Introduction
Sensorineural hearing loss is a type of auditory
impairment that is caused by congenital genetics, such as SCN11A
gene deletion impairing the ribbon synapses and auditory nerves, or
acquired factors, such as ototoxic drugs (1-3).
As a result, the hearing system abnormally perceive and process
sound (4). The main pathological
manifestation of this condition is damage to the inner ear cochlear
hair cells (5). In total, >30%
patients who are deaf are afflicted with drug-induced deafness,
which is mainly caused by the improper use of aminoglycoside
antibiotics, such as neomycin (Neo) and streptomycin, with ~5% of
patients exhibiting significant hearing loss using such antibiotics
(6-8).
In particular, Neo has a high incidence of hearing loss worldwide,
compared with other aminoglycoside antibiotics (3). Since aminoglycoside antibiotics are
widely used in developed countries (9), protecting hair cells and reducing the
damage caused by aminoglycoside antibiotics currently represents a
challenge.
Hair cells are receptors in the ear that produce
auditory impulses (2), which cannot
regenerate if destroyed (10). Ear
damage can lead to irreversible injury of cochlear hair cells and
auditory neurons, leading to permanent sensorineural deafness
(11). Common environmental factors
that cause sensorineural deafness include perinatal infection,
excessive noise, aging and ototoxic drugs (3,12).
Specifically, noise, aging and ototoxic drugs can induce hair cell
death through oxidative stress or activation of apoptotic pathways
(13,14). It has been previously demonstrated
that the mechanism underlying ototoxicity of aminoglycoside
antibiotics is mainly through damage and apoptosis of hair cells,
resulting in permanent hearing loss and vestibular dysfunction
(15). After the drugs enter the
cochlea, the increase in reactive oxygen species (ROS) and calcium
ions in sensory cells and neurons of the inner ear disrupts
physiological mitochondrial protein synthesis and reduces the
mitochondrial membrane potential, in turn promoting the release of
cytochrome c into the cytosol (16). Therefore, local or systemic
application of ROS scavengers may protect the cochlea from
aminoglycoside antibiotics (17).
Calcium-activated protease, such as Calpain, also served an
important role in the destruction of hair cells caused by
aminoglycoside antibiotics (18).
Therefore, inhibiting the activity of calcium-activated proteases
may also effectively protect cochlear hair cells from drug-induced
damage (15).
Fasudil (Fas) is a novel isoquinoline sulfonamide
derivative that was originally developed as an effective Rho kinase
(ROCK) inhibitor and calcium ion antagonist (19). Fas is the only ROCK inhibitor
currently approved for a number of brain disorders in humans,
including ischemia-reperfusion injury, brain edema, and cerebral
microthrombosis (20). It has few
side effects and was approved by Japan in 1995 for the clinical
treatment of subarachnoid hemorrhage caused by cerebral vasospasm
(21). Vasoconstriction and
Ca2+ sensitization induced by ROCK can both be
alleviated and eliminated by Fas, where the pharmacological effects
of this ROCK inhibitor are proposed to be mediated by the
downregulation of ROCK expression/activity under pathobiological
conditions (22,23). Studies have previously shown that
ROS can cause the activation of the RhoA/ROCK signaling pathway
(24-27).
Therefore, using Fas to inhibit RhoA/ROCK signaling reduced
oxidative stress in Neo-induced hair cell damage (7,28),
suggesting that Fas may exert a protective effect against
aminoglycoside-induced damage of the inner ear hair cells. Zhang
et al (7) previously found
that Fas pre-treatment can significantly inhibit the Rho signaling
pathway after exposure to Neo in hair cells, further reducing the
accumulation of ROS and cell apoptosis induced by Neo.
MicroRNAs (miRNAs/miRs) are a class of non-coding
single-stranded RNA molecules with a length of 22 nucleotides
(29). They are involved in the
regulation of gene expression on a post-transcriptional level in
plants and animals, and can therefore regulate various
physiological and pathological processes, including antibacterial
resistance, cell proliferation, autophagy and apoptosis (30-32).
miRNAs recognize and bind to complementary target sequences in the
3'-untranslated region (UTR) region of target mRNA sequences,
leading to mRNA degradation and inhibition of target gene
translation (33). miR-489 has been
shown to inhibit a number of different cancers, including
osteosarcoma, gastric, lung, ovarian, liver and bladder cancer
(34-36).
Soni et al (37) previously
reported that miR-489 could affect the expression of several genes,
such as LAPTM4B involved in autophagy in breast cancer cells. Liao
et al (38) pointed out
miR-489 specifically by inhibiting autophagy by targeting Unc-51
like autophagy activating kinase and lysosomal protein
transmembrane 4β in ovarian cancer cells.
Therefore, the present study was undertaken to
investigate the effects of Fas on the cochlear hair cell line
HEI-OC1 following induction by Neo. In addition, the present study
determined whether its effects were mediated by regulating the
expression of miR-489 and the autophagy-related nuclear dot protein
52 (NDP52) en route to regulating Neo-induced autophagy and hair
cell injury.
Materials and methods
Cell culture
The HEI-OC1 mouse inner ear hair cell line,
originated from House Ear Institute (https://houseinstitute.com/), was purchased from
Biofeng (cat. no. CVCL_D899; http://www.biofeng.com/xibao/xibaozhu/HEI-OC1.html)
and cultured in high-glucose DMEM supplemented with 10% FBS. All
cell culture media and reagents were purchased from Gibco; Thermo
Fisher Scientific, Inc. All cells were incubated in 5%
CO2 at 37˚C. When the cell confluence reached 80%,
subculture was conducted.
Flow cytometry
Flow cytometry was performed with a PI/Annexin V
Cell Apoptosis Detection kit (Sigma-Aldrich; Merck KGaA) according
to the manufacturer's protocols. Briefly, the HEI-OC1 cells were
inoculated into 6-well plates and divided into Control, Neo, Neo +
Fas/Low, Neo + Fas/Mid and Neo + Fas/High groups. When the cells
grew to ~70% confluence, 5 mM Neo was added. Fas was administered
at 5, 10 and 20 µM doses for low, medium and high groups,
respectively. Fas was added to the treatment groups for 1 h prior
to Neo induction. After 24 h of treatment at room temperature, the
cells at a density of 1x105 cell/well were collected and
washed with binding buffer prior to staining with Annexin V-FITC
and PI (5 µl). The percentage of both early and late apoptotic
cells was calculated using flow cytometry (BD FASCanto™ II; BD
Biosciences). The apoptosis analysis was performed by FlowJo V10
software (version 10; Emerald Biotech Co., Ltd).
Cell treatment
Cells were cultured at a density of 1x105
cell/well and 5 mM Neo was added to each well. The medium dose of
Fas (10 µM) was administered to the treatment groups for 1 h prior
to Neo induction, which was used in the following experiments.
Transient transfection assay
Recombinant adenovirus vector plasmid of NDP52
overexpression (Ad5-EGFP-NDP52 OE) and control plasmid (Ad5-EGFP)
were synthesized by Beijing SinoGenoMax Research Center Co., Ltd.
miR-489 mimic (5'-GUGACAUCACAUAUACGGCAGC-3'), miR-negative control
(NC) mimic (5'-UUGUCCGAACGUGUCACGUTT-3'), miR-489 inhibitor (5'-
GCTGCCGTATATGTGATGTCAC-3') and miR-NC inhibitor
(5'-CAGUACUUUUGUGUAGUA CAA-3') were purchased from Guangzhou
RiboBio Co., Ltd.
For adenovirus infection, HEI-OC1 cells were seeded
in 6-well plates at a density of 1x106 cell/well and
infected with 100 MOI adenovirus for 2 days. When ~80% confluence
was reached, HEI-OC1 cells were transfected with 30 nM plasmid
overexpression vectors, miR-489 mimic, miR-489 inhibitor or the
negative control using Lipofectamine® 2000 (Thermo
Fisher Scientific, Inc.) for 2 days at 37˚C, according to the
manufacturer's protocols. Subsequently, cells were treated with 5
mM Neo and/or 10 µM Fas.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from cultured cells by using
TRIzol® reagent (Thermo Fisher Scientific, Inc.) and 1
µg total RNA was reverse-transcribed into cDNA by using AMV Reverse
Transcriptase XL*1 (5 U/µl; Takara Bio, Inc.) and a RT
primer according to the manufacturer's protocols. The temperature
protocol was as follows: 16˚C for 30 min, 42˚C for 30 min and 85˚C
for 5 min. qPCR was performed using a VetMAX™-Plus One-Step RT-PCR
kit (Applied Biosystems; Thermo Fisher Scientific, Inc.) by
following the manufacturer's protocols with U6 as the internal
control. GAPDH was used as reference control for NDP52. The
sequences of the primers were as follows: miR-9 forward,
5'-AGCTTGCTGCACCTTAGTCT-3' and reverse, 5'-TGTGTGCGGCTAGAACATCC-3';
miR-34a forward, 5'-GCGCGCAATCAGCAAGTATAC-3' and reverse,
5'-AGTGCAGGGTCCGAGGTATT-3'; miR-489 forward,
5'-ACACTCCAGCTGGGGTGACATCACATA-3' and reverse,
5'-TGGTGTCGTGGAGTCG-3'; miR-23a forward, 5'-GGGGGTTCCTGGGGATG-3'
and reverse, 5'-AGTGCAG GGTCCGAGGTATT-3'; miR-494 forward,
5'-CGCGTG AAACATACACGGGA-3' and reverse, 5'-AGTGCAGGG
TCCGAGGTATT-3'; miR-93 forward, 5'-CAAAGUGCU GUUCGUGCAGGUAG-3' and
5'-AGTGCAGGGTCCGAG GTATT-3'; miR-214 forward, 5'-GCGACAGCAGGCACA
GACA-3' and reverse, 5'-AGTGCAGGGTCCGAGGTATT-3'; NDP52 forward,
5'-GACAACCCGTGAGTATTACACC-3' and reverse,
5'-TGGAAAGGAATACTTGCTCCC-3'; U6 forward,
5'-GCTTCGGCAGCACATATACTAAAAT-3' and reverse,
5'-CGCTTCACGAATTTGCGTGTCAT-3'; and GAPDH forward,
5'-AGCAGTCCCGTACACTGGCAAAC-3' and reverse,
5'-TCTGTGGTGATGTAAATGTCCTCT-3'. The reactions were performed in a
96-well plate at 95˚C for 10 min, followed by 40 cycles at 95˚C for
10 sec and 60˚C for 1 min. The relative gene expression levels were
normalized to the level of the endogenous control GAPDH, and were
calculated using the 2-ΔΔCT method (39).
