Introduction
Ischemia/reperfusion (I/R) injury is caused by
deficient oxygen supply followed by restoration of blood
circulation. I/R is the primary cause of cerebral damage and is a
major clinical problem in cerebral injury therapy (1,2).
Previous studies have demonstrated that deficient oxygen and
glucose supply may result in neuronal injury during ischemic brain
damage (3,4). Previous studies have also demonstrated
that oxygen-glucose deprivation (OGD) has detrimental effects on
the cells, including oxidative stress and immoderate glutamate
release, resulting in toxic levels (5,6).
However, the specific pathogenesis of cerebral I/R injury and the
possible signaling pathways participating in ischemic neuronal
injury are not completely understood.
A number of studies have indicated that numerous
anesthetic drugs exert neuroprotective effects in cerebral I/R
injury, including isoflurane (7),
propofol (8) and curcumin (9). Lidocaine, a widely used local
anesthetic, may improve the cognitive ability of patients suffering
from cardiopulmonary conditions (10). In addition, the protective effects
of lidocaine on ischemia- or hypoxia-stimulated neuronal injury are
mediated by diverse mechanisms, including vasodilation, improvement
of the microcirculation and suppression of platelet aggregation
(11,12). Although the neuroprotective role of
lidocaine in brain I/R damage has been previously demonstrated in
in vitro and in vivo models (13,14),
the mechanisms underlying the neuroprotective properties of
lidocaine remain to be elucidated. Therefore, the aim of the
present study was to investigate the functions of lidocaine in an
in vitro I/R model.
Various signaling pathways have been associated with
the pathogenesis of cerebral I/R, including mitogen-activated
protein kinase (15), N-methyl
D-aspartate receptor subtype 2B/ERK/cAMP response element-binding
protein (16), Janus kinase
2/STAT3(17) and Wnt/β-catenin
(18) signaling pathways. The Wnt
signaling pathway is the most common signaling pathway that
regulates and mediates a series of cellular processes, including
proliferation and apoptosis (19).
The canonical Wnt/β-catenin signaling pathway serves a key role in
cell survival following cerebral ischemia (18). Previous studies have demonstrated
that activation of the Wnt/β-catenin signaling pathway during I/R
exerts corresponding organ-protective effects (20-22).
However, whether lidocaine affects the Wnt/β-catenin signaling
pathway in I/R injury is not completely understood.
The aim of the present study was to investigate the
roles and mechanisms underlying the action of lidocaine in
OGD/reperfusion (OGD/R)-induced cortical neurons.
Materials and methods
Primary culture of cortical
neurons
Brain cortical neurons were obtained from
Sprague-Dawley rats (age, embryonic day 18; SD rats were obtained
from the Model Animal Research Center of Nanjing University)
according to previous study (23).
In brief, cerebral cortices were separated and digested with
trypsin (Gibco; Thermo Fisher Scientific, Inc.) at 37˚C for 15 min,
re-tempered in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.)
and 1% penicillin/streptomycin (Gibco; Thermo Fisher Scientific,
Inc.) and filtered through a 40-mm nylon mesh. Cells were cultured
in a 6-well poly-L-lysine coated culture plate (Sigma-Aldrich;
Merck KGaA) and maintained at 37˚C in an incubator in DMEM
containing 2% B27 (Gibco; Thermo Fisher Scientific, Inc.), 0.5 mM
glutamine and 100 U/ml penicillin/streptomycin. Following culture
at 37˚C for 24 h, 10 mM cytosine arabinoside (Sigma-Aldrich; Merck
KGaA) was applied to inhibit non-neuronal cell survival and neurons
were cultured at 37˚C for an additional 2 days. The mother rats
(n=3; Model Animal Research Center of Nanjing University, Nanjing,
China) were anaesthetized with 2% halothane prior to sacrifice by
cervical dislocation. The depth of anesthesia was monitored by the
toe pinch method. Death was verified by monitoring cessation of the
heartbeat and breathing. The fetuses (n=6) were also anaesthetized
with 2% halothane prior to sacrifice by cervical dislocation. All
rats were housed under standard conditions at room temperature
(22-24˚C) and 60-65% humidity, on a 12-h light/dark cycle with
ad libitum supply of food and water. The present study was
approved by the Animal Ethics Committee of the First Medical
Center, People's Liberation Army General Hospital. All experiments
conformed to the guidelines for the Care and Use of Laboratory
Animals of the National Institutes of Health (24).
Neuronal I/R injury model
establishment and lidocaine stimulation
The OGD/R model was constructed as previously
described (25). To induce OGD,
brain cortical neurons were cultured in Earle's Balanced Salt
Solution (Thermo Fisher Scientific, Inc.) and transferred into a
hypoxia chamber for 4 h with 95% N2 and 5%
CO2 at 37˚C. Subsequently, cells were cultured in an
environment of 5% CO2 and 95% air at 37˚C for 24 h. To
induce R, following culture in the hypoxic chamber, cells were
cultured for 24 h in HyClone medium 199 (Cytiva) supplemented with
2% B27 and 0.5 mM glutamine in normoxic conditions (95% air/5%
CO2) at 37˚C. For lidocaine stimulation, cultured
cortical neurons were treated with 10 µM lidocaine, as previously
described (26), or saline at 37˚C
for 4 h after exposure to OGD/R injury.
MTT assay
Following treatment, MTT solution was added to each
well (5x104 cells per well) and incubated for 4 h at
37˚C. The supernatant was discarded and 100 µl DMSO was added to
dissolve blue formazan crystals. The absorbance of each well was
measured at a wavelength of 570 nm using a microplate reader
(Jupiter G19060; Montréal Biotech, Inc.).
Lactate dehydrogenase (LDH)
analysis
LDH production in cells was determined using an
LDH-Cytotoxicity assay kit (Sigma-Aldrich; Merck KGaA). Briefly,
the supernatant of cerebral cortical neurons was collected from
each well after treatment through centrifugation (400 x g, 5 min,
4˚C) and subjected to the assay according to the manufacturer's
protocol at room temperature for 15 min. The absorbance of each
sample was measured at a wavelength of 490 nm using the
FLUOstar® Omega Microplate Reader (BMG Labtech
GmbH).
