Introduction
Acute pancreatitis (AP) is a common gastrointestinal
disease caused by a variety of factors, such as excessive drinking,
drug misuse and surgery. In total, 10-15% of patients with AP
develop severe AP (SAP) (1), a
condition that has a 10-30% worldwide mortality rate and is a major
risk factor for pancreatic cancer (2,3). The
pathogenesis of AP is complicated and its mechanisms have not been
fully elucidated, a fact that has caused concern in the medical
community. Although there is abundant literature on the
pathogenesis of AP, the availability of effective evidence-based
treatment methods remains limited (4). In recent years, research has revealed
a new treatment method that alleviates the inflammatory symptoms of
the disease via the transfection of foreign DNA into the cells of
the gastrointestinal tract to produce small RNA (5).
A major development in gene regulation has been the
discovery of microRNA (miRNA/miR), a non-coding, highly-conserved,
single-stranded small RNA averaging 22 bp, which is involved in the
regulation of post-transcriptional gene expression (6). The first confirmed miRNAs were lin-4
and let-7, both found in C. elegans, which can inhibit
protein translation by complementarily binding to the 3'non-coding
region of the target mRNAs, thereby preventing their expression
(7). Subsequently, numerous
research teams have identified >30,000 miRNAs in >200
organisms (8-10),
most of which are highly conserved and differentially expressed
(11). Under normal conditions,
miRNA binding to the complementary sequence of the 3'untranslated
region (UTR) of the target mRNA leads to transcript degradation and
abrogated expression (12). With
the rapid development of miRNA mass spectrometry technology,
evidence the regulatory role of miRNAs in a variety of diseases has
emerged in the literature. In the context of inflammatory diseases,
Liu et al (13) showed that
miR-381 could target HMGB1 expression to alleviate the inflammatory
response of macrophages in polymyositis. Moreover, Wei et al
(14) reported that miR-198 could
act on PTEN in retinoblastoma cells by activating the PI3K/AKT
signaling pathway, while Zhang et al (15) demonstrated that TGF-β could induce
the production of miR-216a to increase the expression of TGF-β
receptor 1 and phosphorylated (p)-AKT via the downregulation of
SMAD7 and PTEN.
Recent evidence has suggested HMGB1 involvement in
the development and progression of AP (16). HMGB1 was first discovered by
Goodwin and John in 1973 who extracted and identified a group of
highly conserved nuclear proteins from the bovine thymus, which
displayed rapid migration during electrophoresis (17). In the HMGB family, HMGB1 is the
most abundant and widely distributed member among tissues in humans
(16). Research has shown that
intracellular HMGB1 inhibits the development of pancreatitis
(18-21),
while extracellular HMGB1 likely promotes the progression of AP
from local inflammation to systemic inflammatory response syndrome
and sepsis (22). In the early
stage of AP, pancreatic acinar cells and peritoneal macrophages
successively release inflammatory factors, such as IL-1, TNFα and
NF-κB (23-25).
These early inflammatory mediators destroy the pancreas and
surrounding tissues, stimulate the secretion of HMGB1 and
subsequently aggravate pancreatitis (20,26,27).
Inflammation is not only part of the early phase of pancreatitis
but also occurs throughout its duration (28). In addition to causing tissue damage
via cell death, these inflammatory factors excessively activate
granulocytes, leading to lysosome release and additional
cytokines/chemokines that increase oxidative stress, injure
vascular endothelium and ultimately cause apoptosis (29). Furthermore, HMGB1 is prone to
binding other pro-inflammatory molecules, including DNA, RNA,
histones, nucleosomes, lipopolysaccharide (LPS), stromal
cell-derived factor 1, IL-1α and IL-1β, amongst others. These
complexes act synergistically to aggravate pancreatitis via cognate
receptors to the HMGB1-partner molecules. Although the list of
reported HMGB1 receptors is fairly extensive, only two receptor
systems, MOK protein kinase and Toll-like receptor (TLR)4, have
been fully confirmed to act as genuine HMGB1 receptors (30). TLR4-deficient mice have been shown
to succumb to endotoxemia in the presence of increased levels of
HMGB1, while caspase11-deficient mice survive (31).