Western blot analysis
Total protein was extracted from cells with RIPA
lysis buffer (Beyotime Institute of Biotechnology). After measuring
the protein concentrations using the Bradford assay, 40 µg protein
was loaded, separated by using 10% SDS-PAGE and then transferred
onto PVDF membranes (Merck KGaA). Next, the membranes were
incubated with 0.5% bovine serum albumin (Gibco; Thermo Fisher
Scientific, Inc.) for 1 h at room temperature, followed by washing
in PBS. The membranes were then incubated with primary antibodies
at 4˚C overnight, followed by washing and incubation in
HRP-conjugated secondary antibodies (1:2,000; cat. no. A0208;
Beyotime Institute of Biotechnology) at room temperature for 1-2 h.
The primary antibodies used were anti-NDP52 (1:1,000, cat. no.
ab68588; Abcam), anti-LC3B (1:1,000, cat. no. ab51520; Abcam) and
anti-Beclin 1 (1:1,000, cat. no. ab210498; Abcam). GAPDH (1:2,000,
cat. no. ab9485; Abcam) served as a loading control. Finally, the
bands were evaluated using scanning densitometry (ImageQuant™
LAS4000; Cytiva) and analyzed using ImageJ software (version
V1.0.8; National Institutes of Health) following Pierce™ ECL Plus
Western Blotting Substrate treatment (Thermo Fisher Scientific,
Inc.).
Dual-luciferase reporter assay
HEI-OC1 cells were cultured in six-well plates at a
density of 1x106 cell/well. TargetScan (http://www.targetscan.org/vert_70/), miRWalk
(http://mirwalk.umm.uni-heidelberg.de/), miRNet
(https://www.mirnet.ca/miRNet/home.xhtml), miRDB
(http://mirdb.org/), microT-CDS (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=microT_CDS/index),
miRSystem (https://tools4mirs.org/software/target_functional_analysis/mirsystem/)
and miRNA MAP (http://mirnamap.mbc.nctu.edu.tw./) were used to
predict microRNAs that may target the NDP52 mRNA 3'-UTR.The
putative binding sites of miR-489 on the NDP52 3'-UTR were
predicted using the TargetScan 7.0 (http://www.targetscan.org/vert_70/) online tool. The
NDP52 3'-UTR sequences were chemically synthesized by Guangzhou
RiboBio Co., Ltd. and introduced into the luciferase reporter
vector (pGL3-Basic) to construct wild-type (WT) luciferase reporter
plasmids (NDP52 WT), and the seed regions of miR-489 in the 3'-UTR
of NDP52 were mutated to construct mutant (MUT) luciferase reporter
plasmids (NDP52 MUT). HEI-OC1 cells were co-transfected with 0.5
µg/μl luciferase reporter plasmids (NDP52 WT or NDP52 MUT in the
pGL3-Basic vector; Guangzhou RiboBio Co., Ltd.), miR-489 mimics or
inhibitors, and their negative control using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.).
After 48 h, the cells were collected and measured using the
Dual-Luciferase Reporter Assay (Promega Corporation) according to
the manufacturer's protocols. The dual-luciferase activity of the
target gene was normalized to Renilla luciferase
activity.
MitoTracker mitochondrial
staining
MitoTracker (Thermo Fisher Scientific, Inc.) was
added to serum-free DMEM (Gibco; Thermo Fisher Scientific, Inc.)
and diluted to a concentration of 200 nM, before the cell culture
medium was replaced with this serum-free medium containing 200 nM
MitoTracker, followed by incubation at 37˚C for 25 min. After
incubation, the cells were washed with serum-free medium and
staining was observed under a fluorescence microscope
(magnification, x200; Zeiss 710; Carl Zeiss AG).
Mitochondrial membrane potential
detection
The mitochondrial membrane potential was detected
using JC-1 fluorescence mitochondrial imaging (40). JC-1 (200X; cat. no. C2006; Beyotime
Institute of Biotechnology) was diluted by adding ultra-pure water
(ddH2O) following the addition of JC-1 buffer (5X) to
prepare JC-1 staining buffer (1X). The cells at a density of
1x105 cell/well were incubated with JC-1 staining
solution (1X) at 37˚C for 20 min. After incubation, the supernatant
was removed and cells were washed with JC-1 staining buffer (1X)
for two times. DAPI (100 ng/ml; Beijing Solarbio Science &
Technology Co., Ltd.) was used for counterstaining of the cell
nuclei at 37˚C for 10 min and the cells were observed under a
fluorescence microscope (magnification, x200; ZEISS 710; Carl Zeiss
AG). When detecting JC-1 monomer (green), excitation was set at 490
nm and the emission was set at 530 nm. For the detection of JC-1
aggregation (red), excitation light was set at 525 nm and the
emission was set at 590 nm. The ratio of red to green fluorescence
represented the mitochondrial membrane potential.
Mitochondrial autophagy detection
Mitochondrial autophagy was detected by transfection
of the green fluorescent protein GFP-LC3B plasmid synthesized by
Nanjing GenScript Biotechnology Co., Ltd. and combined with
MitoTracker Red (Beyotime Institute of Biotechnology) fluorescence
staining. HEI-OC1 cells at a density of 1x105 cell/well
were cultured in 24-well plates. The cells were divided into the
following three groups: Control group, Neo group and Neo + Fas
group. The GFP-LC3B plasmid (500 ng), P3000™ reagent (1 µl; Thermo
Fisher Scientific, Inc.; used to enhance the cell transfection
efficiency of Lipofectamine 3000) and Lipofectamine 3000 (1 µl;
Thermo Fisher Scientific, Inc.) were added into the fresh culture
medium (50 µl) for 20 min at room temperature and the
aforementioned medium was added into each well, and cultured for a
further 8 h, according to the manufacturer's protocol. MitoTracker
Red working solution (PBS diluted) with a concentration of 25 nM
was prepared and put into the cell culture incubator for
preheating. After rinsing the cells with PBS three times, the
MitoTracker Red working solution was added to the cells at 500
µl/well (final concentration, 12.5 µM) and incubated at 37˚C for 20
min. After the incubation, the cells were washed three times with
pre-cooled PBS, and DAPI (100 ng/ml; Beijing Solarbio Science &
Technology Co., Ltd.) was used for counterstaining of the cell
nuclei at 37˚C for 10 min. Then, cells were observed under a
fluorescence microscope at x200 magnification (ZEISS 710; Carl
Zeiss AG). Green light represents the position of GFP-LCB and red
light represents the position of mitochondria. The superposition of
green fluorescence and red fluorescence is regarded as autophagy in
mitochondria.
Fluorescence detection of
intracellular ROS levels
The intracellular ROS levels were examined using
2,7-dichlorfluoresceindiacetate staining (DCFH-DA; Beyotime
Institute of Biotechnology) according to the manufacturer's
protocols. Briefly, the number of cells in each group after drug
treatment was adjusted to 1x106-2x107.
DCFH-DA was diluted in serum-free DMEM at 1:1,000 to a final
concentration of 10 µM. The cells were collected and suspended in
the DCFH-DA-containing serum-free medium to a cell concentration of
1x106-2x107/ml. The cells were incubated at
37˚C for 20 min. The cells were then washed three times with
serum-free DMEM to fully remove DCFH-DA that did not enter the
cells. Flow cytometry (BD Biosciences) was used for detection of
the fluorescence intensity of intracellular ROS using 488 nm
excitation wavelength and 525 nm emission wavelength. The
fluorescence analysis was performed by FlowJo V10 software (version
10; Emerald Biotech Co., Ltd)
Statistical analysis
All values are presented as the mean ± SEM from
three independent experiments. Statistical analysis was performed
by using one-way ANOVA followed by Bonferroni's test for selected
pairs using the GraphPad Prism 5 statistical software (GraphPad
Software, Inc.). P<0.05 was considered to indicate statistically
significant difference.
Results
Fas inhibits Neo-induced ototoxicity
at the cellular level
Neo exhibits strong ototoxicity and can induce hair
cell death (41). To investigate
the effect of Fas on ototoxicity induced by Neo at the cellular
level, flow cytometry was used to analyze Fas- and Neo-treated
HEI-OC1 hair cells. The results demonstrated that Neo significantly
increased HEI-OC1 cell apoptosis, whilst Fas significantly
inhibited Neo-induced apoptosis of HEI-OC1 cells in a
dose-dependent manner (Fig. 1A and
B). Since Fas at 5, 10 and 20 µM
all exerted satisfactory effects, the medium dose of Fas (10 µM)
was selected for subsequent experiments.