Flow cytometry analysis
Following OGD/R and lidocaine or saline treatment,
the cerebral cortical neurons were collected for Annexin V-FITC/PI
(BD Biosciences) double staining, according to the manufacturer's
instructions. Neuronal cell apoptosis (early + late apoptosis) was
evaluated by flow cytometry using a BD FACSCalibur flow cytometer
(BD Biosciences) and FlowJo software (version 7.2.4; FlowJo
LLC).
Determination of caspase-3
activity
To determine caspase3 activity, a Caspase-3 Assay
kit (cat. no. ab39401; Abcam) was used according to the
manufacturer's protocol. Briefly, cells were resuspended in 50 µl
of chilled Cell Lysis Buffer (Abcam) and incubated the cells on ice
for 10 min. Then cells were centrifuged at 10,000 x g for 1 min.
Subsequently, supernatant (cytosolic extract) was transferred to a
fresh tube and put on ice. Subsequently, an automatic micro-plate
reader (ELX800; BioTek Instruments, Inc.) was used to
spectrophotometrically determine caspase-3 activity levels at a
wavelength of 405 nm according to the manufacturer's protocol.
Reverse transcription-quantitative PCR
(RT-qPCR) analysis
Following the indicated treatment, total RNA was
extracted from cerebral cortical neurons (107 cells per
well) using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. Total
RNA was reverse transcribed into cDNA using the PrimeScript™ RT
reagent kit (Takara Bio, Inc.). The temperature protocol for
reverse transcription was as follows: 70˚C for 5 min, 37˚C for 5
min and 42˚C for 1 h. Subsequently, qPCR was performed using an ABI
7000 Real-Time PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.) with SYBR-Green PCR Master Mix Kit (Takara Bio,
Inc.). The thermocycling conditions used for qPCR were as follows:
Initial denaturation at 95˚C for 5 min; 40 cycles at 95˚C for 15
sec and 60˚C for 60 sec; and a final extension at 72˚C for 30 sec.
The following primers were used for qPCR (Sangon Biotech Co.,
Ltd.): Wnt3a forward, 5'-AACTGCACCACCGTCCAC-3' and reverse,
5'-AAGGCCGACTCCCTGGTA-3'; β-catenin forward,
5'-AACAGGGTCTGGGACATTAGTC-3' and reverse,
5'-CGAAAGCCAATCAAACACAAAC-3'; cyclin D1 forward,
5'-AACTACCTGGACCGCTTCCT-3' and reverse, 5'-CCACTTGAGCTTGTTCACCA-3';
and GAPDH forward, 5'-TGTTGCCATCAATGACCCCTT-3' and reverse,
5'-CTCCACGACGTACTCAGCG-3'. mRNA expression levels were quantified
using the 2-ΔΔCq method (27) and normalized to the internal
reference gene GAPDH.
Western blotting
The cerebral cortical neurons were treated as
indicated. Total protein was extracted from cerebral cortical
neurons using RIPA lysis buffer (Sigma-Aldrich; Merck KGaA). Total
protein was quantified using the BCA Protein assay kit
(Sigma-Aldrich; Merck KGaA). Proteins (40 µg/lane) were subjected
to 10% SDS-PAGE and transferred onto PVDF membranes (EMD
Millipore). The membranes were blocked in 5% fat-free milk at room
temperature for 1.5 h. Subsequently, the membranes were incubated
overnight at 4˚C with primary antibodies targeted against: Wnt3a
(cat. no. ab219412; 1:1,000; Abcam), β-catenin (cat. no. ab32572;
1:1,000; Abcam), cyclin D1 (cat. no. ab16663; 1:1,000; Abcam),
Bcl-2 (cat. no. ab196495; 1:1,000; Abcam), Bax (cat. no. ab32503;
1:1,000; Abcam), Bcl-xl (cat. no. ab32370; 1:1,000; Abcam) and
GAPDH (cat. no. ab181602; 1:1,000; Abcam). Subsequently, the
membranes were incubated with a horseradish peroxidase-conjugated
secondary antibody (cat. no. ab7090; 1:2,000; Abcam) at room
temperature for 2 h. Finally, the protein bands were observed by
chemiluminescence using the ECL Advance Western Blotting Detection
kit (Cytiva). Protein expression levels were quantified using
ImageJ software (version 1.52s; National Institutes of Health).
Statistical analysis
SPSS (version 21.0; IBM Corp.) was used to perform
statistical analyses. All data are presented as the mean ± standard
deviation from three independent experiments. Comparisons between
two groups were analyzed using the unpaired Student's t-test.
