Introduction
Neurofibromatosis type 1 (NF1) was first described
in the 13th century in the literature by Madigan, Schaw and Masello
in ‘Neurofibromatosis in the 13th century and report of NF-like
case-Monstrorum History’, but there are descriptions of individuals
presumed of having neurofibromatosis, recovered in manuscripts from
1,000 B.C. However, only in 1882 was it recognized by the German
pathologist Freidrich von Recklinghausen as a distinct disorder and
he initiated the term ‘neurofibroma’, as he observed the appearance
of tumors from the sheath of the peripheral nerve (1-5).
NF1 is a multisystem, autosomal dominant genetically transmitted
disease, also known as von Recklinghausen disease, which influences
the cell growth of neuronal tissues. NF1 is an inherited disorder
relatively common, family history being present in half of the
cases, the rest developing a new genetic mutation, and it affects 1
in 2,500-3,000 births worldwide, regardless of sex and ethnicity
(6-12).
Neurofibromin 1, the gene of NF1, was first
discovered in 1990 located on chromosome 17, band q11.2 and is a
large gene which codes the protein neurofibromin, with the highest
rates of spontaneous mutations in the entire human genome (2,3,5,13-18).
Neurofibromin is omnipresent during development in human tissue and
later is identified exclusively in the nervous system, neurons,
Schwann and glial cells in high concentrations (1,3,15,19).
NF1 genetic testing is reserved for making reproductive decisions
or for unusual forms of disease, the diagnosis finally being made
based on clinical criteria (4,20).
The patients with clinical manifestations can be
diagnosed by performing a careful examination. The signs may be
dermatologic/cutaneous: café-au-lait spots/macules (CALM), axillary
freckling; ocular: Lisch nodules, optic nerve glioma; endocrine:
precocious/delayed puberty; neurologic: brain tumors, epilepsy,
headache, macrocephaly, mental retardation, learning disabilities;
cardiovascular: vascular defects, hypertension; orthopaedic:
pseudarthrosis, scoliosis, pectus excavatum, genu valgum/varum.
Each category can have variable severity (3,18,21).
For the diagnosis of NF1, 7 cardinal diagnostic criteria (Table I) have been delineated, taking into
consideration the most common cutaneous, neurologic, ocular and
skeletal manifestations with the addition of the genetic component,
and at least 2 of the listed features. The diagnostic criteria were
defined in 1988 during the Neurofibromatosis Conference Statement
of the National Institutes of Health Consensus Development
Conference, in Bethesda, MD, USA (1,3,12,18,20).
Diagnosis can be delayed due to signs/symptoms that appear at
variable ages and due to the patient having some dermatologic
features but without meeting enough diagnostic criteria (1,3).
 | Table IDiagnostic criteria for
neurofibromatosis type 1. |
Table I
Diagnostic criteria for
neurofibromatosis type 1.
|
Characteristics | Criteria |
|---|
| 1. Six or more
café-au-lait macules | 5 mm in diameter
prepubertal 15 mm in diameter postpubertal |
| 2. Two or more
neurofibromas or one plexiform neurofibroma | Of any type |
| 3. Freckling | Axillary |
| | Inguinal (Crowe
sign) |
| 4. Optic nerve
glioma | - |
| 5. Two or more
Lisch nodules (iris hamartomas) | Identified by an
ophthalmologist through slit-lamp examination |
| 6. A bone
lesion | Sphenoid wing
dysplasia |
| | Typical long bone
abnormalities - |
| |
Pseudarthrosis/thinning of cortex |
| 7. A first-degree
relative with NF1 | Parent |
| | Sibling |
| | Offspring |
Café-au-lait spots are the most common and among the
silent features of NF1. They are present in 99% of patients at
birth or appear in the first two years of life, with multiple spots
being very suggestive for NF1. The spots are flat brownish macules,
uniform colored from tan to dark brown, 10-100 mm in diameter,
ovoid shape, with well-defined borders. These lesions tend to
darken with sun exposure, lighten with age and have non-malignant
potential (12,17,22,23).
The spots with the same characteristics but smaller in diameter are
named freckles or ephelides and are localized inguinal or, more
common, in axillary zones (Crowe sign), in almost 80% of the
children starting at 3-5 years old, following the appearance of
café-au-lait spots (3,4,12,18).
Neurofibromas are benign tumors composed by neoplastic Schwann
cells, mast cells, endothelial cells, macrophages, located along
the nerves, with an accelerated proliferation during puberty and
pregnancy. The types of neurofibromas are: cutaneous (superficial);
subcutaneous (deeper); nodular plexiform; and diffuse plexiform
neurofibromas that penetrate deep into bones, muscle and viscera.
They usually become apparent after puberty and increase in size and
number continuously during adulthood and they should be
differentiated from other skin lesions (3-5,12,24,25).
In addition to neurofibromas, neurofibromatosis may be associated
with other tumors of the central nervous system including glioma of
the optic nerve, hamartoma of the iris, meningioma, and
glioblastoma (4,12). Neurofibrosarcomas, malignant
peripheral nerve sheath tumors (MPNSTs), are a severe complication
with an elusive nature and high rate of recurrence (3,5,26).
The cardiovascular abnormalities developed in NF1
include congenital heart disease; vasculopathy (aortic
coarctation), renal and cerebral artery stenosis, arteriovenous
malformations; as well as, pulmonary hypertension which may be a
rare, but formidable complication of NF1 (4,5,27). NF1
patients often present with hypertension due to renal artery
stenosis or in association with pheochromocytoma or paragangliomas
(28). The neurological
manifestations associated with the disease are macrocephaly without
hydrocephalus, mild epilepsy, cognitive problems with a low average
IQ, and learning difficulties. Behavioral problems include impaired
socialization, sleep disturbance, anxiety and depression (4,5,12).