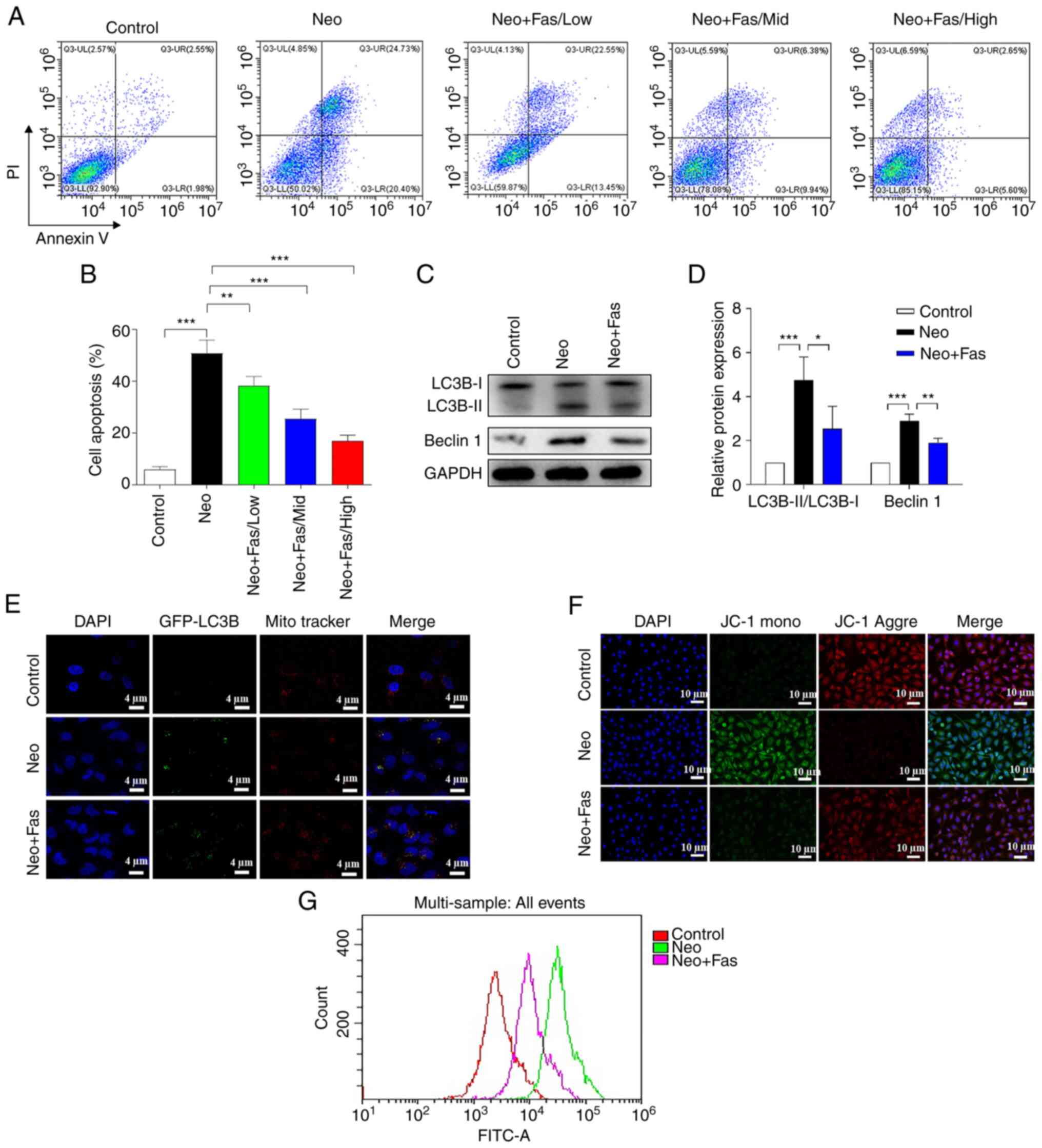 | Figure 1Fas inhibits Neo-induced ototoxicity
at the cellular level. HEI-OC1 cells were treated with 5 mM Neo to
induce apoptosis. Fas at 10 µM was added 1 h before induction.
Testing was performed after 24 h of treatment. (A) Cell apoptosis
was detected with flow cytometry after HEI-OC1 cells were treated
with Neo and Fas, (B) which was quantified. (C) Western blot
analysis of autophagy-associated protein expression s (LC3B and
Beclin1). The gray analysis of LC3B and Beclin1 in the control
group was classified as 1. (D) Reverse transcription-quantitative
PCR was performed to detect the mRNA level of autophagy-associated
proteins. (E) Fluorescence images of LC3B-GFP and mitochondria.
Red, mitochondria; green, autophagosomes; blue, nucleus. Scale bar,
4 µm. (F) Fluorescence images of HEI-OC1 with JC-1. Red, JC-1
aggregates; green, JC-1 monomers; blue, nucleus. Scale bar, 10 µm.
(G) Reactive oxygen species levels were detected with flow
cytometry after 2,7-dichlorfluoresceindiacetate staining. Scale
bar, 10 µm. n=6 in each group, each independent experiment was
repeated three times. Data are expressed as mean ± SEM.
*P<0.05, **P<0.01,
***P<0.001. Neo, neomycin; Fas, fasudil; GFP, green
fluorescent protein. |
Previous studies have shown that Neo induces
autophagy in hair cells, which plays an important role in their
survival (42,43). In addition, Fas has been reported to
inhibit angiotensin-II-induced autophagy of podocytes as a ROCK
inhibitor (44). Therefore, the
effects of Neo and Fas on autophagy in hair cells of the inner ear
were examined. Western blotting was used to detect the protein
expression of the autophagy-related markers LC3B and Beclin 1. Neo
significantly increased the LC3B-II/LC3B-I ratio and Beclin 1
expression in HEI-OC1 cells, suggesting that Neo induced activation
of inner ear hair cell autophagy (Fig.
1C and D). Fas markedly
reversed Neo-induced increases of LC3B-II/LC3B-I ratio and Beclin 1
expression (Fig. 1C and D), suggesting that Fas inhibited
Neo-induced activation of autophagy in hair cells. Cell autophagy
can be divided into non-selective autophagy or the specific
autophagy of organelles, including mitochondrial, peroxidase,
endoplasmic reticulum and ribosomal autophagy (45-47).
Furthermore, it has been previously reported that mitochondrial
fragmentation and reductions in the membrane potential are
conditions for mitochondrial autophagy (48-50).
Therefore, it was explored in the present study whether the effect
of Fas on Neo-induced autophagy was selective or non-selective. As
displayed in Fig. 1E, following Neo
treatment, GFP-LC3B was increased and co-localized with the
mitochondria, highlighted by Mito Tracker, compared with the
control group. These findings suggested mitochondrial autophagy was
activated. However, green fluorescence was reduced in the Neo + Fas
group compared with the Neo group, suggesting that Neo-induced
mitochondrial autophagy was suppressed by Fas.
Subsequently, JC-1 and the fluorescent probe DCFH-DA
were used to detect the effects of Neo and Fas on the mitochondrial
membrane potential and the level of ROS in hair cells,
respectively. Following treatment with Neo, green fluorescence
increased and red fluorescence decreased, suggesting that Neo
induced a reduction in the mitochondrial membrane potential of hair
cells (Fig. 1F). By contrast, in
cells treated with Fas, the effect of Neo on the mitochondrial
membrane potential of hair cells was reversed (Fig. 1F). The fluorescent probe DCFH-DA was
then used to detect the effects of Neo and Fas on ROS levels in
hair cells. As shown in Fig. 1G,
Neo increased ROS levels in hair cells, but Fas inhibited the
Neo-induced increase in ROS levels (Fig. 1G).
Fas inhibits Neo-induced autophagy and
cellular injury by reducing NDP52 expression
In the present study, the effect of Neo on the
expression of the NDP52 protein in hair cells was investigated.
Results of western blotting demonstrated that Neo induced a
significant increase in NDP52 protein expression, but Fas
significantly inhibited the Neo-induced increase in NDP52 protein
levels (Fig. 2A).