Comparisons among multiple groups were analyzed using one-way ANOVA
followed by Tukey's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
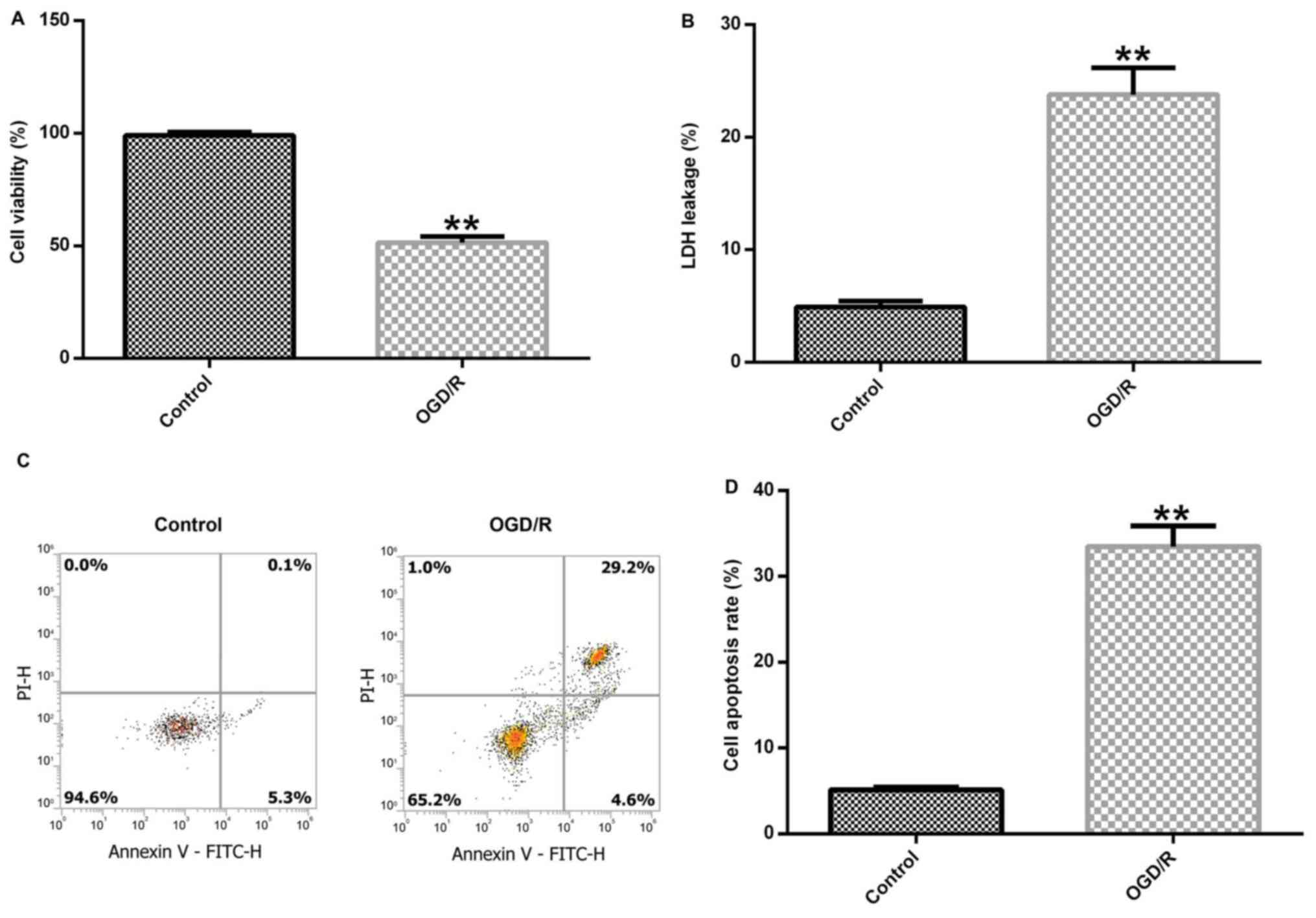
OGD/R successfully induces a cerebral
I/R injury model
Brain cortical neurons were cultured under OGD
conditions for 4 h and exposed to R conditions for 24 h to induce
cerebral I/R injury in vitro. Subsequently, cell viability,
LDH release and cell apoptosis were detected to assess neuronal
injury. Cerebral cortical neuronal cell viability was significantly
decreased in the OGD/R group compared with the control group
(Fig. 1A). In addition,
OGD/R-exposed cortical neurons displayed significantly increased
LDH release compared with control cortical neurons (Fig. 1B). Furthermore, the level of
apoptosis in control and OGD/R-stimulated cells was determined.
OGD/R treatment notably enhanced cell apoptosis and significantly
increased the percentage of apoptotic neurons in the OGD/R group
compared with the control group (Fig.
1C and D). In summary, the
results demonstrated that OGD/R successfully induced the neuronal
I/R injury model in vitro.
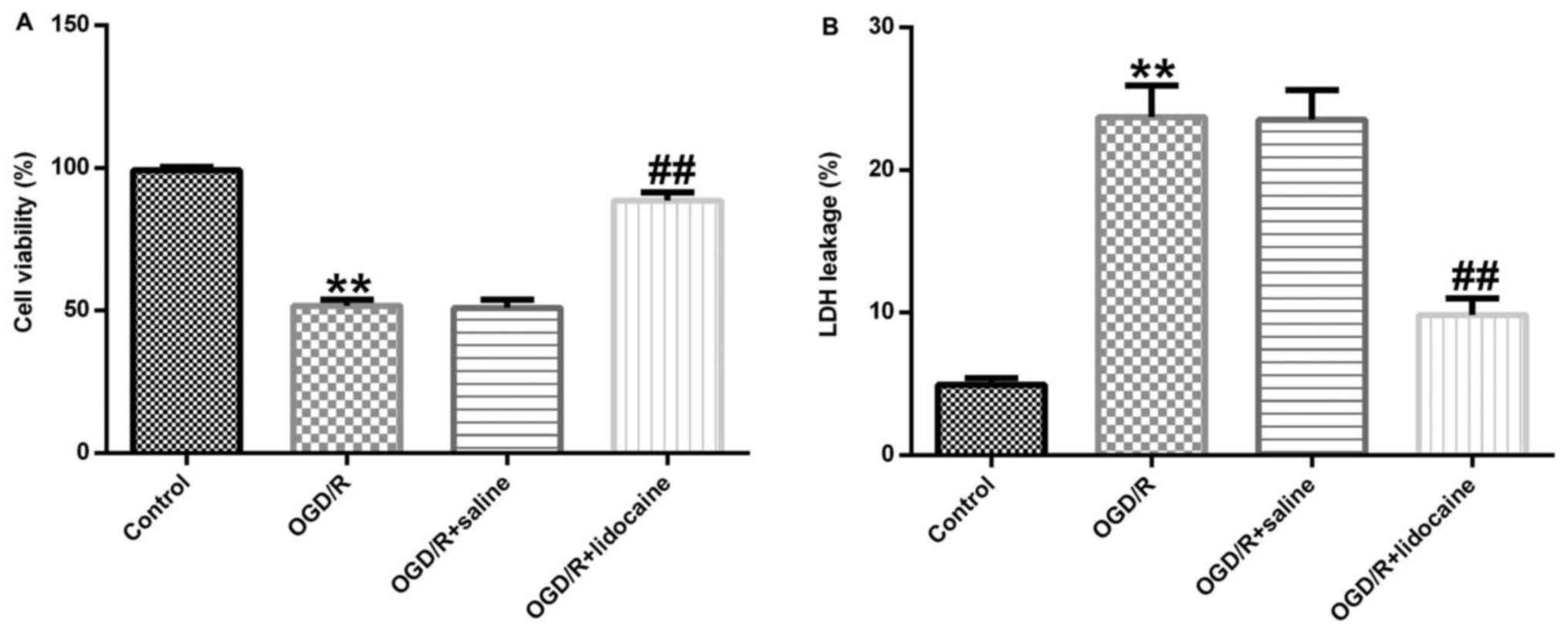
Lidocaine exerts neuroprotective
effects against OGD/R-induced neuronal injury
The role of lidocaine in OGD/R-stimulated neuronal
damage was investigated. Cerebral cortical neurons were treated
with 10 µM lidocaine or saline for 4 h following exposure to OGD/R.