Other associated autoimmune diseases have been previously described
as uncommon occurrences, such as lichen sclerosus or vitiligo
(29-34).
Case report
A 16 year-old boy living in a rural area was
admitted on July 2016 to the clinical Department of
Neuropsychomotor Rehabilitation, ‘Sf.Ioan’ Clinical Hospital for
Children (Galati, Romania) for clinico-functional evaluation and
specific treatment. The family history revealed the mother and two
maternal uncles with NF1 skin markers which were uninvestigated and
undiagnosed, as well as a sister and a brother diagnosed with NF1.
From his personal history it was recorded that the patient had
undergone 3 febrile seizures until the age of 3 for which he had
not received treatment, and café-au-lait spots that appeared after
the age of 2. At the age of 5 years the patient was hospitalized
with the diagnosis of NF1-associated congenital pseudarthrosis in
both left leg bones and the surgical treatment of pseudarthrosis,
osteosynthesis with Steinmann brooch and Ilizarov external fixative
implant (removed after one year) were performed. At the age of 7
years the patient received surgical treatment for pseudarthrosis
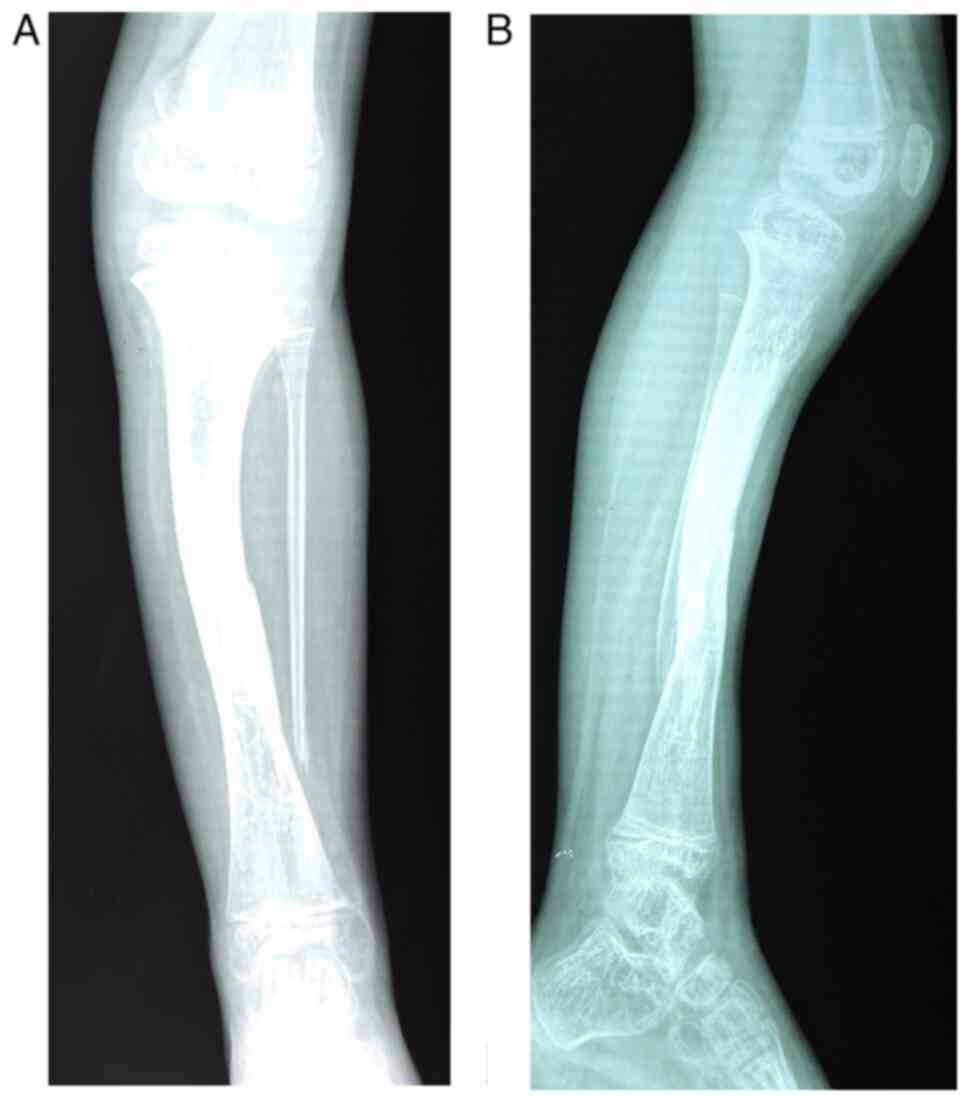
again by Phemister procedure. Fig.
1 reveals the anterior and the lateral view of the radiological
images of the left leg of the patient obtained at age of 10, that
reveal an old fracture 1/3 lower of the left tibial shaft
strengthened by osteosynthesis, lack of bone lower extremity left
fibula, and intense changes in osteoporosis in both left leg
bones.