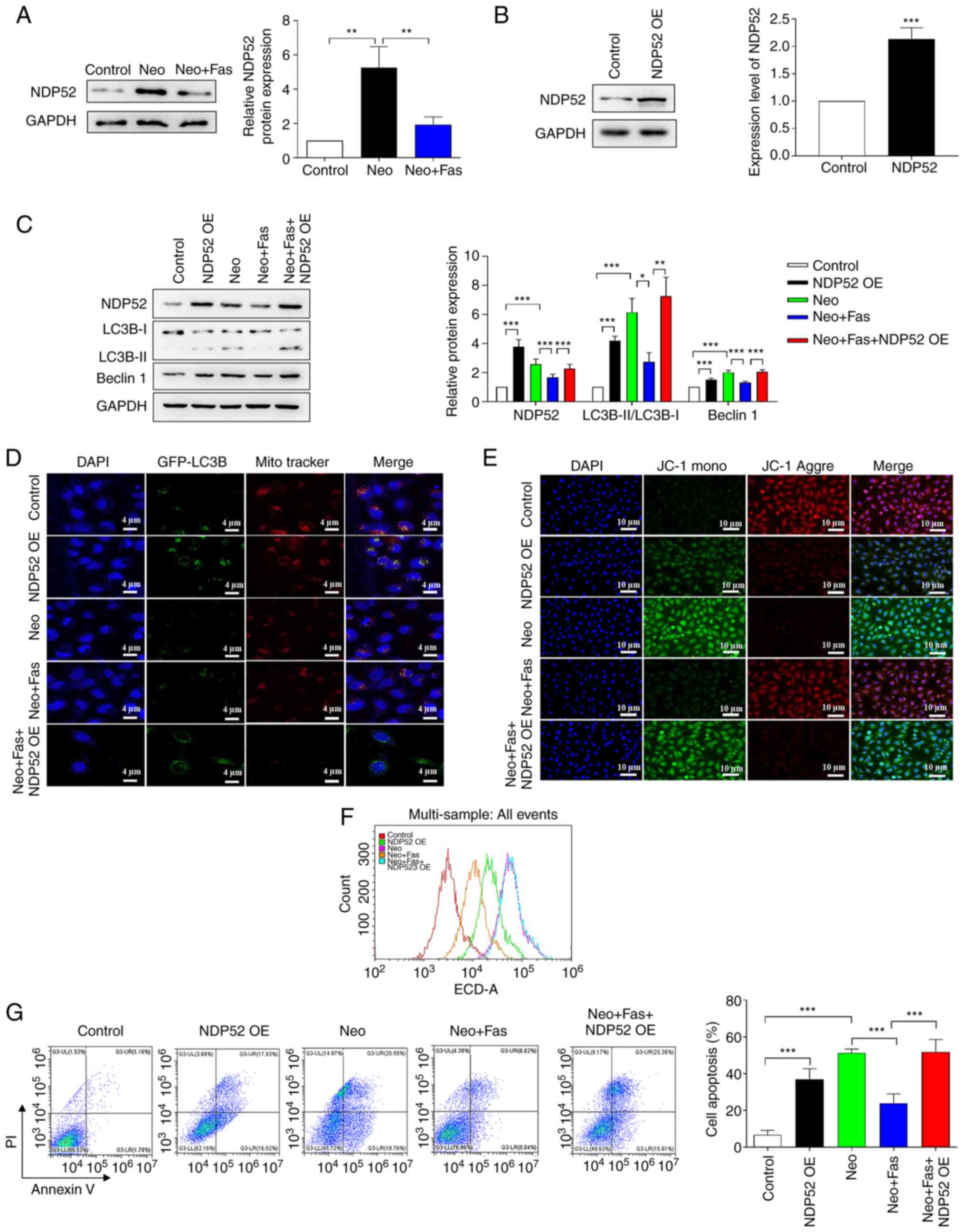 | Figure 2Fas inhibits Neo-induced autophagy
and cellular damage by reducing NDP52 expression. (A) Western blot
analysis of NDP52 protein expression and grayscale analysis. (B)
NDP52 expression was measured in HEI-OC1 cells following
transfection with NDP52 plasmids. (C) Western blot analysis of
autophagy-associated protein expression and grayscale analysis. The
gray analysis of the control group was classified as 1 in western
blot analysis. (D) Fluorescence images of GFP-LC3B and
mitochondria. Red, mitochondria; green, autophagosome; blue,
nucleus. Scale bar, 4 µm. (E) Fluorescence images of HEI-OC1 with
JC-1. Red, JC-1 aggregates; green, JC-1 monomer; blue, nucleus.
Scale bar, 10 µm. (F) Reactive oxygen species levels were detected
with flow cytometry after 2,7-dichlorfluoresceindiacetate staining.
(G) Cell apoptosis was detected with flow cytometry after NDP52
plasmid-transfected HEI-OC1 cells were treated with Neo and Fas.
n=6 in each group, each independent experiment was repeated three
times. Data are expressed as mean ± SEM. *P<0.05,
**P<0.01, ***P<0.001. Neo, neomycin;
Fas, fasudil; NDP52, nuclear dot protein 52; OE, overexpression;
GFP, green fluorescent protein. |
To determine whether Fas regulated mitochondrial
autophagy by regulating NDP52, NDP52 was overexpressed in hair
cells. As shown in Fig. 2B, the
results demonstrated that NDP52 expression was significantly
increased in cells following transfection with NDP52 overexpression
plasmids. The effect of NDP52 overexpression on mitochondrial
autophagy was then investigated. As shown in Fig. 2C, overexpression of NDP52 or Neo
treatment significantly promoted the activation of autophagy,
characterized as the increase of Beclin-1 expression and LC3B
II/LC3B I ratio, but Fas significantly inhibited Neo-induced
activation of autophagy (Fig. 2C).
However, following overexpression of NDP52, the inhibitory effect
of Fas on autophagy was weakened or even lost (Fig. 2C).
Mitochondrial colocalization experiments revealed
that overexpression of NDP52 and Neo treatment promoted the
activation of autophagy. Fas inhibited the Neo-induced activation
of autophagy indicated by autophagosomes (green fluorescence)
colocalized with mitochondria (red fluorescence). However,
following overexpression of NDP52, the inhibitory effect of Fas on
autophagy was lost (Fig. 2D),
suggesting that Fas may inhibit mitochondrial autophagy by
inhibiting NDP52 expression. Measurements of mitochondrial membrane
potential revealed that green fluorescence was enhanced after
overexpression of NDP52 or Neo treatment, whilst the red
fluorescence was decreased, suggesting that Neo induced a decrease
in the mitochondrial membrane potential of hair cells (Fig. 2E). Fas reversed the reduction in
mitochondrial membrane potential in hair cells induced by Neo
(Fig. 2E). However, in cells
overexpressing NDP52, Fas failed to inhibit the Neo-induced
decrease in the mitochondrial membrane potential in hair cells
(Fig. 2E). Therefore, these
observations suggested that overexpression of NDP52 blocked the
inhibitory effects of Fas on the Neo-induced reduction of hair cell
mitochondrial membrane potential.
To verify that Fas inhibited Neo-induced hair cell
injury by upregulating the expression of NDP52, ROS levels were
measured and the results demonstrated that the overexpression of
NDP52 or Neo treatment increased the intracellular ROS levels
(Fig. 2F). By contrast, ROS levels
in cells treated with Fas were markedly lower compared with those
induced by Neo (Fig. 2F). However,
Fas failed to inhibit the Neo-induced increase in ROS in
NDP52-overexpressing cells (Fig.
2F). To verify that the overexpression of NDP52 blocked the
inhibitory effect of Fas on Neo-induced hair cell apoptosis, flow
cytometry was used to detect apoptosis. Compared with those in the
control group, the apoptosis of NDP52-overexpressing and
Neo-treated cells was increased significantly (Fig. 2G). Compared with the Neo group, cell
apoptosis in the Neo + Fas group was significantly reduced.
However, Fas could not inhibit Neo-induced apoptosis in
NDP52-overexpressing cells (Fig.
2G).
Fas post-transcriptionally regulates
NDP52 expression through miR-489
Thus far, results from the present study suggested
that NDP52 is an important factor in mitochondrial autophagy
activation, whereas Fas could inhibit mitochondrial autophagy
activation by reducing the expression of NDP52. To explore the
mechanism through which Fas regulated NDP52 levels, RT-qPCR was
used to detect the effect of Neo and Fas on NDP52 mRNA expression.
As shown in Fig. 3A, the mRNA
levels of NDP52 both in the Neo and Neo + Fas groups did not change
significantly compared with those in control group. As western
blotting results (Fig. 2A)
demonstrated that Fas could inhibit the Neo-induced increase of
NDP52 protein expression, Fas is suggested to regulate the
expression of NDP52 at the post-transcriptional level.