Cell viability was evaluated by performing the MTT assay. Cell
viability in the OGD/R group was significantly decreased compared
with the control group, whereas cell viability was significantly
enhanced in the OGD/R + lidocaine group compared with the OGD/R +
saline group (Fig. 2A). In
addition, LDH release was measured to assess cell toxicity. The
results indicated that OGD/R exposure significantly increased LDH
release from cerebral cortical neurons compared with the control
group, whereas lidocaine reversed this effect (Fig. 2B). The results demonstrated that
lidocaine increased cell viability and reduced LDH secretion in the
OGD/R-induced neuronal injury model, indicating that lidocaine
suppressed neuronal injury.
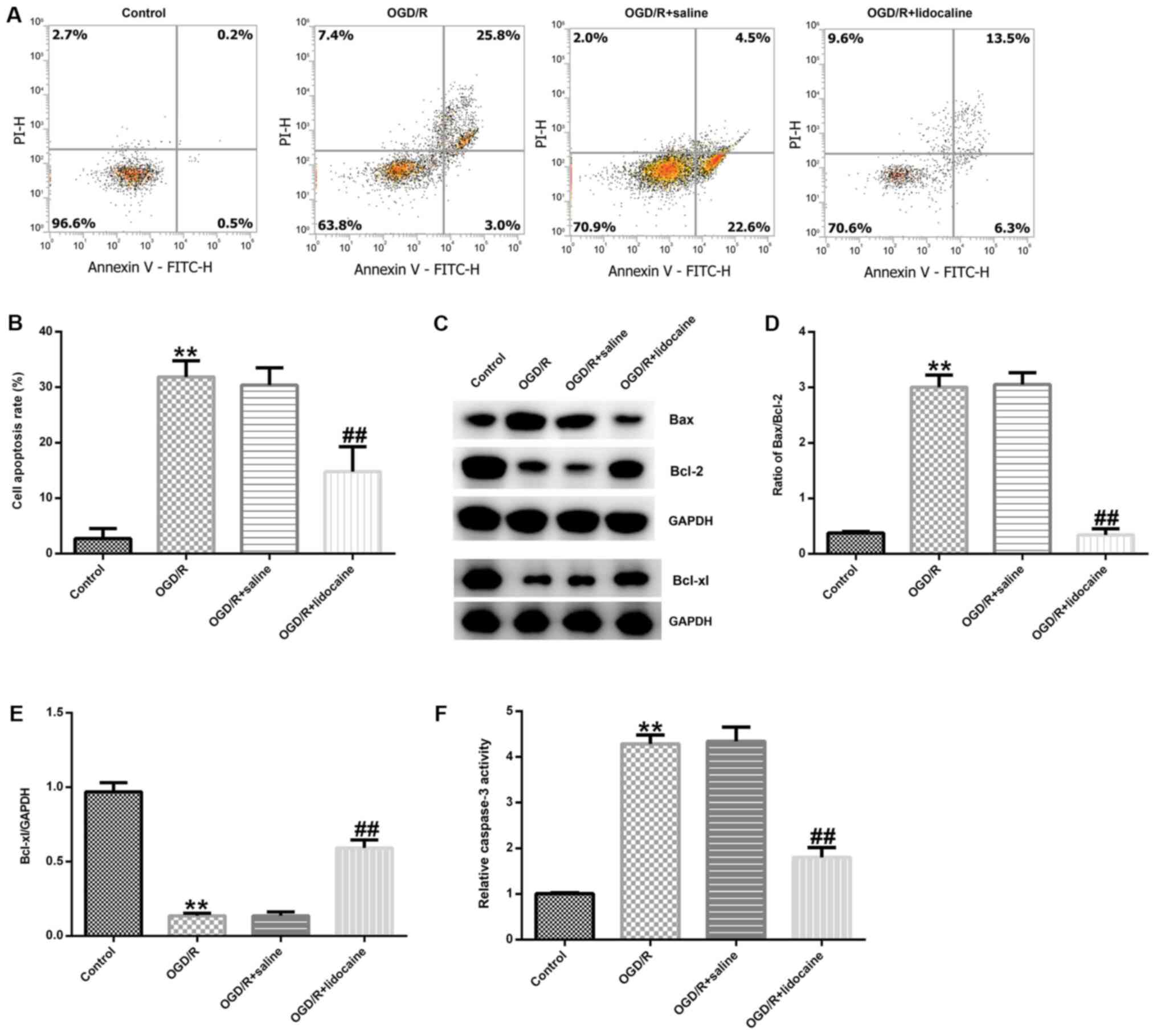
Lidocaine inhibits OGD/R-induced
neuronal cell apoptosis
To further elucidate the mechanism underlying the
effects of lidocaine in OGD/R-triggered neuronal injury, neuronal
cells were subjected to OGD/R and then treated with 10 µM lidocaine
or saline. Neuronal cell apoptosis was assessed by flow cytometry.