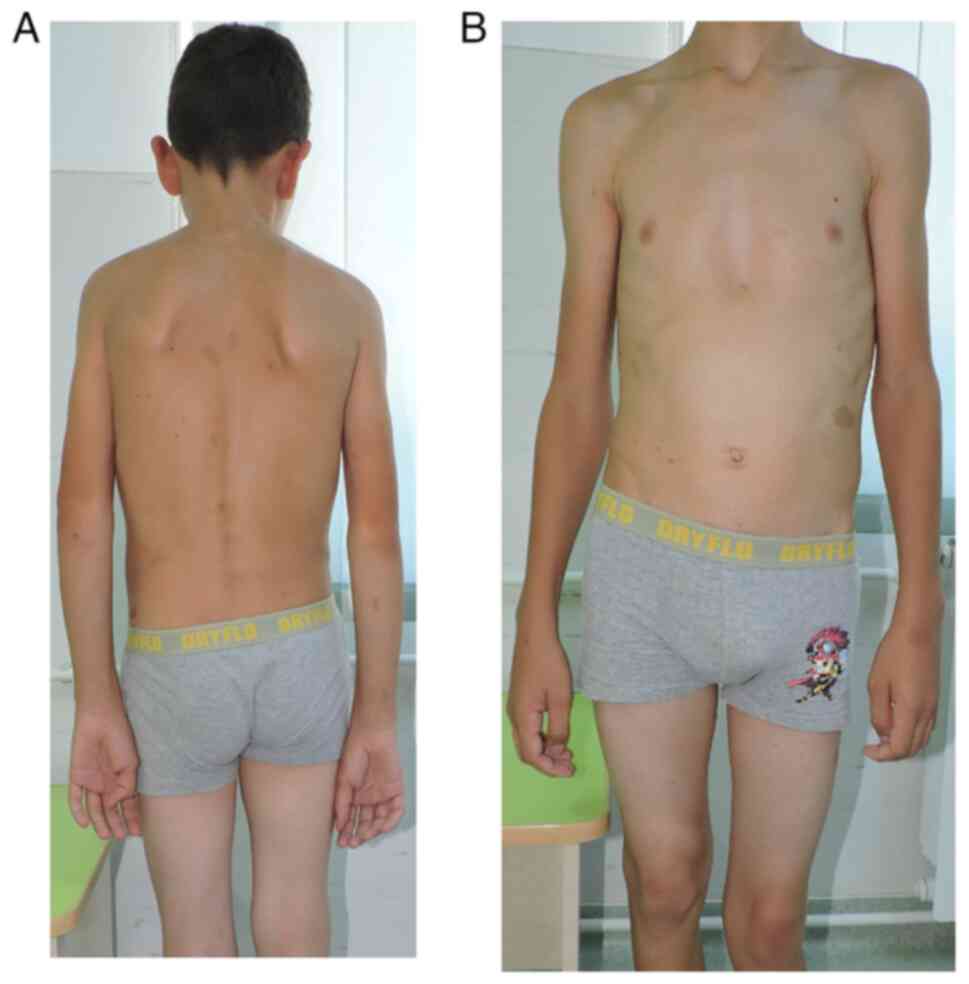
From the clinical examination the following were
observed: i) a weight of 49 kg and a height of 154 cm; ii)
autonomous walking, limping on the left; iii) spinal deviation in
the frontal plane to the right (dorsal) and to the left (lumbar);
iv) humeral imbalance, with the left shoulder ascended; v) the
waist triangle erased on the left (Fig.
2A); vi) sternal depression in the lower 1/3 and flared ribs
(Fig. 2B); vii) normal spine
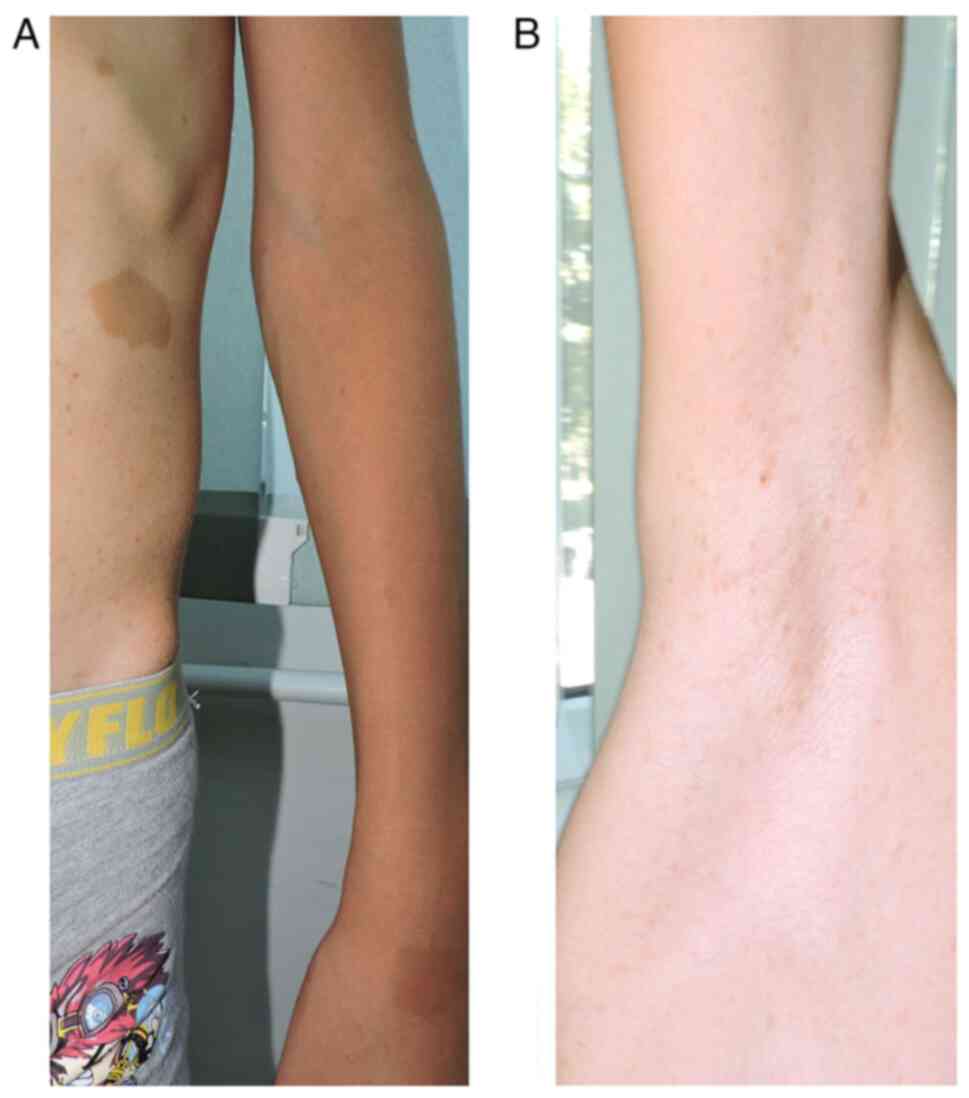
mobility; viii) multiple café-au-lait spots (over 6 in number), the
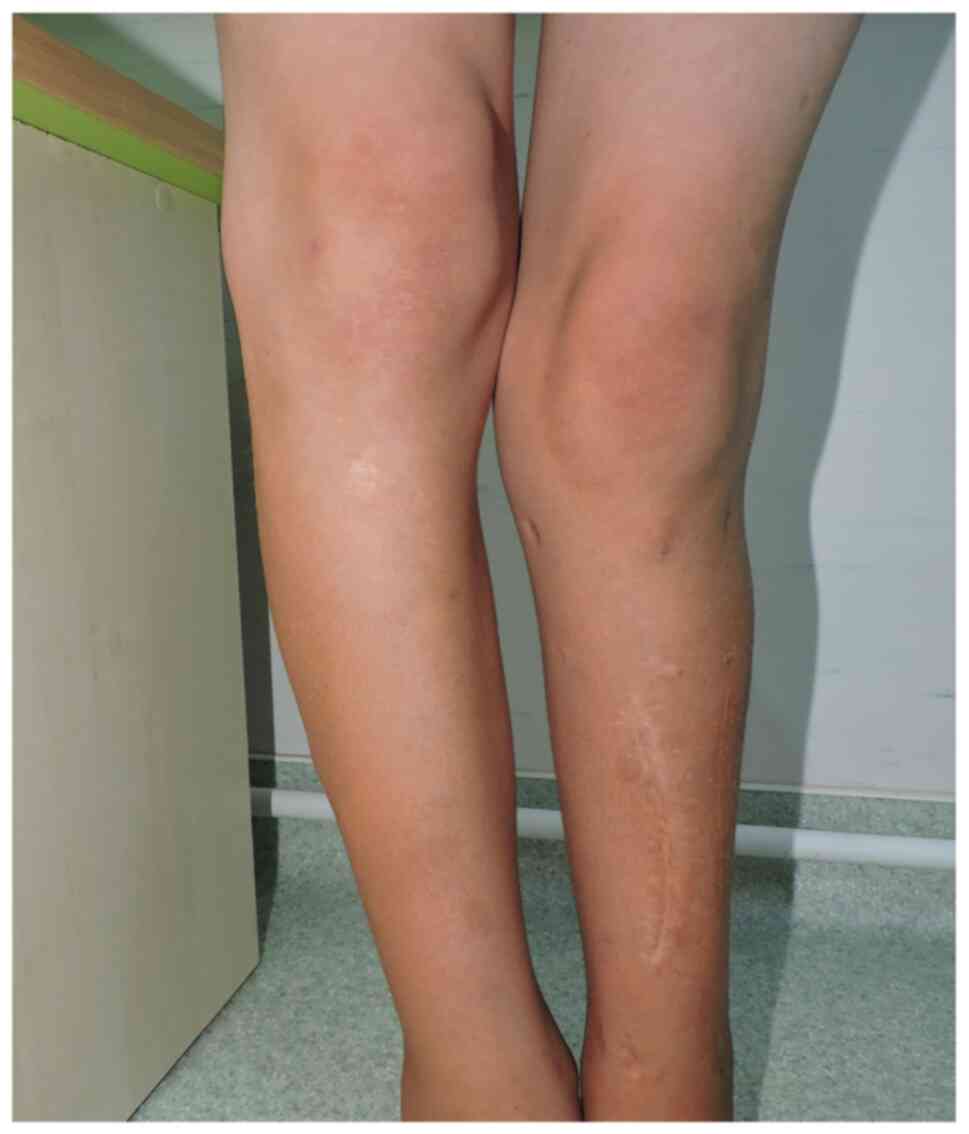
largest on the left flank, with a diameter of 3.5/3 cm (Fig. 3A); iv) axillary freckles (Fig. 3B); x) two surgical scars present on
the left pretibial area, one measuring 13 cm, and the other 4 cm,
respectively; xi) left genum valgum; xii) lower limb inequality,
left leg shorter by 5 cm; xiii) left hypotrophy on the leg (3 cm)
and on the thigh (2 cm) (Fig. 4);
xiv) macrocephaly; xv) language disorder; and xvi) mild
intellectual deficit.
Discussion
NF1 is a multisystem, autosomal dominant genetically
transmitted disease, which influences the cell growth of neuronal
tissues. NF1 is an inherited disorder relatively common, with
family history being present in half of the cases, while the
remaining cases develop a new genetic mutation, and it influences 1
in 2,500-3,000 births worldwide (6-12).
The patients with clinical manifestations can be diagnosed by
performing a careful examination. The signs may be cutaneous,
ocular, endocrine, neurologic, cardiovascular, and orthopaedic:
pseudarthrosis, scoliosis, pectus excavatum, genu valgum/varum
(3,18,21).