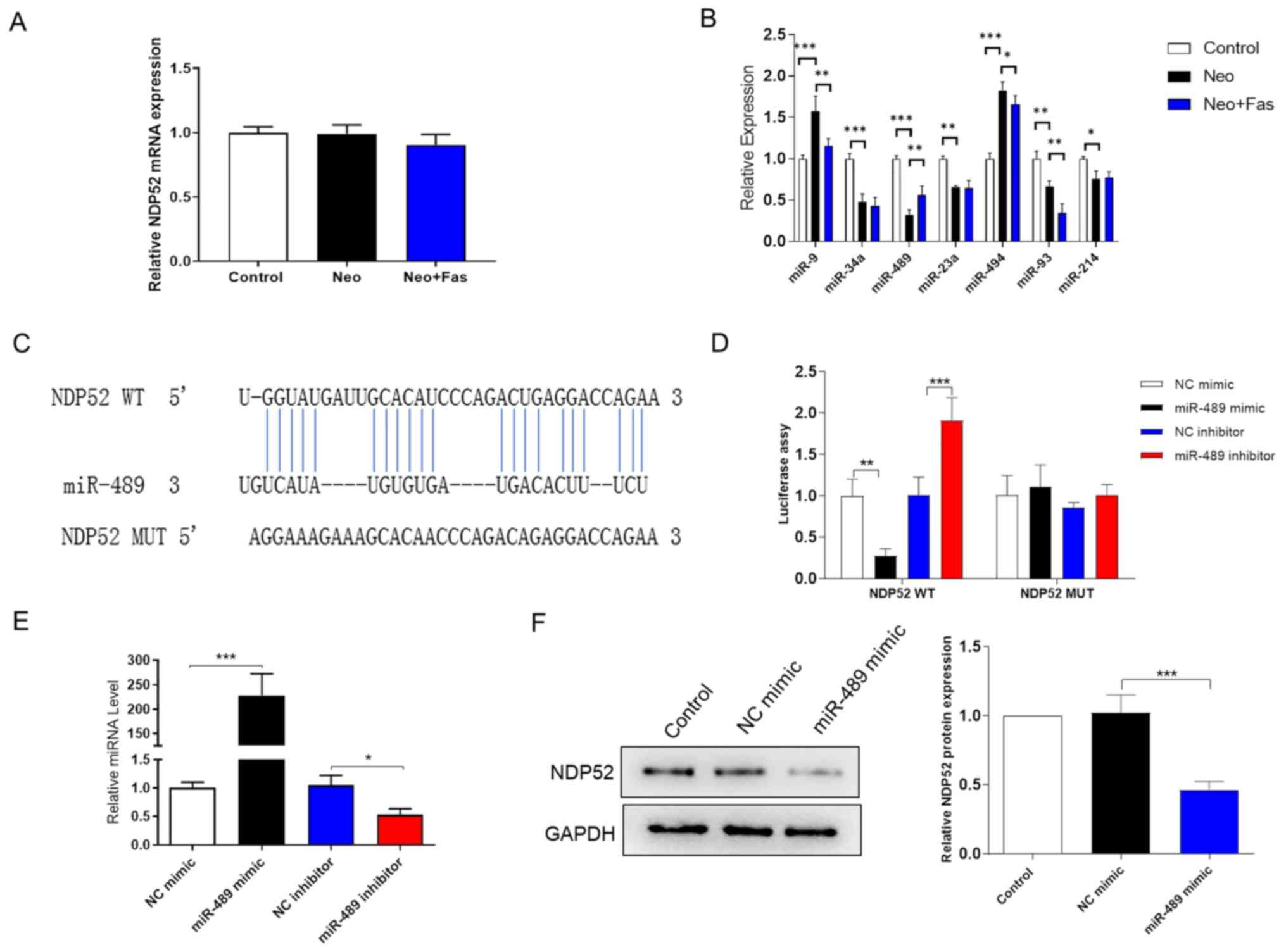 | Figure 3Post-transcriptional regulation of
NDP52 by Fas is at least in part mediated by miR-489. (A) Reverse
transcription-quantitative PCR was used to detect the effects of
Neo and Fas on the mRNA level of NDP52. (B) Expression levels of a
panel of miRNAs were detected by reverse transcription-quantitative
PCR. (C) Bioinformatics analysis predicted the 3’-UTR binding site
of miR-489 on the NDP52 mRNA sequence. (D) Luciferase report assay
was used to assess the binding of miR-489 to the 3’-UTR of NDP52
mRNA. (E) Transfection efficiency of the miR-489 mimic and miR-489
inhibitor. (F) Western blotting was used to detect the expression
of NDP52 after overexpression of miR-489 in HEI-OC1 cells. The gray
analysis of the control group was classified as 1. n=6 in each
group, each independent experiment was repeated three times. Data
are expressed as mean ± SEM. *P<0.05,
**P<0.01, ***P<0.001. NDP52, nuclear
dot protein 52; UTR, untranslated region; WT, wild-type; MUT,
mutant; NC mimic, negative control mimic; miR, microRNA; Neo,
neomycin; Fas, fasudil. |
Inhibition of target gene expression by miRNAs is a
common post-transcriptional regulatory mechanism (51). It is known that the expression level
of the miRNA is inversely associated with the expression level of
the target protein (31).
Therefore, TargetScan, miRWalk, miRNet, miRDB, microT-CDS,
miRSystem and miRNA MAP were used to predict microRNAs that may
target the NDP52 mRNA 3'-UTR. In total, seven miRNAs (miR-9,
miR-34a, miR-489, miR-23a, miR-494, miR-93 and miR-214) were
screened out by intersecting with the reported articles associated
with deafness and autophagy (31,52-55).
RT-qPCR was then used to detect the effect of Fas and Neo on miRNA
expression in HEI-OC1 cells. The results shown in Fig. 3B demonstrated that Neo significantly
inhibited the expression of miR-489 compared with that in the
control group, whilst Fas inhibited the effect of Neo on miR-489
expression. However, the expressions of miR-9, miR-34a, miR-23a,
miR-93, and miR-214 were not in accordance with the above
trend.
As shown in Fig. 3C,
the NDP52 gene was found to contain two putative sites on the
3'-UTR that matched the miR-489 seed region. As shown in Fig. 3D, the miR-489 mimic could inhibit
the luciferase activity compared with that transfected with the
mimic NC, while the miR-489 inhibitor group enhanced the luciferase
activity compared with that in the inhibitor NC group. However,
after mutating the binding site of miR-489 in the 3'-UTR region of
NDP52, neither the miR-489 mimic nor the miR-489 inhibitor could
significantly affect the luciferase activity (Fig. 3D). These results suggested that
miR-489 can directly bind to the NDP52 3'-UTR region. The
transfection efficiency of the miR-489 mimic and inhibitor was
verified by RT-qPCR (Fig. 3E).
Western blotting was then used to detect the expression of NDP52 in
HEI-OC1 cells after miR-489 overexpression. The results
demonstrated that the miR-489 mimic significantly reduced the
protein expression of NDP52 compared with that in cells transfected
with the mimic NC (Fig. 3F).
Fas inhibits Neo-induced autophagy and
cellular damage by increasing miR-489 levels
To explore the role of miR-489 in the regulation of
autophagy by Fas, miR-489 expression was either overexpressed
(miR-489 mimic) or knocked down (miR-489 inhibitor) in hair cells,
before the cells were treated with Neo and Fas. RT-qPCR was first
used to detect the expression of miR-489 in the cells. Neo
treatment significantly decreased the expression of miR-489,
whereas both Fas and miR-489 mimic significantly increased the
level of miR-489 in Neo-treated cells (Fig. 4A). The level of miR-489 expression
was significantly decreased in the Neo + Fas + miR-489 inhibitor
group compared with the Neo + Fas group. As shown in Fig. 4B and C, the levels of LC3B-II/LC3B-I and Beclin
1 in Neo-treated cells were significantly increased compared with
the control group, suggesting enhanced autophagy. Both miR-489
mimic and Fas inhibited the activation of Neo-induced autophagy
(Fig. 4B and C). However, compared with the Neo + Fas
group, following treatment with the miR-489 inhibitor, Fas could
not inhibit Neo-activated autophagy in the Fas + Neo + miR-489
inhibitor group (Fig. 4B and
C).
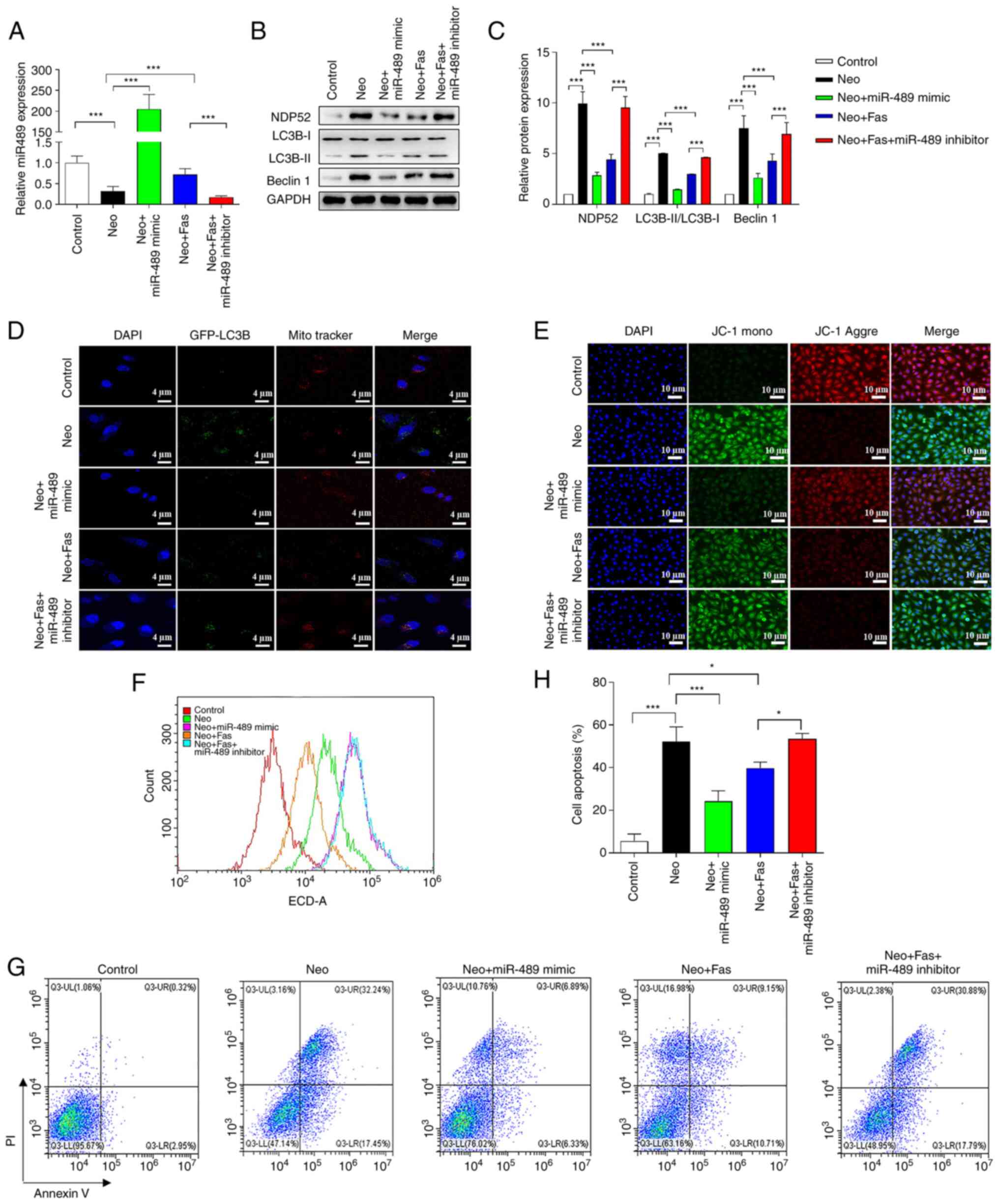 | Figure 4Fas inhibits Neo-induced autophagy
and cellular injury by increasing miR-489 levels. (A) miR-489
expression in each treatment group was detected by reverse
transcription-quantitative PCR. (B) Western blotting was used to
detect the expression of autophagy-related proteins in HEI-OC1
cells after overexpression or knockdown of miR-489. (C) ImageJ
software grayscale analysis. The gray analysis of the control group
was classified as 1. (D) Fluorescence images of GFP-LC3B and
mitochondria. Red, mitochondria; green, autophagosome; blue,
nucleus. Scale bar, 4 µm. (E) Fluorescence images of HEI-OC1 cells
with JC-1. Red, JC-1 aggregates; green, JC-1 monomer; blue,
nucleus. Scale bar, 10 µm. (F) Reactive oxygen species levels were
detected with flow cytometry after 2,7-dichlorfluoresceindiacetate
staining. (G) Cell apoptosis was detected with flow cytometry after
NDP52 plasmid-transfected HEI-OC1 cells were treated with Neo and
Fas. (H) Statistical analysis of flow cytometry. n=6 in each group,
each independent experiment was repeated three times. Data are
expressed as mean ± SEM. *P<0.05,
***P<0.001. NDP52, nuclear dot protein 52; miR,
microRNA; Neo, neomycin; Fas, fasudil. |
Mitochondrial co-localization experiments
demonstrated that mitochondrial autophagy was enhanced in
Neo-treated cells, whereas overexpression of miR-489 using the
miR-489 mimic and Fas could inhibit the activation of mitochondrial
autophagy (Fig. 4D). However,
compared with the Neo + Fas group, Fas could not inhibit the
activation of mitochondrial autophagy by Neo in the Neo + Fas +
miR-489 inhibitor group (Fig. 4D).