The PI3K/AKT signaling pathway plays a key role in
the regulation of cell apoptosis and proliferation. Activated (i.e.
phosphorylated) AKT promotes cell proliferation and inhibits
apoptosis, leading to both blunted and exacerbated inflammatory
responses, depending on the context (32).
Based on prior reports and an online search
regarding the association between miR-340-5p and HMGB1 expression
in pancreatic acinar cells, we hypothesized that the upregulation
of miR-340-5p could cause the downregulation of HMGB1, in turn
inhibiting the activation of TLR4 and restraining cellular
inflammation and apoptosis (Fig.
S1).
Materials and methods
Primary culture of pancreatic acinar
cells and treatment
Pancreatic acinar cells were isolated from healthy
adult male 4-6 weeks-old C57BL/6J mice (weight, 25-30 g; Beijing
Vital River Laboratory Animal Technology Co., Ltd.). Animals were
housed in specific pathogen-free conditions under a standard
temperature (22±1˚C), humidity (50-60%) and light cycle (12 h
light/dark cycle), with ad libitum access to food and water.
The animal experiments were approved by the Ethics Committee
(approval no. 2021-1523) of Xi'an Jiaotong University (Xi'an,
China). The number of animals used was 20. Mouse death was
confirmed by the stopping of the heartbeat. Preparation of mouse
pancreatic acinar cells was carried out by using previously
described methods (33). Briefly,
mice were sacrificed by exsanguination under deep anesthesia
(sodium pentobarbital intraperitoneal injection, 50 mg/kg), the
pancreas was immediately removed from the sacrificed mouse and
incubated in buffer solution (Thermo Scientific Fisher, Inc.) at
37˚C for 10 min in a shaking bath (100 cycles/min). The buffer
solution contained: 130 mM NaCl; 4.7 mM KCl; 1.3 mM
CaCl2; 1 mM MgCl2; 1.2 mM
KH2PO4; 10 mM glucose; 10 mM HEPES; 0.01%
trypsin inhibitor (soybean) and 0.2% BSA (pH adjusted to 7.4 with
NaOH, Sigma-Aldrich; Merck KGaA). Then, the cell suspension was
centrifuged at 335 x g for 5 min at 4˚C. Next, the acinar cell
pellets were resuspended in HEPES buffer without collagenase and
centrifuged at 335 x g for 5 min at 4˚C, following which the
supernatant was removed. Primary pancreatic cells were maintained
in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.) with
10% FBS (Gibco; Thermo Fisher Scientific, Inc.), 1% antibiotics
(penicillin and streptomycin; Gibco; Thermo Fisher Scientific,
Inc.), 2 mM L-glutamine and 25 µg/ml gentamicin at 37˚C with 5%
CO2. LPS (100 ng/ml; Sigma-Aldrich; Merck KGaA) was
added to cells for 0, 2, 6, 12, 24, 48 h at 37˚C.
Cell viability analysis
Pancreatic acinar cells (1x104) were
cultured on a 96-well plate and treated with increasing
concentrations of LPS (0-200 ng/ml) for 48 h at 37˚C. Cell
viability was then measured using a Cell Counting Kit (CCK)-8 assay
for 1 h (Beyotime Institute of Biotechnology) according to the
manufacturer's instructions.
Western blotting
Briefly, cell pellets were lysed in the ice-cold RIPA
buffer (Xi'an Hat Biotechnology Co., Ltd.). Proteins were extracted
from the pancreatic cells and quantified using a BCA protein kit
(Thermo Fisher Scientific, Inc.). Proteins (30 µg) were loaded on
12% SDS-PAGE gels per lane, separated via electrophoresis and
transferred to PVDF membranes (MilliporeSigma). The membranes were
blocked with 5% non-fat milk diluted with TBS-0.1% Tween-20 (TBST)
buffer at room temperature for 1 h and then incubated with primary
antibodies overnight at 4˚C. Antibodies against HMGB1 (1:500;
Abcam; cat. no. ab18256), TLR4 (1:1,000; Abcam; cat. no. ab13556),
pan-AKT (1:1,000; Abcam; cat. no. ab8805), p-AKT (1:500; Abcam;
cat. no. ab38449), Bcl2 (1:500; Abcam; cat. no. ab182858),
cleaved-caspase3 (1:500; Invitrogen; cat. no. PA5-114687; Thermo
Fisher Scientific, Inc.) and β-actin (1:2,000; Invitrogen; cat. no.