The skeletal manifestations of von Recklinghausen
disease (NF1) can be generalized, most commonly, and with mild
clinical implications such as osteopenia or osteoporosis, or short
stature; and can also be focal, less commonly, with significant
morbidity, such as scoliosis, long bone dysplasia, and sphenoid
wing dysplasia. The most frequent bones manifestations of patients
with NF1 are listed in Table
II.
| Table IIFrequent bone manifestations of
patients with neurofibromatosis type 1. |
Table II
Frequent bone manifestations of
patients with neurofibromatosis type 1.
| Bone
manifestations | Types |
|---|
| Bone
deformities | Long bone
dysplasia |
| | Congenital bone
bowing |
| | Pseudarthrosis |
| | Genu
varum/algum |
| | Sphenoid wing
dysplasia |
| | Scoliosis |
| | Kyphoscoliosis |
| |
Spondylolisthesis |
| | Cervical spine
disorders |
| | Abnormalities of
the rib cage |
| | Macrocephaly |
| | Short stature |
| Bone metabolism
disorders | Osteopenia |
| | Osteoporosis |
| | Impaired bone
healing |
| | Hypophosphatic
rickets |
Only 2-4% of patients with NF1 develop long bone
dysplasia, tibia being more often involved than other long bones,
which are less affected (5,35-37).
Congenital tibial dysplasia, also known as congenital
pseudarthrosis of the tibia (CPT), is unusual in the general
population, but is more common in individuals with NF1 (37-39).
CPT present in NF1 is described as the antero-lateral bowing of
tibia that appear at birth or in the first year of life (40,41).
Histopathological analysis of the resected tissue of the
pseudarthrosis revealed a hard whitish fibrous tissue, surrounded
by fatty lobules. Specifically between the bone ends, thick bands
of dense fibrous tissue intervene, that prevent the union and
constitute a true focus of pseudarthrosis, which is not a
neurofibroma, but an unspecified cell fibrous overgrowth (36,42).
Clinically, there is a varus with anterior bowing mid-distal third
of the leg, with a thin, sclerotic and fragile bone, producing a
spontaneous fracture of tibia or both leg bones intrauterine,
perinatally or in the first years of life due to functional stress
or microtrauma (5,43). Once the fracture has occurred, the
absence of union through the formation of the callus is
characteristic. Therefore, this usually progresses to
pseudarthrosis when a spontaneous fracture occurs in children under
4 years of age, with tibial bowing and not exhibiting adequate bony
callus with treatment (44). Due to
the rigid immobilization during treatment, stiffness of the ankle
is frequently observed (44,45).
The gait and the muscular strength of the patients are altered.
Early appearance followed by early surgical treatment, frequently
requiring fixation of the ankle, leads to an abnormal gait
(46,47). The deformity with anterior bowing is
usual, as well as its progressive increase with the consequent
shortening of the posterior myotendinous structures of the leg and
ankle (44,48).
Several classifications have been proposed,
considering the morphology of the lesion, but with limited
prognostic value due to the changes that arise during the disease
(49,50). The most determining factor is the
moment when the fracture appears: after 4 years of age suggests a
more benign behavior, while before 4 years of age entails a more
rebellious evolution (44,45,48,51).
The management of congenital dysplasia of the tibia associated with
NF1 may be frustrating, with frequent and severe complications,
fractures and refractures, common after different treatments.
Surgical treatment performed without adequate biomechanical
criteria does not appear to be particularly successful (45,46,51-53).
A non-surgical method consists of bracing with an ankle-foot or
knee-ankle-foot orthosis, with the first method used until weight
bearing begins, switching after that to a knee-ankle-foot method.
Bracing is used to prevent fractures in dysplastic bones and if the
fracture occurs to delay surgical intervention (3,53). The
objective of surgical treatment is to maximize the union through
the resection head, following the specific methods: external
fixation (Ilizarov technique), bone grafting with intramedullary
fixation, and free vascularized fibular grafting. When multiple
attempts have failed, and with the leg remaining extremely short,
amputation may be the best solution (3,5,50,53).
Intramedullary fixation with the iliac bone, while leaving the
device in place, even after healing of the fracture, is considered
the first line treatment since it is a relatively easy procedure,
provides stable fixation, has minimal postoperative complications
and reduces the risk of refracture, compared with alternative
procedures (3,54). The most severe complications are
residual angular deformity, limb length inequality, refracture,
ankle stiffness and chronic pain (53).
The Ilizarov external fixative implant provides
numerous advantages in the treatment of congenital dysplasia of the
tibia and its associated problems. This method allows the surgeon
to manage limb dismetria, angular deformity, proximal migration and
nonunion of the fibula, ankle valgus and foot contractures
(55). Refractures are extremely
common, although this method initially produces a high binding
rate. Another disadvantage of the Ilizarov method is the external
device, which is not well tolerated by pediatric patients. The
multiple complications with this procedure include dorsiflexion
contracture of the ankle with calcaneo-valgus deformity, joint
stiffness, cystic bone lesions, nail infections, cartilage
necrosis, injury to a nerve and possible appearance of compartment
syndromes (56).
The scoliosis associated with NF1 can be dystrophic,
less common and more severe, with rapid progression, and
non-dystrophic, with the same rate and a similar clinical
appearance, but an earlier onset and poorer prognosis. Early
scoliosis screening is required for patients with
neurofibromatosis. After diagnosis all individuals should be MRI/CT
scan-evaluated to assess the deformity and the dystrophic changes,
if present, and to detect the intra/extraspinal neoplasia (3,57).
Dystrophic scoliosis leads to severe curves, 4-6 sharply angulated
vertebra, accompanied by osseous abnormalities (3,5,58). The
management of nondystrophic scoliosis depends on the degree of
curvature. For curves <20˚ the observation is enough, for curves
between 20-40˚ or for documented progression, bracing is
recommended, for >40˚ posterior fusion is recommended, and
finally for curves >90˚ anterior-posterior fusion is recommended
(3,59). In the early forms of the disease,
other pathologies in differential diagnosis such as multiple
symmetrical lipomatosis or diabetes and its comorbidities, may also
be considered (60,61).