These results suggested that Fas inhibited mitochondrial autophagy
by increasing the level of miR-489 expression. The detection
results of mitochondrial membrane potential suggested that both Fas
and miR-489 mimic inhibited the Neo-induced reduction in the
mitochondrial membrane potential (Fig.
4E). However, the mitochondrial membrane potential in the Neo +
Fas group was higher than that in in the Neo + Fas + miR-489
inhibitor group (Fig. 4E).
ROS detection results demonstrated that
intracellular ROS levels increased after Neo treatment, whilst ROS
levels in cells treated with Fas or miR-489 mimic were markedly
lower compared with those induced by Neo alone (Fig. 4F). In the Neo + Fas + miR-489
inhibitor group, Fas failed to inhibit the Neo-induced increase in
ROS levels in hair cells (Fig. 4F).
The results from flow cytometry showed that the level of apoptosis
after Neo treatment was significantly increased, but the level of
apoptosis of cells treated with Fas or miR-489 mimic was
significantly lower compared with that induced by Neo (Fig. 4G and H). Furthermore, Fas could not inhibit
Neo-induced apoptosis in hair cells with miR-489 inhibitor
(Fig. 4G and H).
Discussion
Fas is a new isoquinoline sulfonamide derivative, an
effective ROCK-II inhibitor and a calcium antagonist (56). Fas is mainly used for clinically
treating subarachnoid hemorrhage caused by cerebral vasospasm
(57). The present study revealed
the inhibitory effects of Fas on Neo-induced inner ear hair cell
damage. Further mechanistic studies suggested that Fas reduced the
expression of the autophagy-related protein NDP52 at the
post-transcriptional level by upregulating the expression of
miR-489, thereby inhibiting the activation of Neo-induced autophagy
whilst preventing Neo-induced hair cell injury.
The mechanism through which ototoxic drugs cause
hair cell damage and apoptosis is complex, though the accumulation
of ROS in the cell has been reported to activate multiple signaling
pathways associated with apoptosis, such as the ROS/JNK pathway and
the caspase-independent apoptosis pathway (58,59),
and can serve an important role in inducing hair cell death. For
example, mitochondrial calcium uptake is based on ROS generation
during aminoglycoside-induced hair cell death (60). Cell survival requires the
maintenance of balance between oxidative stress and antioxidant
defense systems. When ROS production exceeds the limit of cell
repair, cell damage ensues, where ROS accumulation leads to cell
death (61,62).
Autophagy is a mechanism for the orderly degradation
and renewal of non-essential or damaged cellular components in all
eukaryotes (63,64). Autophagosomes engulf organelles or
other cytoplasmic components that must be degraded into a bilayer
membrane, which then fuse with lysosomes; the contents are degraded
through lysosomal hydrolysis (65-67).
In certain cases, autophagy can induce autophagic cell death, where
the overactivation of autophagy can cause cell death through a
variety of degradation pathways, such as macro-autophagy and
peroxisome degradation pathways (68). Autophagy attenuates noise-induced
hearing loss by reducing oxidative stress (69). Neo can induce hair cell injury,
possibly through the increased levels of autophagy in hair cells
(43,70). When autophagy occurs, cytoplasmic
LC3-I enzymatically cleaves a small polypeptide fragment to become
membrane-type LC3-II (48).
Therefore, the value of the LC3-II/I ratio may be used to estimate
changes in autophagy levels (71).
Results of a previous study demonstrated that Fas
regulates autophagy to alleviate cell damage. For example, Fas
suppressed TGF-ß-mediated autophagy in urethra fibroblasts of mice
to attenuate traumatic urethral stricture (72). Moreover, autophagy inhibition
stimulated apoptosis in oesophageal squamous cell carcinoma treated
with Fas (73). Thus, the present
study aimed to further explore the potential effects of Fas on
ototoxicity induced by Neo, and determine whether this was
associated with autophagy. HEI-OC1 cells were divided into the
control group, Neo group and Neo + Fas group, following which the
dose-dependent effects of Fas on Neo-induced apoptosis in cells
were assessed via flow cytometry. Further experiments demonstrated
that Fas inhibited the increase of mitochondrial autophagy induced
by Neo, inhibited the decrease in mitochondrial membrane potential
induced by Neo, reduced ROS levels in HEI-OC1 cells and inhibited
cell apoptosis.
The mechanism through which Fas regulates autophagy
was then investigated. NDP52 is a newly discovered
autophagy-related protein that can bind to the LC3 protein on the
surface of autophagosomes to promote autophagy (74,75).
Gibbings et al (75,76) previously found that the expression
level of miR-489 was decreased following induction by Neo. The
inhibitory effects of Fas on Neo-induced mitochondrial autophagy,
mitochondrial membrane potential decline, ROS levels and apoptosis
were blocked by the overexpression of NDP52, which was verified in
HEI-OC1 cells. It was subsequently explored further how Fas
regulated the reduction in NDP52 protein expression. Several
bioinformatic analysis software packages were used to predict
miRNAs that may target the NDP52 mRNA 3'-UTR. In total, seven
miRNAs (miR-9, miR-34a, miR-489, miR-23a, miR-494, miR-93 and
miR-214) were screened out by intersecting with previous articles
related to deafness and autophagy (31,52-55,77-79).
It was predicted and verified that miR-489 could bind to the 3'-UTR
of NDP52 mRNA to negatively regulate the expression of NDP52. A
series of overexpression and knockdown experiments were also
conducted to verify that Fas can inhibit Neo-induced autophagy
activation by upregulating the expression of miR-489. This reduced
the expression of the autophagy-related protein NDP52 at the
post-transcriptional level, thereby inhibiting Neo-induced
autophagy activation and preventing Neo-induced hair cell
damage.
In conclusion, Fas is a low toxicity drug, which has
been widely used in the clinical treatment of postoperative
cerebral vasospasm in patients with subarachnoid hemorrhage and
acute ischemic stroke (80,81). The findings of the present study
suggested that Fas may hold promise as a candidate drug for the
prevention and treatment of sensorineural deafness caused by Neo.
Furthermore, investigation into the underlying mechanism uncovered
that miR-489 may be a potential target for the treatment of
drug-induced deafness.
Acknowledgements
Not applicable.
Funding
Funding: The present study was funded by the National Natural
Science Foundation of China (grant nos. 81970884 and 81771019).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WL conceived and designed the current study,
performed all experiments and prepared the manuscript. YZ, JC and
JX contributed to acquisition, and analysis and interpretation of
data. XG assisted in experimental design, provided general
supervision and was responsible for all experimental processes. WL
and XG contributed to drafting, critical revision and editing. WL
and XG confirmed the authenticity of all the raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ng M and Horlbeck DM: Sensorineural
hearing loss-congenital-genetics. In: Encyclopedia of
Otolaryngology, Head and Neck Surgery. Kountakis SE (ed). Springer,
Berlin, Heidelberg, 2013.
|
|
2
|
Lin RJ, Krall R, Westerberg BD, Chadha NK
and Chau JK: Systematic review and meta-analysis of the risk
factors for sudden sensorineural hearing loss in adults.