The number of apoptotic neurons was notably higher in the OGD/R
group compared with the control group (Fig. 3A). However, the number of apoptotic
neurons was obviously reduced in the OGD/R + lidocaine group
compared with the OGD/R + saline group (Fig. 3A). The percentage of apoptotic
neurons was also quantified in the different groups. The number of
apoptotic neurons was significantly higher in the OGD/R group
compared with the control group, but significantly lower in the
ODG/R + lidocaine group compared with the ODG/R + saline group
(Fig. 3B). Neuronal apoptosis is
regulated by apoptosis-specific proteins, including Bcl-2, Bax and
Bcl-xl (28); thus, the expression
of apoptosis-related proteins in the different groups was detected
by western blotting. The western blotting results indicated that
OGD/R markedly decreased Bcl-2 and Bcl-xl protein expression
levels, but increased Bax protein expression levels compared with
the control group. However, lidocaine reversed OGD/R-mediated
apoptosis-related protein expression (Fig. 3C). The Bax/Bcl-2 ratio was also
significantly increased by OGD/R treatment compared with the
control group, whereas lidocaine treatment reduced the effect of
OGD/R on the Bax/Bcl-2 ratio (Fig.
3D). OGD/R-mediated reductions in Bcl-xl protein expression
were inhibited by lidocaine treatment (Fig. 3E). Compared with the control group,
caspase-3 activity was also significantly increased by OGD/R
treatment, but lidocaine treatment reversed OGD/R-mediated
alterations to caspase-3 activity (Fig.
3F). The results indicated the potential role of lidocaine in
OGD/R-mediated neuronal injury.
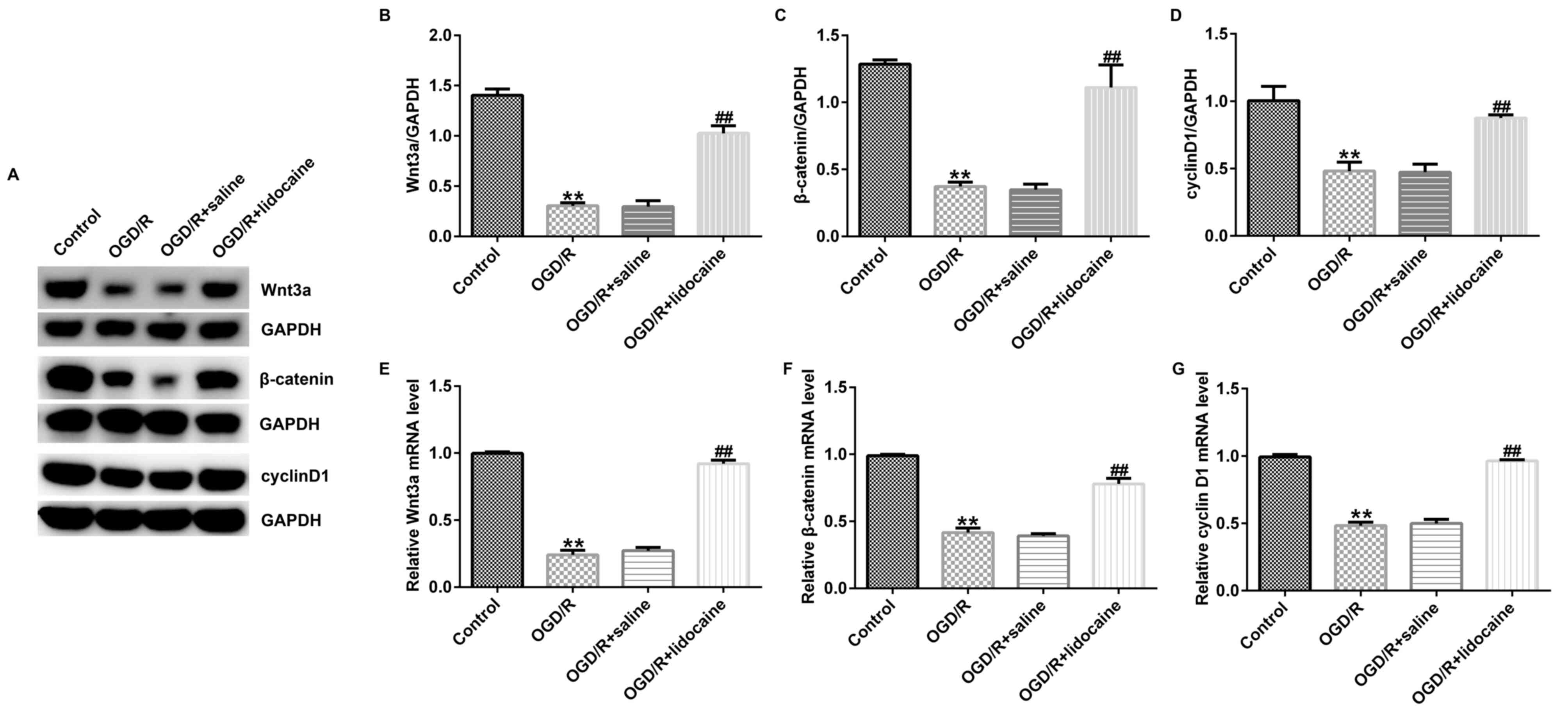
Lidocaine regulates the activation of
the Wnt/β-catenin signaling pathway in OGD/R-induced neurons
Finally, the potential mechanism underlying the
effects of lidocaine on OGD/R-triggered neurons was investigated.
The western blotting results indicated that Wnt3a, β-catenin and
cyclin D1 protein expression levels were significantly reduced in
OGD/R-stimulated neurons compared with control neurons. However,
lidocaine significantly upregulated the expression of the three
proteins in OGD/R-exposed neurons compared with the OGD/R + saline
group (Fig. 4A-D). Similar results
were obtained for mRNA expression levels of Wnt3a, β-catenin and
cyclin D1 (Fig. 4E-G).