MA5-15739-HRP; Thermo Fisher Scientific, Inc.) were used. On the
following day, the PVDF membranes were washed with TBST buffer and
then incubated with HRP-conjugated secondary antibody (1:10,000;
cat. no. TA130004; OriGene Technologies, Inc.) at room temperature
for 2 h. Western blots were developed using an ECL reagent
(MilliporeSigma) plus a western blotting detection system (Bio-Rad
Laboratories, Inc.). Densitometry was performed using Universal
Hood III software (version no. 731BR03155; Bio-Rad Laboratories,
Inc.).
Reverse transcription-quantitative
(RT-q) PCR analysis
Total RNA was extracted using TRIzol®
reagent (MilliporeSigma) following the manufacturer's instructions.
cDNA was synthesized by using a FastKing RT kit (Qiagen China Co.,
Ltd.) in accordance with the manufacturer's protocol. RT-qPCR were
carried out with a SuperReal Premix Plus kit (Vazyme Biotech Co.,
Ltd.). The thermocycling conditions for qPCR were as follows: 15
min at 95˚C to activate the chemically modified
hot-start Taq DNA polymerase, followed by 40 cycles of duration for
15 sec at 95˚C and 30 sec of annealing and extension at
60˚C. The primer sequences were as follows: HMGB1
forward (F), 5'-CGCGGAGGAAAATCAACTAA-3' and reverse (R),
5'-TCATAACGAGCCTTGTCAGC-3'; TNFα F, 5'-TCCCAGGTTCTCTTCAAGGGA-3' and
R, 5'-GGTGAGGAGCACGTAGTCGG-3'; IL-1β F, 5'-TGGAAAAGCGGTTTGTCTTC-3'
and R, 5'-TACCAGTTGGGGAACTCTGC-3'; IL-6 F,
5'-GCTGGTGACAACCACGGCCT-3' and R, 5'-AGCCTCCGACTTGTGAAGTGGT-3'; and
GAPDH F, 5'-CGTCCCGTAGACAAAATGGT-3' and R,
5'-TTGATGGCAACAATCTCCAC-3'. GAPDH was amplified as the internal
control. The miR-340-5p primer was the Bulge-Loop™ miRNA
RT-PCR primer (Guangzhou RiboBio Co., Ltd.), with U6 snRNA
(forward, 5'-CCGCCCGCCGCCAGGCCCC-3' and reverse,
5'-ATATGGAACGCTTCACGAATT-3') as the miRNA quantitative internal
reference. The original Ct values of the sample (cycle of the
threshold) were adjusted to internal control and relative
transcript levels were analyzed using the 2-ΔΔCq method
(34).