In conclusion, NF1 (von Recklinghausen disease)
influences multiple organs and the diagnosis could be made
clinically, based on careful multidisciplinary examination. While
certain characteristics are present at birth, others are
age-related, and all of them require periodical monitoring. Other
examinations (histopathologic, radiological, MRI, ophthalmologic
and neurologic) are important for tracking the complications and
the evolution of this disorder. The bones are affected more often
in NF1, having an increased morbidity and even profound invalidism.
A careful strategy for the management of musculoskeletal
disabilities may improve the quality of life of patients with this
disorder.
In the present study, the case of a 16 year-old male
living in a rural area was presented, who at 5 years of age was
diagnosed with NF1-associated congenital pseudarthrosis in both
left leg bones and received the surgical treatment for
pseudarthrosis, osteosynthesis with Steinmann brooch and Ilizarov
external fixative implant. At the age of 7 years the patient
received surgical treatment again for pseudarthrosis by Phemister
procedure.
The patients with NF1 develop multiple
complications, since it affects multiple systems and benefit most
from a multidisciplinary strategy. If the musculoskeletal
disabilities are carefully managed, the quality of life in
individuals with NF1 can be improved.
The case presented was associated with frequent
complications including pseudarthrosis of both bones of the leg,
kyphoscoliosis, chest deformities, macrocephaly, language disorder
and mild intellectual deficit. Tibial dysplasia with pseudarthrosis
is a challenging complication, since the patient, after all the
surgical interventions, still has a marked functional
disability.
Despite advances of continuous research for the
diagnosis and monitoring of NF1, there is no medical treatment
available for it. Medical care should be focused on genetic
counseling and the early detection of complications.
Acknowledgements
Not applicable.
Funding
Funding: The present work was supported by the ‘Dunarea de Jos’
University of Galati, Romania, through the research center,
Multidisciplinary Integrated Center of Dermatological Interface
Research MIC-DIR [Centrul Integrat Multidisciplinar de Cercetare de
Interfata Dermatologica (CIM-CID)].
Availability of data and materials
The information generated and analyzed during the
current study is available from the corresponding author on
reasonable request.
Authors' contributions
FN and ALT were major contributors in writing the
manuscript. FN, DSR, EN, ALT, AVB, MCV, SF and LB were involved in
all the stages of the study. DSR, AlN, AVB, VC, AL, LCN and EN
contributed to the conception and design of the work, as well as
the revision of the study. AVB, AuN, VC, LCN, EDP, SF and LB helped
analyze the data for the work. AVB, MCV, LA and EDP revised it for
important intellectual content. ALT and AlN approved the final
version to be published. FN and AVB confirm the authenticity of all
the raw data. All authors have participated equally and have equal
rights to this study. All authors read and approved the final
manuscript and agree to be accountable for all aspects of the work
in ensuring that questions related to the accuracy or integrity of
any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
Ethical approval was obtained from the Ethics
Committee of the Emergency Clinical Hospital for Children ‘Sf.
Ioan’ (Glati, Romania), with the approval no. 2774, from
18.02.2021. The guardians of the patient provided written informed
consent.
Patient consent for publication
The guardians of the patient provided consent for
publication and it is included in the medical chart of the
patient.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sehgal NV, Verma P and Chatterjee K: Type
1 neurofibromatosis (von Recklinghausen disease). Cutis. 96:e23–26.
2015.PubMed/NCBI
|
|
2
|
Leroy K, Dumas V, Martin-Garcia N, Falzone
MC, Voisin MC, Wechsler J, Revuz J, Creange A, Levy E, Lantieri L,
et al: Malignant peripheral nerve sheath tumors associated with
neurofibromatosis type 1: A clinicopathologic and molecular study
of 17 patients. Arch Dermatol. 137:908–913. 2001.PubMed/NCBI
|
|
3
|
Feldman DS, Jordan C and Fonseca L:
Orthopaedic manifestations of neurofibromatosis type 1. J Am Acad
Orthop Surg. 18:346–357. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Hirbe AC and Gutman DH: Neurofibromatosis
type 1: A multidisciplinary approach to care. Lancet Neurol.
13:834–843. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ferner RE, Huson SM, Thomas N, Moss C,
Willshaw H, Evans DG, Upadhyaya M, Towers R, Gleeson M, Steiger C
and Kirby A: Guidelines for the diagnosis and management of
individuals with neurofibromatosis 1. J Med Genet. 44:81–88.
2007.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Boyd KP, Korf BR and Theos A:
Neurofibromatosis type 1. J Am Acad Dermatol. 61:1–14.
2009.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Guler M, Aydin T and Poyraz E:
Neurofibromatosis type 1 with invasive spinal cord compression
(case report). Afr J Pharm Pharmacol. 7:1615–1618. 2013.
|
|
8
|
Friedman JM: Epidemiology of
neurofibromatosis type 1. Am J Med Genet. 89:1–6. 1999.PubMed/NCBI
|
|
9
|
Lammert M, Friedman JM, Kluwe VF and
Mautner VF: Prevalence of neurofibromatosis 1 in German children at
elementary school enrollment. Arch Dermatol. 141:71–74.
2005.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Huson SM, Compston DA, Clark P and Harper
PS: A genetic study of von Recklinghausen neurofibromatosis in
south east Wales. Prevalence, fitness, mutaton rate and effect of
parental transmision on severity. J Med Genet. 26:704–711.
1989.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Gutmann DH, Ferner RE, Listernick RH, Korf
BR, Wolters PL and Johnson KJ: Neurofibromatosis type 1. Nat Rev
Dis Primers. 3(17004)2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Nica SC, Mihailescu G, Nica SM, Baetu C,
Clatici VG and Buruga I: Neurofibromatosis-one disease for a
multidisciplinary team. RoJCED. 3:38–49. 2016.
|
|
13
|
Viskochil D, Buchberg AM, Xu G, Cawthon
RM, Stevens J, Wolff RK, Culver M, Carey JC, Copeland NG and
Jenkins NA: Deletions and a translocation interrupt a cloned gene
at the neurofibromatosis type 1 locus. Cell. 62:187–192.