Laryngoscope. 122:624–635. 2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Arkaravichien W and Schacht J: Drug
induced hearing loss: A worldwide problem. Int Med J. 4:243–251.
1997.
|
|
4
|
Cone BK, Wake M, Tobin S, Poulakis Z and
Rickards FW: Slight-mild sensorineural hearing loss in children:
Audiometric, clinical, and risk factor profiles. Ear Hear.
31:202–212. 2010.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Xiong H, Lai L, Ye Y and Zheng Y: Glucose
protects cochlear hair cells against oxidative stress and
attenuates noise induced hearing loss in mice. Neurosci Bull.
37:657–668. 2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ware SL: Human Hearing Loss. PeerJ
PrePrints. 2(e378v1)2014.
|
|
7
|
Zhang Y, Li W, He Z, Wang Y, Shao B, Cheng
C, Zhang S, Tang M, Qian X, Kong W, et al: Pre treatment with
fasudil prevents neomycin induced hair cell damage by reducing the
accumulation of reactive oxygen speciec. Front Mol Neurosci.
12(264)2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Nakagawa T, Yamane H, Takayama M, Sunami K
and Nakai Y: Time-dependent response of vestibular hair cells of
guinea pigs following high-dose applications of streptomycin. Acta
Otolaryngol Suppl. 538:32–35. 1998.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Becker B and Cooper MA: Aminoglycoside
antibiotics in the 21st century. ACS Chem Biol. 8:105–115.
2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Brignull HR, Raible DW and Stone JS:
Feathers and fins: Non-mammalian models for hair cell regeneration.
Brain Res. 1277:12–23. 2009.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Chen J, Guan L, Zhu H, Xiong S, Zeng L and
Jiang H: Transplantation of mouse-induced pluripotent stem cells
into the cochlea for the treatment of sensorineural hearing loss.
Acta Otolaryngol. 137:1136–1142. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kujawa SG and Liberman MC: Synaptopathy in
the noise-exposed and aging cochlea: Primary neural degeneration in
acquired sensorineural hearing loss. Hear Res. 330 (Pt B):191–199.
2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Liberman MC: Noise-induced and age-related
hearing loss: New perspectives and potential therapies. F1000Res.
6(927)2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Géléoc GS and Holt JR: Sound strategies
for hearing restoration. Science. 344(1241062)2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Chen Y, Huang WG, Zha DJ, Qiu JH, Wang JL,
Sha SH and Schacht J: Aspirin attenuates gentamicin ototoxicity:
From the laboratory to the clinic. Hear Res. 226:178–182.
2007.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Kamogashira T, Fujimoto C and Yamasoba T:
Reactive oxygen species, apoptosis, and mitochondrial dysfunction
in hearing loss. BioMed Res Int. 2015(617207)2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Jiang H, Sha SH and Schacht J: NF-kappaB
pathway protects cochlear hair cells from aminoglycoside-induced
ototoxicity. J Neurosci Res. 79:644–651. 2005.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Momiyama J, Hashimoto T, Matsubara A,
Futai K, Namba A and Shinkawa H: Leupeptin, a calpain inhibitor,
protects inner ear hair cells from aminoglycoside ototoxicity.
Tohoku J Exp Med. 209:89–97. 2006.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Yamashita K, Kotani Y, Nakajima Y,
Shimazawa M, Yoshimura S, Nakashima S, Iwama T and Hara H: Fasudil,
a Rho kinase (ROCK) inhibitor, protects against ischemic neuronal
damage in vitro and in vivo by acting directly on neurons. Brain
Res. 1154:215–224. 2007.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Greathouse KM, Henderson BW, Gentry EG and
Herskowitz JH: Fasudil or genetic depletion of ROCK1 or ROCK2
induces anxiety-like behaviors. Behav Brain Res.
373(112083)2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Masaoka H, Takasato Y, Nojiri T, Hayakawa
T, Akimoto H, Yatsushige H, Toumori H, Miyazaki Y and Honma M:
Clinical effect of Fasudil hydrochloride for cerebral vasospasm
following subarachnoid hemorrhage. Acta Neurochir Suppl (Wien).
77:209–211. 2001.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Scherer EQ, Arnold W and Wangemann P:
Pharmacological reversal of endothelin-1 mediated constriction of
the spiral modiolar artery: A potential new treatment for sudden
sensorineural hearing loss. BMC Ear Nose Throat Disord.
5(10)2005.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Bonnevier J, Fässler R, Somlyo AP, Somlyo
AV and Arner A: Modulation of Ca2+ sensitivity by cyclic
nucleotides in smooth muscle from protein kinase G-deficient mice.
J Biol Chem. 279:5146–5151. 2004.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Lesniak W, Pecoraro VL and Schacht J:
Ternary complexes of gentamicin with iron and lipid catalyze
formation of reactive oxygen species. Chem Res Toxicol. 18:357–364.
2005.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Pirvola U, Xing-Qun L, Virkkala J, Saarma
M, Murakata C, Camoratto AM, Walton KM and Ylikoski J: Rescue of
hearing, auditory hair cells, and neurons by CEP-1347/KT7515, an
inhibitor of c-Jun N-terminal kinase activation. J Neurosci.
20:43–50. 2000.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wang J, Van De Water TR, Bonny C, de
Ribaupierre F, Puel JL and Zine A: A peptide inhibitor of c-Jun
N-terminal kinase protects against both aminoglycoside and acoustic
trauma-induced auditory hair cell death and hearing loss. J
Neurosci. 23:8596–8607. 2003.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Eshraghi AA, Wang J, Adil E, He J, Zine A,
Bublik M, Bonny C, Puel JL, Balkany TJ and Van De Water TR:
Blocking c-Jun-N-terminal kinase signaling can prevent hearing loss
induced by both electrode insertion trauma and neomycin
ototoxicity. Hear Res. 226:168–177. 2007.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Yuan B, Yu WY, Dai LS, Gao Y, Ding Y, Yu
XF, Chen J and Zhang JB: Expression of microRNA 26b and
identification of its target gene EphA2 in pituitary tissues in
Yanbian cattle. Mol Med Rep. 12:5753–5761. 2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Beisel K, Hansen L, Soukup G and Fritzsch
B: Regenerating cochlear hair cells: Quo vadis stem cell. Cell
Tissue Res. 333:373–379. 2008.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Geng W and Liu L: miR-494 alleviates
lipopolysaccharide (LPS)-induced autophagy and apoptosis in PC-12
cells by targeting IL-13. Adv Clin Exp Med. 28:85–94.
2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Kuhn S, Johnson SL, Furness DN, Chen J,
Ingham N, Hilton JM, Steffes G, Lewis MA, Zampini V, Hackney CM, et
al: miR-96 regulates the progression of differentiation in
mammalian cochlear inner and outer hair cells. Proc Natl Acad Sci
USA. 108:2355–2360. 2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ghasemi-Dehkordi P, Allahbakhshian-Farsani
M, Abdian N, Mirzaeian A, Saffari-Chaleshtori J, Heybati F, Mardani
G, Karimi-Taghanaki A, Doosti A, Jami MS, et al: Comparison between
the cultures of human induced pluripotent stem cells (hiPSCs) on
feeder-and serum-free system (Matrigel matrix), MEF and HDF feeder
cell lines. J Cell Commun Signal. 9:233–246. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zhang B, Ji S, Ma F, Ma Q, Lu X and Chen
X: miR-489 acts as a tumor suppressor in human gastric cancer by
targeting PROX1. Am J Cancer Res. 6:2021–2030. 2016.PubMed/NCBI
|
|
35
|
Patel Y, Shah N, Lee JS, Markoutsa E, Jie
C, Liu S, Botbyl R, Reisman D, Xu P and Chen H: A novel
double-negative feedback loop between miR-489 and the
HER2-SHP2-MAPK signaling axis regulates breast cancer cell
proliferation and tumor growth. Oncotarget. 7:18295–18308.
2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Li J, Qu W, Jiang Y, Sun Y, Cheng Y, Zou T
and Du S: miR-489 suppresses proliferation and invasion of human
bladder cancer cells. Oncol Res. 24:391–398. 2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Soni M, Patel Y, Markoutsa E, Jie C, Liu
S, Xu P and Chen H: Autophagy, cell viability, and chemoresistance
are regulated by miR 489 in breast cancer. Mol Cancer Res.
16:1348–1360. 2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Liao CC, Ho MY, Liang SM and Liang CM:
Autophagic degradation of SQSTM1 inhibits ovarian cancer motility
by decreasing DICER1 and AGO2 to induce MIRLET7A-3P. Autophagy.
14:2065–2082. 2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-ΔΔC(T)) method. Methods. 25:402–408. 2001.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Fischel-Ghodsian N: Genetic factors in
aminoglycoside toxicity. Pharmacogenomics. 6:27–36. 2005.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zheng Z, Tang D, Zhao L, Li W, Han J, Hu
B, Nie G and He Y: Liproxstatin 1 protects hair cell like HEI OC1
cells and cochlear hair cells against neomycin ototoxicity. Oxid
Med Cell Longev. 2020(1782659)2020.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Mortimore GE and Pösö AR: Intracellular
protein catabolism and its control during nutrient deprivation and
supply. Annu Rev Nutr. 7:539–564. 1987.PubMed/NCBI View Article : Google Scholar
|
|
43
|
He Z, Guo L, Shu Y, Fang Q, Zhou H, Liu Y,
Liu D, Lu L, Zhang X, Ding X, et al: Autophagy protects auditory
hair cells against neomycin-induced damage. Autophagy.