Collectively, the results suggested that Wnt/β-catenin signaling
may be involved in OGD/R-mediated neuron injury, and lidocaine
displayed neuroprotective activity in OGD/R-exposed neurons, at
least partly by activating the Wnt/β-catenin signaling pathway.
Discussion
I/R injury is a major health concern in the clinic
(29); therefore, designing an
effective treatment strategy for cerebral I/R injury is of clinical
significance. According to previous studies, deficient oxygen and
glucose supply may contribute to neuronal injury during ischemic
brain damage (3,4,30). OGD
may have detrimental effects on cell function, such as oxidative
stress and immoderate glutamate release, resulting in toxic levels
(31). In previous studies,
cerebral cortical neurons were stimulated with OGD/R to induce an
in vitro cerebral I/R injury model (32,33).
In the present study, cortical neurons were isolated from
Sprague-Dawley rat embryos and OGD/R was used to establish an in
vitro I/R injury model. Subsequently, cell viability, LDH
release and cell apoptosis were detected to evaluate neuronal cell
damage. The results indicated that OGD/R exposure significantly
decreased cerebral cortical neuronal viability, increased LDH
release and induced cell apoptosis compared with control neurons.
The results also indicated that OGD/R successfully established an
in vitro I/R injury model. However, the staining of
NSE of primary cerebral cortical neurons was not conducted in the
present study, which was a key limitation.
Previous studies have demonstrated that various
elements serve neuroprotective roles in cerebral I/R injury models,
such as tamibarotene (34),
daucosterol (35) and orally
administered crocin (36). Zhang
et al (36) reported that
orally administered crocin exerted protective effects against
cerebral I/R injury via metabolic transformation of crocetin by the
gut microbiota. Tian et al (34) observed that tamibarotene improves
hippocampal injury stimulated by focal cerebral I/R via the
PI3K/Akt signaling pathway in rats. Lidocaine, a local anesthetic
that is widely used in surgery, is frequently used to treat
neurovascular diseases and serves pivotal roles in the prevention
and treatment of ischemic brain injury (37). However, the neuroprotective effects
of lidocaine and the underlying mechanisms in OGD/R-stimulated
injury are not completely understood. Therefore, the present study
investigated the neuroprotective mechanisms underlying the effects
of lidocaine in cerebral I/R neurons in vitro.
To evaluate the role of lidocaine in OGD/R-induced
neuronal cell injury, MTT and LDH release assays were performed.
The results demonstrated that 10 µM lidocaine significantly
enhanced cell viability and reduced LDH release in OGD/R-induced
cerebral cortical neurons compared with the OGD/R + saline group.
Therefore, the results indicated that lidocaine relieved
OGD/R-induced neuronal cell injury. It was previously suggested
that lidocaine may be beneficial to the nervous system by
regulating cell apoptosis (38). In
the present study, flow cytometry was conducted to evaluate
apoptosis in the OGD/R-induced neuronal cell injury model and the
results indicated that the number of apoptotic neurons was
significantly decreased in OGD/R-induced neurons following
lidocaine treatment compared with saline treatment. Neuronal cell
apoptosis is usually triggered by interactions between Bcl-2 family
members via endogenic apoptotic cascades (39,40).
In addition, the Bax/Bcl-2 ratio is considered a key indicator of
apoptotic stimulation (41). In the
present study, lidocaine enhanced Bcl-2 and Bcl-xl protein
expression levels, but decreased Bax expression levels, the
Bax/Bcl-2 ratio and caspase-3 activity in OGD/R-stimulated neurons
compared with the OGD/R + saline group. Multiple signaling pathways
are associated with the pathogenesis of cerebral I/R, including the
Wnt/β-catenin signaling pathway (18,42).
To further investigate the mechanism underlying the neuroprotective
effects of lidocaine, RT-qPCR and western blotting were conducted
to evaluate the relative mRNA and protein levels of Wnt/β-catenin
signaling pathway components. The results demonstrated that the
mRNA and protein expression levels of Wnt3a, β-catenin and cyclin
D1 were significantly increased in OGD/R-exposed neurons treated
with 10 µM lidocaine compared with OGD/R-exposed neurons treated
with saline; however, immunofluorescence staining was not performed
to verify the results, which may be a limitation of the present
study. Taken together, the results indicated that lidocaine
treatment promoted neuronal viability, and reduced LDH release and
neuronal cell apoptosis by regulating the Wnt/β-catenin signaling
pathway. However, the effects of lidocaine on I/R injury were not
studied in vivo in the present study, which was a further a
limitation of the present study and requires further
investigation.
The findings of the present study may provide novel
insight into the mechanism underlying the neuroprotective effects
of lidocaine against OGD/R-induced neuronal damage via activating
the Wnt/β-catenin signaling pathway. However, the present study was
only a preliminary in vitro study of the effects of
lidocaine on I/R injury. In order to verify the results of the
present study, further in-depth research is required. For example,
an inhibitor of Wnt/β-catenin signaling pathway should be applied
to investigate whether lidocaine directly regulates the
Wnt/β-catenin signaling pathway. Furthermore, only one dose of
lidocaine (10 µg/ml) was used in the present study to investigate
the effect of lidocaine on I/R injury in vitro, which is a
further limitation of the present study. Therefore, future studies
should investigate the effect of different doses of lidocaine on
I/R injury. In addition, the effect of lidocaine on I/R injury
should also be investigated in vivo.
