveolar septal cell destruction (29). It is unclear whether the
pathobiological mechanisms underlying modeling by CSE injection are
the same as those of conventional inhalation of cigarette smoke for
several months. However, following i.p. injection of CSE for four
weeks, mice developed emphysema, which was accompanied by alveolar
dilatation and destruction, and confirmed the efficiency of CSE
injection in the establishment of a tobacco-related model of
emphysema in mice. Furthermore, TF-3 suppressed these emphysematous
changes. Therefore, these results suggested that TF-3 may inhibit
the progression of pulmonary emphysema in CSE-induced mice.
In conclusion, the present study demonstrated that
TF-3 attenuates lung tissue architecture damage, oxidative stress,
inflammatory responses and necroptosis in the lung tissue of mice
with CSE-induced pulmonary emphysema. Furthermore, it was indicated
that TF-3 reduced ROS and proinflammatory cytokine generation and
inhibited necroptosis via the p38 MAPK/RIPK3/MLKL signaling
pathways in CSE-treated BEAS-2B cells. Thus, there may be signaling
pathways that link oxidative stress, inflammation and necroptosis
in the pathogenesis of emphysema. The evidence demonstrated that
TF-3 serves an important role in protecting against pulmonary
emphysema by alleviating oxidative stress, inflammation and
necroptosis. However, the protective effect of TF-3 against
pulmonary emphysema needs to be further explored. The current study
still presents certain limitations, such as the lack of positive
controls and the failure to explore the optimal concentration of
TF-3. More experiments should be carried out to reveal the
underlying mechanisms in vitro. Furthermore, the biological
activity of TF-3 in other animal models, different treatment cycles
and differences in its effect in humans and animals, need to be
investigated.
Acknowledgements
Not applicable.
Funding
This work was supported by a grant from the National Natural
Science Foundation of China (grant no. 82004432). The funders had
no role in the study design, in the collection, analysis and
interpretation of data, in the writing of the report, or in the
decision to submit the article for publication.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GL, ZZ and KW performed the experiments and analyzed
the data. GL participated in the experimental design. GL and SY
designed the study, analyzed the data and wrote the manuscript. GL
and SY confirm the authenticity of all the raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was conducted according to the
revised Declaration of Helsinki and was approved by the
Institutional Review Board and Animal Ethics Committee of Shanghai
University of Medicine and Health Science (Shanghai, China). All
the animal experiments were carried out following the National
Institutes of Health Guide for the Care and Use of Laboratory
Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Labaki WW and Rosenberg SR: Chronic
obstructive pulmonary disease. Ann Intern Med. 173:ITC17–ITC32.
2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Tuder RM, Yoshida T, Arap W, Pasqualini R
and Petrache I: State of the art. Cellular and molecular mechanisms
of alveolar destruction in emphysema: An evolutionary perspective.
Proc Am Thorac Soc. 3:503–510. 2006.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bedoui S, Herold MJ and Strasser A:
Emerging connectivity of programmed cell death pathways and its
physiological implications. Nat Rev Mol Cell Biol. 21:678–695.
2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Mizumura K, Cloonan SM, Nakahira K,
Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko
GR, et al: Mitophagy-dependent necroptosis contributes to the
pathogenesis of COPD. J Clin Invest. 124:3987–4003. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Balentine DA, Wiseman SA and Bouwens LC:
The chemistry of tea flavonoids. Crit Rev Food Sci Nutr.
37:693–704. 1997.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ukil A, Maity S and Das PK: Protection
from experimental colitis by theaflavin-3,3'-digallate correlates
with inhibition of IKK and NF-kappaB activation. Br J Pharmacol.
149:121–131. 2006.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wu Y, Jin F and Wang Y, Li F, Wang L, Wang
Q, Ren Z and Wang Y: In vitro and in vivo anti-inflammatory effects
of theaflavin-3,3'-digallate on lipopolysaccharide-induced
inflammation. Eur J Pharmacol. 794:52–60. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ai Z, Wu Y, Yu M, Li J and Li S:
Theaflavin-3, 3'-digallate suppresses RANKL-induced
osteoclastogenesis and attenuates ovariectomy-induced bone loss in
mice. Front Pharmacol. 11(803)2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Pan H, Kim E, Rankin GO, Rojanasakul Y, Tu
Y and Chen YC: Theaflavin-3, 3'-digallate inhibits ovarian cancer
stem cells via suppressing Wnt/β-Catenin signaling pathway. J Funct
Foods. 50:1–7. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Council NR: Guide for the Care and Use of
Laboratory Animals: Eighth Edition. Washington, DC, The National
Academies Press, 246, 2011.
|
|
11
|
Chen Y, Hanaoka M, Chen P, Droma Y,
Voelkel NF and Kubo K: Protective effect of beraprost sodium, a
stable prostacyclin analog, in the development of cigarette smoke
extract-induced emphysema. Am J Physiol Lung Cell Mol Physiol.