13:1884–1904. 2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Djavaheri-Mergny M, Amelotti M, Mathieu J,
Besançon F, Bauvy C, Souquère S, Pierron G and Codogno P: NF-kappaB
activation represses tumor necrosis factor-alpha-induced autophagy.
J Biol Chem. 281:30373–30382. 2006.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J,
Lee J, Provost P, Rådmark O, Kim S, et al: The nuclear RNase III
Drosha initiates microRNA processing. Nature. 425:415–419.
2003.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Chendrimada TP, Gregory RI, Kumaraswamy E,
Norman J, Cooch N, Nishikura K and Shiekhattar R: TRBP recruits the
Dicer complex to Ago2 for microRNA processing and gene silencing.
Nature. 436:740–744. 2005.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Ameres SL and Zamore PD: Diversifying
microRNA sequence and function. Nat Rev Mol Cell Biol. 14:475–488.
2013.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Forman JJ and Coller HA: The code within
the code: microRNAs target coding regions. Cell Cycle. 9:1533–1541.
2010.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Lytle JR, Yario TA and Steitz JA: Target
mRNAs are repressed as efficiently by microRNA-binding sites in the
5' UTR as in the 3' UTR. Proc Natl Acad Sci USA. 104:9667–9672.
2007.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Meister G, Landthaler M, Patkaniowska A,
Dorsett Y, Teng G and Tuschl T: Human Argonaute2 mediates RNA
cleavage targeted by miRNAs and siRNAs. Mol Cell. 15:185–197.
2004.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Mizushima N: Autophagy: Process and
function. Genes Dev. 21:2861–2873. 2007.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Li W, Yang Y, Ba Z, Li S, Chen H, Hou X,
Ma L, He P, Jiang L, Li L, et al: MicroRNA-93 regulates
hypoxia-induced autophagy by targeting ULK1. Oxid Med Cell Longev.
2017(2709053)2017.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Liu L, Ren W and Chen K: miR-34a promotes
apoptosis and inhibits autophagy by targeting HMGB1 in acute
myeloid leukemia cells. Cell Physiol Biochem. 41:1981–1992.
2017.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Si X, Cao D, Chen J, Nie Y, Jiang Z, Chen
MY, Wu JF and Guan XD: miR 23a downregulation modulates the
inflammatory response by targeting ATG12 mediated autophagy. Mol
Med Rep. 18:1524–1530. 2018.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Zhou DM, Sun LL, Zhu J, Chen B, Li XQ and
Li WD: miR-9 promotes angiogenesis of endothelial progenitor cell
to facilitate thrombi recanalization via targeting TRPM7 through
PI3K/Akt/autophagy pathway. J Cell Mol Med. 24:4624–4632.
2020.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Dulon D, Hiel H, Aurousseau C, Erre JP and
Aran JM: Pharmacokinetics of gentamicin in the sensory hair cells
of the organ of Corti: Rapid uptake and long term persistence. C R
Acad Sci III. 316:682–687. 1993.PubMed/NCBI
|
|
57
|
Lemasters JJ: Selective mitochondrial
autophagy, or mitophagy, as a targeted defense against oxidative
stress, mitochondrial dysfunction, and aging. Rejuvenation Res.
8:3–5. 2005.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Holze C, Michaudel C, Mackowiak C, Haas
DA, Benda C, Hubel P, Pennemann FL, Schnepf D, Wettmarshausen J,
Braun M, et al: Oxeiptosis, a ROS-induced caspase-independent
apoptosis-like cell-death pathway. Nat Immunol. 19:130–140.
2018.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Wang Z, Yu K, Hu Y, Su F, Gao Z, Hu T,
Yang Y, Cao X and Qian F: Schisantherin A induces cell apoptosis
through ROS/JNK signaling pathway in human gastric cancer cells.
Biochem Pharmacol. 173(113673)2020.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Esterberg R, Linbo T, Pickett SB, Wu P, Ou
HC, Rubel EW and Raible DW: Mitochondrial calcium uptake underlies
ROS generation during aminoglycoside-induced hair cell death. J
Clin Invest. 126:3556–3566. 2016.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Platini F, Pérez-Tomás R, Ambrosio S and
Tessitore L: Understanding autophagy in cell death control. Curr
Pharm Des. 16:101–113. 2010.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Su Z, Yang Z, Xu Y, Chen Y and Yu Q:
Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol
Cancer. 14(48)2015.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Sheth S, Mukherjea D, Rybak LP and
Ramkumar V: Mechanisms of cisplatin induced ototoxicity and
otoprotection. Front Cell Neurosci. 11(338)2017.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Tabuchi K, Nishimura B, Nakamagoe M,
Hayashi K, Nakayama M and Hara A: Ototoxicity: Mechanisms of
cochlear impairment and its prevention. Curr Med Chem.
18:4866–4871. 2011.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Niwa K, Matsunobu T, Kurioka T, Kamide D,
Tamura A, Tadokoro S, Satoh Y and Shiotani A: The beneficial effect
of Hangesha-shin-to (TJ-014) in gentamicin-induced hair cell loss
in the rat cochlea. Auris Nasus Larynx. 43:507–513. 2016.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Choung YH, Taura A, Pak K, Choi SJ, Masuda
M and Ryan AF: Generation of highly-reactive oxygen species is
closely related to hair cell damage in rat organ of Corti treated
with gentamicin. Neuroscience. 161:214–226. 2009.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Vernon PJ and Tang D: Eat-me: Autophagy,
phagocytosis, and reactive oxygen species signaling. Antioxid Redox
Signal. 18:677–691. 2013.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Wang X, Wang P, Zhang Z, Farré JC, Li X,
Wang R, Xia Z, Subramani S and Ma C: The autophagic degradation of
cytosolic pools of peroxisomal proteins by a new selective pathway.
Autophagy. 16:154–166. 2020.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Yuan H, Wang X, Hill K, Chen J, Lemasters
J, Yang SM and Sha SH: Autophagy attenuates noise-induced hearing
loss by reducing oxidative stress. Antioxid Redox Signal.
22:1308–1324. 2015.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Oh KH, Rah YC, Hwang KH, Lee SH, Kwon SY,
Cha JH and Choi J: Melatonin mitigates neomycin-induced hair cell
injury in zebrafish. Drug Chem Toxicol. 40:390–396. 2017.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Lee Y, Jeon K, Lee JT, Kim S and Kim VN:
MicroRNA maturation: Stepwise processing and subcellular
localization. EMBO J. 21:4663–4670. 2002.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Feng H, Huang X, Fu W, Dong X, Yang F, Li
L and Chu L: A Rho kinase inhibitor (Fasudil) suppresses TGF-β
mediated autophagy in urethra fibroblasts to attenuate traumatic
urethral stricture (TUS) through re-activating Akt/mTOR pathway: An
in vitro study. Life Sci. 267(118960)2021.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Xie FJ, Zheng QQ, Qin J, Zhang LL, Han N
and Mao WM: Autophagy inhibition stimulates apoptosis in
oesophageal squamous cell carcinoma treated with fasudil. J Cancer.
9:1050–1056. 2018.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Falcon B, Noad J, McMahon H, Randow F and
Goedert M: Galectin-8-mediated selective autophagy protects against
seeded tau aggregation. J Biol Chem. 293:2438–2451. 2018.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Gibbings D, Mostowy S and Voinnet O:
Autophagy selectively regulates miRNA homeostasis. Autophagy.
9:781–783. 2013.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Gibbings D, Mostowy S, Jay F, Schwab Y,
Cossart P and Voinnet O: Selective autophagy degrades DICER and
AGO2 and regulates miRNA activity. Nat Cell Biol. 14:1314–1321.
2012.PubMed/NCBI View Article : Google Scholar : Erratum in: Nat
Cell Biol 17: 1088, 2015.
|
|
77
|
Hu JL, He GY, Lan XL, Zeng ZC, Guan J,
Ding Y, Qian XL, Liao WT, Ding YQ and Liang L: Inhibition of
ATG12-mediated autophagy by miR-214 enhances radiosensitivity in
colorectal cancer. Oncogenesis. 7(16)2018.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Sivakumaran TA, Resendes BL, Robertson NG,
Giersch AB and Morton CC: Characterization of an abundant COL9A1
transcript in the cochlea with a novel 3' UTR: Expression studies
and detection of miRNA target sequence. J Assoc Res Otolaryngol.
7:160–172. 2006.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Xiong H, Chen S, Lai L, Yang H, Xu Y, Pang
J, Su Z, Lin H and Zheng Y: Modulation of miR-34a/SIRT1 signaling
protects cochlear hair cells against oxidative stress and delays
age-related hearing loss through coordinated regulation of
mitophagy and mitochondrial biogenesis. Neurobiol Aging. 79:30–42.
2019.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Masumoto A, Mohri M, Shimokawa H, Urakami
L, Usui M and Takeshita A: Suppression of coronary artery spasm by
the Rho-kinase inhibitor fasudil in patients with vasospastic
angina. Circulation. 105:1545–1547. 2002.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Shibuya M, Hirai S, Seto M, Satoh S and
Ohtomo E: Fasudil Ischemic Stroke Study Group. Effects of fasudil
in acute ischemic stroke: Results of a prospective
placebo-controlled double-blind trial. J Neurol Sci. 238:31–39.
2005.PubMed/NCBI View Article : Google Scholar
|