In conclusion, the present study indicated that
lidocaine suppressed OGD/R-induced neuronal cell injury and
apoptosis by stimulating the Wnt/β-catenin signaling pathway,
thereby exerting a protective effect against OGD/R-induced neuronal
injury. Therefore, lidocaine may serve as a potential therapeutic
candidate for I/R injury.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XL contributed to study design, data collection,
statistical analysis, data interpretation and manuscript
preparation. YX contributed to data collection and statistical
analysis and manuscript preparation. XL and YX confirmed the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Animal Ethics
Committee of the First Medical Center, People's Liberation Army
General Hospital. All experiments conformed to the guidelines for
the Care and Use of Laboratory Animals of the National Institutes
of Health.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Papadakis M and Buchan A: Approaches to
neuroprotective and reperfusion injury therapy. Handb Clin Neurol.
94:1205–1223. 2009.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Zhào H, Liu Y, Zeng J, Li D, Zhang W and
Huang Y: Troxerutin and cerebroprotein hydrolysate injection
protects neurovascular units from oxygen-glucose deprivation and
reoxygenation-induced injury in vitro. Evid Based Complement
Alternat Med. 2018(9859672)2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Zhuonan Z, Sen G, Zhipeng J, Maoyou Z,
Linglan Y, Gangping W, Cheng J, Zhongliang M, Tian J, Peijian Z and
Kesen X: Hypoxia preconditioning induced HIF-1alpha promotes
glucose metabolism and protects mitochondria in liver I/R injury.
Clin Res Hepatol Gastroenterol. 39:610–619. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Yin X, Feng L, Ma D, Yin P, Wang X, Hou S,
Hao Y, Zhang J, Xin M and Feng J: Roles of astrocytic connexin-43,
hemichannels, and gap junctions in oxygen-glucose
deprivation/reperfusion injury induced neuroinflammation and the
possible regulatory mechanisms of salvianolic acid B and
carbenoxolone. J Neuroinflammation. 15(97)2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Park SJ, Jung YJ, Kim YA, Lee-Kang JH and
Lee KE: Glucose/oxygen deprivation and reperfusion upregulate
SNAREs and complexin in organotypic hippocampal slice cultures.
Neuropathology. 28:612–620. 2008.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Jung YJ, Park SJ, Park JS and Lee KE:
Glucose/oxygen deprivation induces the alteration of synapsin I and
phosphosynapsin. Brain Res. 996:47–54. 2004.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wang S, Yin J, Ge M, Dai Z, Li Y, Si J, Ma
K, Li L and Yao S: Transforming growth-beta 1 contributes to
isoflurane postconditioning against cerebral ischemia-reperfusion
injury by regulating the c-Jun N-terminal kinase signaling pathway.
Biomed Pharmacother. 78:280–290. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Yu W, Gao D, Jin W, Liu S and Qi S:
Propofol prevents oxidative stress by decreasing the ischemic
accumulation of succinate in focal cerebral ischemia-reperfusion
injury. Neurochem Res. 43:420–429. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hou Y, Wang J and Feng J: The
neuroprotective effects of curcumin are associated with the
regulation of the reciprocal function between autophagy and
HIF-1alpha in cerebral ischemia-reperfusion injury. Drug Des Devel
Ther. 13:1135–1144. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Zeng ZW, Zhang YN, Lin WX, Zhang WQ and
Luo R: A meta-analysis of pharmacological neuroprotection in
noncardiac surgery: Focus on statins, lidocaine, ketamine, and
magnesium sulfate. Eur Rev Med Pharmacol Sci. 22:1798–1811.
2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wen X, Xu S, Zhang Q, Li X, Liang H, Yang
C, Wang H and Liu H: Inhibitory gene expression of the Cav3.1
T-type calcium channel to improve neuronal injury induced by
lidocaine hydrochloride. Eur J Pharmacol. 775:43–49.
2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Mitchell SJ and Merry AF: Lignocaine:
Neuro-protective or wishful thinking? J Extra Corpor Technol.
41:P37–P42. 2009.PubMed/NCBI
|
|
13
|
Naito H, Takeda Y, Danura T, Kass IS and
Morita K: Effect of lidocaine on dynamic changes in cortical
reduced nicotinamide adenine dinucleotide fluorescence during
transient focal cerebral ischemia in rats. Neuroscience. 235:59–69.
2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zhang Y and Lipton P: Cytosolic
Ca2+ changes during in vitro ischemia in rat hippocampal
slices: Major roles for glutamate and Na+-dependent Ca2+
release from mitochondria. J Neurosci. 19:3307–3315.
1999.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hsu HC, Tang NY, Liu CH and Hsieh CL:
Antiepileptic effect of Uncaria rhynchophylla and rhynchophylline
involved in the initiation of c-Jun N-terminal kinase
phosphorylation of MAPK signal pathways in acute seizures of kainic
acid-treated rats. Evid Based Complement Alternat Med.
2013(961289)2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhao P, Yang JM, Wang YS, Hao YJ, Li YX,
Li N, Wang J, Niu Y, Sun T and Yu JQ: Neuroprotection of cytisine
against cerebral ischemia-reperfusion injury in mice by regulating
NR2B-ERK/CREB signal pathway. Neurochem Res. 43:1575–1586.
2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Liu X, Zhang X, Zhang J, Kang N, Zhang N,
Wang H, Xue J, Yu J, Yang Y, Cui H, et al: Diosmin protects against
cerebral ischemia/reperfusion injury through activating JAK2/STAT3
signal pathway in mice. Neuroscience. 268:318–327. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Zhang GX, Ge MY, Han ZW, Wang S, Yin J,
Peng L, Xu F, Zhang Q, Dai Z, Xie L, et al: Wnt/β-catenin signaling
pathway contributes to isoflurane postconditioning against cerebral
ischemia-reperfusion injury and is possibly related to the
transforming growth factorβ1/Smad3 signaling pathway. Biomed
Pharmacother. 110:420–430. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Anastas JN and Moon RT: WNT signalling
pathways as therapeutic targets in cancer. Nat Rev Cancer.
13:11–26. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Chen X, Wang CC, Song SM, Wei SY, Li JS,
Zhao SL and Li B: The administration of erythropoietin attenuates
kidney injury induced by ischemia/reperfusion with increased
activation of Wnt/β-catenin signaling. J Formos Med Assoc.