296:L648–L56. 2009.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Sun L, Wang H, Wang Z, He S, Chen S, Liao
D, Wang L, Yan J, Liu W, Lei X and Wang X: Mixed lineage kinase
domain-like protein mediates necrosis signaling downstream of RIP3
kinase. Cell. 148:213–227. 2012.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sun W, Wu X, Gao H, Yu J, Zhao W, Lu JJ,
Wang J, Du G and Chen X: Cytosolic calcium mediates RIP1/RIP3
complex-dependent necroptosis through JNK activation and
mitochondrial ROS production in human colon cancer cells. Free
Radic Biol Med. 108:433–444. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Cabrera J, Negrín G, Estévez F, Loro J,
Reiter RJ and Quintana J: Melatonin decreases cell proliferation
and induces melanogenesis in human melanoma SK-MEL-1 cells. J
Pineal Res. 49:45–54. 2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Kilkenny C, Browne WJ, Cuthi I, Emerson M
and Altman DG: Improving bioscience research reporting: The ARRIVE
guidelines for reporting animal research. PLoS Biol.
8(e1000412)2010.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhang Y, Cao J, Chen Y, Chen P, Peng H,
Cai S, Luo H and Wu SJ: Intraperitoneal injection of cigarette
smoke extract induced emphysema, and injury of cardiac and skeletal
muscles in BALB/C mice. Exp Lung Res. 39:18–31. 2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Bahramsoltani R, Ebrahimi F, Farzaei MH,
Baratpourmoghaddam A, Ahmadi P, Rostamiasrabadi P, Rasouli
Amirabadi AH and Rahimi R: Dietary polyphenols for atherosclerosis:
A comprehensive review and future perspectives. Crit Rev Food Sci
Nutr. 59:114–132. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Tuder RM and Petrache I: Pathogenesis of
chronic obstructive pulmonary disease. J Clin Invest.
122:2749–2755. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Polverino F, Rojas-Quintero J, Wang X,
Petersen H, Zhang L, Gai X, Higham A, Zhang D, Gupta K, Rout A, et
al: A disintegrin and metalloproteinase Domain-8: A novel
protective proteinase in chronic obstructive pulmonary disease. Am
J Respir Crit Care Med. 198:1254–1267. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Yang CS, Kim JJ, Lee SJ, Hwang JH, Lee CH,
Lee MS and Jo EK: TLR3-triggered reactive oxygen species contribute
to inflammatory responses by activating signal transducer and
activator of transcription-1. J Immunol. 190:6368–6377.
2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Martinon F: Signaling by ROS drives
inflammasome activation. Eur J Immunol. 40:616–619. 2010.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Galluzzi L, Kepp O, Chan FK and Kroemer G:
Necroptosis: Mechanisms and relevance to disease. Annu Rev Pathol.
12:103–130. 2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Pearson JS, Giogha C, Mühlen S, Nachbur U,
Pham CL, Zhang Y, Hildebrand JM, Oates CV, Lung TW, Ingle D, et al:
EspL is a bacterial cysteine protease effector that cleaves RHIM
proteins to block necroptosis and inflammation. Nat Microbiol.
2(16258)2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wang Y, Liu J, Zhou JS, Huang HQ, Li ZY,
Xu XC, Lai TW, Hu Y, Zhou HB, Chen HP, et al: MTOR suppresses
cigarette smoke-induced epithelial cell death and airway
inflammation in chronic obstructive pulmonary disease. J Immunol.
200:2571–2580. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ryter SW, Rosas IO, Owen CA, Martinez FJ,
Choi ME, Lee CG, Elias JA and Choi AMK: Mitochondrial Dysfunction
as a pathogenic mediator of chronic obstructive pulmonary disease
and idiopathic pulmonary fibrosis. Ann Am Thorac Soc. 15 (Suppl
4):S266–S272. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Newton K and Manning G: Necroptosis and
Inflammation. Annu Rev Biochem. 85:743–763. 2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Weinlich R, Oberst A, Beere HM and Green
DR: Necroptosis in development, inflammation and disease. Nat Rev
Mol Cell Biol. 18:127–136. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Sullivan AK, Simonian PL, Falta MT,
Cosgrove GP, Brown KK, Kotzin BL, Voelkel NF and Fontenot AP:
Activated oligoclonal CD4+ T cells in the lungs of patients with
severe emphysema. Proc Am Thorac Soc. 3(486)2006.PubMed/NCBI View Article : Google Scholar
|