1990.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Li Y, O'Connell P, Breidenbach HH, Cawthon
R, Stevens J, Xu G, Neil S, Robertson M, White R and Viskochil D:
Genomic organization of the neurofibromatosis 1 gene (Nf1).
Genomics. 25:9–18. 1995.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Daston MM, Scrable H, Nordlund M, Sturbaum
AK, Nissen LM and Ratner N: The protein product of the
neurofibromatosis type 1 gene is expressed at highest abundance in
neurons, Schwann cells and oligodendrocytes. Neuron. 8:415–428.
1992.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Gutmann DH, Parada LF, Silva AJ and Ratner
N: Neurofibromatosis type 1: Modeling CNS dysfunction. J Neurosci.
32:14087–14093. 2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Peltonen S: Neurofibromatosis type 1:
Dermatologists should take an active role. Forum for Nord Derm Ven.
13:74–77. 2008.
|
|
18
|
Antônio JR, Goloni-Bertollo EM and Trídico
LA: Neurofibromatosis: Chronological history and current issues. An
Bras Dermatol. 88:329–343. 2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Stocker KM, Baizer L, Coston T, Sherman L
and Ciment G: Regulated expression of neurofibromin in migrating
neural crest cells of avian embryos. J Neurobiol. 27:535–552.
1995.PubMed/NCBI View Article : Google Scholar
|
|
20
|
National Institutes of Health Consensus
Development Conference Statement: Neurofibromatosis. Bethesda, Md.,
USA July 13-15, 1987. Neurofibromatosis. 1:172–178. 1988.PubMed/NCBI
|
|
21
|
Tonsgard JH: Clinical manifestations and
management of neurofibromatosis type 1. Semin Pediatr Neurol.
13:2–7. 2006.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Ferner RE: Neurofibromatosis 1. Eur J Hum
Genet. 15:131–138. 2007.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Williams VC, Lucas J, Babcock MA, Gutmann
DH, Korf B and Maria BL: Neurofibromatosis type 1 revisited.
Pediatrics. 123:124–133. 2009.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kolanczyk M, Mautner V, Kossler N, Nguyen
R, Kuhnisch J, Zemojtel T, Jamsheer A, Wegener E, Thurisch B,
Tinschert S, et al: MIA is a potential biomarker for tumour load in
neurofibromatosis type 1. BMC Med. 9(82)2011.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Tatu AL: Umbilicated blue black lesion on
the lateral thorax. J Cutan Med Surg. 21(252)2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Valeyrie-Allanore L, Ortonne N, Lentieri
L, Ferkal S, Wechsler J, Bagot M and Wolkenstein P:
Histopathologically dysplastic neurofibromas in neurofibromatosis
1: Diagnostic criteria, prevalence and clinical signifiance. Br J
Dermatol. 158:1008–1012. 2008.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Montani D, Coulet F, Girerd B, Eyries M,
Bergot E, Mal H, Biondi G, Dromer C, Hugues T, Marquette C, et al:
Pulmonary hypertension in patients with neurofibromatosis type I.
Medicine (Baltimore). 90:201–211. 2011.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wang J, Wei G, Wang Z and Huang H:
Detection of severe hypertension in a patient with
neurofibromatosis type 1 during anesthesia induction: A case
report. J Med Case Rep. 13(349)2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Nanda A: Autoimmune diseases associated
with neurofibromatosis type 1. Pediatr Dermatol. 25:392–393.
2008.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Tatu AL and Ionescu MA: Multiple
autoimmune syndrome type III-thyroiditis, vitiligo and alopecia
areata. Acta Endocrinol (Buchar). 13:124–125. 2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Tandon S, Singh A, Arora P and Gautam RK:
Neurofibromatosis with vitiligo: An uncommon association rather
than coexistence? An Bras Dermatol. 94:624–626. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Miraglia E, Calvieri S and Giustini S:
Neurofibromatosis type 1 and lichen sclerosus: An uncommon
association. G Ital Dermatol Venereol. 152:83–84. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Mihăilă B, Dinică RM, Tatu AL and Buzia
OD: New insights in vitiligo treatments using bioactive compounds
from Piper nigrum. Exp Ther Med. 17:1039–1044.
2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Tatu AL and Nwabudike LC: The treatment
options of male genital lichen sclerosus et atrophicus: Treatments
of genital lichen sclerosus. In: Proceedings of the 14th National
Congress of Urogynecology and the National Conference of the
Romanian Association for the Study of Pain, pp262-264, 2017.
|
|
35
|
Korf BR: Diagnostic outcome in children
with cafe au lait spots. Pediatrics. 90:924–927. 1992.PubMed/NCBI
|
|
36
|
Stevenson DA, Moyer-Mileur LJ, Murray M,
Slater H, Sheng X, Carey JC, Dube B and Viskochil DH: Bone mineral
density in children and adolescents with neurofibromatosis type 1.
J Pediatr. 150:83–88. 2007.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Friedman JM and Birch PH: Type I
neurofibromatosis: A descriptive analysis of the disorder in 1728
patients. Am J Med Genet. 70:138–143. 1997.
|
|
38
|
Gilbert A and Brockman R: Congenital
pseudarthrosis of the tibia: Long-term followup of 29 cases treated
by microvascularbone transfer. Clin Orthop Relat Res. 314:37–44.