114:430–437. 2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kuncewitch M, Yang WL, Molmenti E,
Nicastro J, Coppa GF and Wang P: Wnt agonist attenuates liver
injury and improves survival after hepaticischemia/reperfusion.
Shock. 39:3–10. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
He X, Mo Y, Geng W, Shi Y, Zhuang X, Han
K, Dai Q, Jin S and Wang J: Role of Wnt/β-catenin in the tolerance
to focal cerebral ischemia induced by electroacupuncture
pretreatment. Neurochem Int. 97:124–132. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Tremblay R, Hewitt K, Lesiuk H, Mealing G,
Morley P and Durkin JP: Evidence that brain-derived neurotrophic
factor neuroprotection is linked to its ability to reverse the
NMDA-induced inactivation of protein kinase C in cortical neurons.
J Neurochem. 72:102–111. 1999.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Bayne K: Revised guide for the care and
use of laboratory animals available. American Physiological
Society. Physiologist. 39:199208–199211. 1996.PubMed/NCBI
|
|
25
|
Wu X, Li X, Liu Y, Yuan N, Li C, Kang Z,
Zhang X, Xia Y, Hao Y and Tan Y: Hydrogen exerts neuroprotective
effects on OGD/R damaged neurons in rat hippocampal by protecting
mitochondrial function via regulating mitophagy mediated by
PINK1/Parkin signaling pathway. Brain Res. 1698:89–98.
2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhang Y, Tao GJ, Hu L, Qu J, Han Y, Zhang
G, Qian Y, Jiang CY and Liu WT: Lidocaine alleviates morphine
tolerance via AMPK-SOCS3-dependent neuroinflammation suppression in
the spinal cord. J Neuroinflammation. 14(211)2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2011.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wang L, Zhang L and Chow BKC: Secretin
prevents apoptosis in the developing cerebellum through Bcl-2 and
Bcl-xL. J Mol Neurosci. 68:494–503. 2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Cai Y, Xu H, Yan J, Zhang L and Lu Y:
Molecular targets and mechanism of action of dexmedetomidine in
treatment of ischemia/reperfusion injury. Mol Med Rep. 9:1542–1550.
2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhang H, Cao HJ, Kimelberg HK and Zhou M:
Volume regulated anion channel currents of rat hippocampal neurons
and their contribution to oxygen-and-glucose deprivation induced
neuronal death. PLoS One. 6(e16803)2011.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Khaspekov L, Friberg H, Halestrap A,
Viktorov I and Wieloch T: Cyclosporin A and its
nonimmunosuppressive analogue N-Me-Val-4-cyclosporin A mitigate
glucose/oxygen deprivation-induced damage to rat cultured
hippocampal neurons. Eur J Neurosci. 11:3194–3198. 1999.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Hou QL, Wang Y, Li YB, Hu XL and Wang SL:
Protective effect of notoginsenoside R1 on neuron injury induced by
OGD/R through ATF6/Akt signaling pathway. Zhongguo Zhong Yao Za
Zhi. 42:1167–1174. 2017.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
33
|
Wei R, Zhang R, Li H, Li H, Zhang S, Xie
Y, Shen L and Chen F: MiR-29 Targets PUMA to suppress oxygen and
glucose deprivation/reperfusion (OGD/R)-induced cell death in
hippocampal neurons. Curr Neurovasc Res. 15:47–54. 2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Tian X, An R, Luo Y, Li M, Xu L and Dong
Z: Tamibarotene improves hippocampus injury induced by focal
cerebral ischemia-reperfusion via modulating PI3K/Akt pathway in
rats. J Stroke Cerebrovasc Dis. 28:1832–1840. 2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhang HY, Song YM and Feng C: Improvement
of cerebral ischemia/reperfusion injury by daucosterol
palmitate-induced neuronal apoptosis inhibition via PI3K/Akt/mTOR
signaling pathway. Metab Brain Dis: May 4, 2020 (Epub ahead of
print).
|
|
36
|
Zhang Y, Geng J, Hong Y, Jiao L, Li S, Sun
R, Xie Y, Yan C, Aa J and Wang G: Orally administered crocin
protects against cerebral ischemia/reperfusion injury through the
metabolic transformation of crocetin by gut microbiota. Front
Pharmacol. 10(440)2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Seyfried FJ, Adachi N and Arai T:
Suppression of energy requirement by lidocaine in the ischemic
mouse brain. J Neurosurg Anesthesiol. 17:75–81. 2005.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zheng X, Chen L, Du X, Cai J, Yu S, Wang
H, Xu G and Luo Z: Effects of hyperbaric factors on
lidocaine-induced apoptosis in spinal neurons and the role of p38
mitogen-activated protein kinase in rats with diabetic neuropathic
pain. Exp Ther Med. 13:2855–2861. 2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Wu J, Yang J, Liu Q, Wu S, Ma H and Cai Y:
Lanthanum induced primary neuronal apoptosis through mitochondrial
dysfunction modulated by Ca2+ and Bcl-2 family. Biol
Trace Elem Res. 152:125–134. 2013.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Ghosh AP, Walls KC, Klocke BJ, Toms R,
Strasser A and Roth KA: The proapoptotic BH3-only, Bcl-2 family
member, Puma is critical for acute ethanol-induced neuronal
apoptosis. J Neuropathol Exp Neurol. 68:747–756. 2009.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Khodapasand E, Jafarzadeh N, Farrokhi F,
Kamalidehghan B and Houshmand M: Is Bax/Bcl-2 ratio considered as a
prognostic marker with age and tumor location in colorectal cancer?
Iran Biomed J. 19:69–75. 2015.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Jin Z, Ke J, Guo P, Wang Y and Wu H:
Quercetin improves blood-brain barrier dysfunction in rats with
cerebral ischemia reperfusion via Wnt signaling pathway. Am J
Transl Res. 11:4683–4695. 2019.PubMed/NCBI
|