1995.PubMed/NCBI
|
|
39
|
Sulaiman AR, Nordin S, Faisham WI, Zulmi W
and Halim AS: Residual nonunion following vascularized fibular
graft treatment for congenital pseudarthosis of the tibia: A report
of two cases. J Orthop Surg (Hong Kong). 14:64–66. 2006.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Stevenson DA, Birch PH, Friedman JM,
Viskochil DH, Balestrazzi P, Boni S, Buske A, Korf BR, Niimura M,
Pivnick EK, et al: Descriptive analysis of tibial pseudarthrosis in
patients with neurofibromatosis 1. Am J Med Genet. 84:413–419.
1999.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Kong LD, Cheng HX and Nie T: Treat the
congenital pseudarthrosis of the tibia with Ilizarov technology.
Case report. Medicine (Baltimore). 97(e13384)2018.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Sakamoto A, Yoshida T, Yamamoto H, Oda Y,
Tsuneyoshi M and Iwamoto Y: Congenital pseudarthrosis of the tibia:
Analysis of the histology and the NF1 gene. J Orthop Sci.
12:361–365. 2007.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Tuncay IC, Johnston CE II and Birch JG:
Spontaneous resolution of congenital anterolateral bowing of the
tibia. J Pediatr Orthop. 14:599–602. 1994.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Crawford AH and Schorry EK:
Neurofibromatosis in children: The role of the orthopaedist. J Am
Acad Orthop Surg. 7:217–230. 1999.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Traub JA, O'Connor W and Masso PD:
Congenital pseudarthrosis of the tibia: A retrospective review. J
Pediatr Orthop. 19:735–738. 1999.PubMed/NCBI
|
|
46
|
Karol LA, Haideri NF, Halliday SE,
Smitherman TB and Johnston CE II: Gait analysis and muscle strength
in children with congenital pseudarthrosis of the tibia: The effect
of treatment. J Pediatr Orthop. 18:381–386. 1998.PubMed/NCBI
|
|
47
|
Vanderstappen J, Lammens J, Berger P and
Laumen A: Ilizarov bone transplant as a treatment of congenital
pseudarthrosis of the tibia: A long-term follow-up study. J Child
Orthop. 9:319–324. 2015.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Ippolito E, Corsi A, Grill F, Wientroub S
and Bianco P: Pathology of bone lesions associated with congenital
pseudarthrosis of the leg. J Pediatr Orthop B. 9:3–10.
2000.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Hefti F, Bollini C, Dungl P, Fixsen J,
Grill F, Ippolito E, Romanus B, Tudisco C and Wientroub S:
Congenital pseudarthrosis of the tibia: History, etiology,
classification and epideiologic data. J Pediatr Orthop B. 9:11–15.
2000.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Lehman WB, Atar D, Feldman DS, Gordon JC
and Grant AD: Congenital pseudarthrosis of the tibia. J Pediatr
Orthop B. 9:103–107. 2000.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Tudisco C, Bollini C, Dungel P, Fixen J,
Grill F, Hefti F, Romanus B and Wientroub S: Functional results at
the end of skeletal growth in 30 patients affected by congenital
pseudarthrosis of the tibia. J Pediatr Orthop B. 9:94–102.
2000.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Grill F, Bollini C, Dungl P, Fixsen J,
Hefti F, Ippolito E, Romanus B, Tudisco C and Wientroub S:
Treatment approaches for congenital psedarthrosis of tibia: Results
of the EPOS multicenter study. European Paediatric Orthopaedic
Society (EPOS). J Pediatr Orthop B. 9:75–89. 2000.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Elefteriou F, Kolnczyk M, Schindeler A,
Viskochil DH, Hock JM, Schorry EK, Crawford AH, Friedman JM, Little
D, Peltonen J, et al: Skeletal abnormalities in neurofibromatosis
type 1: Approaches to therapeutic options. Am J Med Genet A. 149A.
2327–2338. 2009.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Dobbs MB, Rich MM, Gordon JE, Szymanski DA
and Schoenecker PL: Use of an intramedullary rod for treatment of
congenital pseudarthrosis of the tibia: A long-term follow-up
study. J Bone Joint Surg Am. 86:1186–1197. 2004.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Boero S, Catagni M, Donzelli O, Facchini R
and Frediani PV: Congenital pseudarthrosis of the tibia associated
with neurofibromatosis 1: Treatment with Ilizarov's device. J
Pediatr Orthop. 17:675–684. 1997.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Velan CJ, Katz K and Hendel D: Failed
treatment of congenital pseudarthrosis of the tibia-a case of
Ilizarov transportation of proximal tibia with artrodesis to talus.
Acta Orthop Scand. 69:433–434. 1998.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Ramachandran M, Tsirikos AI, Lee J and
Saifuddin A: Whole-spine magnetic resonance imaging in patients
with neurofibromatosis type 1 and spinal deformity. J Spinal Disord
Tech. 17:483–491. 2004.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Singh K, Samartzis D and An HS:
Neurofibromatosis type 1 with severe dystrophic kyphoscoliosis and
its operative management via a simultaneous anterior-posterior
approach: A case report and review of the literature. Spine J.
5:461–466. 2005.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Li M, Fang X, Li Y, Ni J, Gu S and Zhu X:
Succesful use of posterior instrumented spinal fusion alone for
scoliosis in 19 patients with neurofibromatosis type-1 followed up
for at least 25 months. Arch Orthop Trauma Surg. 129:915–921.
2009.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Ardeleanu V, Chicoş SC, Tutunaru D and
Georgescu C: Multiple benign symmetric lipomatosis-a differential
diagnosis of obesity. Chirurgia (Bucur). 108:580–583.
2013.PubMed/NCBI
|
|
61
|
Ardeleanu V, Toma A, Pafili K, Papanas N,
Motofei I, Diaconu CC, Rizzo M and Stoian AP: Current
pharmacological treatment of painful diabetic neuropathy: A
narrative review. Medicina (Kaunas). 56(25)2020.PubMed/NCBI View Article : Google Scholar
|