Luciferase assay and LPS
treatment
A lentivirus vector (pGCL-GFP; Promega Corporation)
containing a U6 promoter and a green fluorescent protein (GFP)
reporter was used for cloning HMGB1 short hairpin RNAs
(shRNAs/shR). The shRNA sequences were as follows: shR-HMGB1,
5'-GGCTCGTTATGAAAGAGAAAT-3'; and shR-control,
5'-GTTCTCCGAACGTGTCACGT-3'. 293T cells were inoculated in T75
culture flasks (Nalge Nunc International) at a density of
2x106 cells and were left to reach 70-80% confluence the
day prior to infection. The lentiviral plasmid PGCL-GFP (6 µg) and
packaging plasmids (pHelper 1.0 4.5 µg and pHelper 2.0; 2.4 µg; all
Invitrogen; Thermo Fisher Scientific Inc.) were transfected into
293T cells using the X-tremeGENE™ HP DNA Transfection
Reagent (Roche Diagnostics GmbH) for 16 h at 37˚C in 5%
CO2, according to the manufacturer's protocol. Following
48-72 h, supernatants containing lentiviral particles were
harvested and filtered through a 0.45-µm filter (EMD Millipore) to
remove cell debris. The supernatants were concentrated by
ultracentrifugation at 50,000 x g at 4˚C for 90 min,
after which the lentiviral particle pellet was resuspended in 100%
FBS and stored at -80˚C. The viral titers of
concentrated lentiviral particles were measured by infecting 293T
cells that were seeded at a density of 1x105 cells/well
in a 12-well plate with viral serial dilutions
(10-10-8). After 3 days, GFP expression was detected
using flow cytometry and the viral titer was calculated using the
following equation: Viral titer (Tu/µl)=(% GFP + cells x number of
cells transduced)/virus volume. For lentiviral transduction,
Pancreatic acinar cells were seeded at 1x106 cells/ml in
six-well plates and infected with lentivirus at a multiplicity of
infection of 10 for 24 h at 37˚C and 5% CO2.
After 2 days of culture, the cells were collected, and the
expression of GFP was detected using fluorescence microscopy. Then,
cells were treated with 100 ng/ml LPS for 24 h at 37˚C, and then
lysates were harvested and analyzed with a Luciferase Reporter
Assay system (Promega Corporation).
Construction of luciferase reporter
gene vector and dual-luciferase reporter gene assay
The miR-340-5p mimics and negative control duplex
were synthesized by Shanghai GenePharma Co., Ltd. The sequence of
the miR-340-5p mimic was 5'-UUAGUCAGAGUAACGAAAUAUU-3'; and that of
the NC mimic was 5'-GCCUGAGUCUGGCAUCCGGGGC-3'. The microRNA.org online (http://www.targetscan.org/cgibin/targetscan/vert_72/targetscan.cgi?species=Mouse&gid=HMGB1&mir_sc=&mir_c=&mir_nc=&mir_vnc=&mirg=miR-340-5p)
target gene prediction tool predicted the association between
miR-340-5p and the 3'UTR of HMGB1 mRNA. HMGB1 3'UTR full length
fragment and mutation-containing fragments were cloned into the
luciferase reporter plasmid (pSiCHECK2 vectors; Promega
Corporation) to transform DH5α competent cells (Sangon Biotech Co.,
Ltd.; cat. no. B528413). The efficient plasmids were sequenced,
screened and named wild-type (WT)-HMGB1 and mutated (MUT)-HMGB1.
The sequences were as follows: WT-HMGB1,
5'-AUACAUUUGCUUUUUCUUUAUAA-3'; and MUT-HMGB1,
5'-AUACAUUUGCUUUUUGAAAUAUU-3'. The WT-HMGB1 or MUT-HMGB1 and
miR-340-5p mimics were co-transfected into 293T cells using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) following the manufacturer's instructions. 293T
cells were seeded into 6-well plates at a density of 105
cells/well. A total of 10 µl transfection reagent was mixed with
100 µl serum-free culture. Meanwhile, 10 µl miR-340-5p mimics,
WT-HMGB1 and MUT-HMGB1 was mixed as aforementioned. Next, the two
mixtures were mixed and incubated at room temperature for 10 min.
Subsequently, the mixture was added to the 6-well plate at a final
concentration of 20 nM. The reporter luciferase activities were
detected via a thermoplate reader (Rayto Life and Analytical
Science Co., Ltd.) after 48 h. Firefly luciferase activity was then
normalized to that of Renilla luciferase. All the
transfection experiments were performed in triplicate and repeated
at least three times independently.
Statistical analysis
All data are presented as the mean ± SD, and were
analyzed using SPSS statistical software version 18.0 (SPSS, Inc.).
Each experiment was repeated in triplicate and the statistical
difference was analyzed using an unpaired Student's t-test for
comparisons of two groups or one-way ANOVA for two factor
experiments or one-way ANOVA followed by Tukey's test for
comparisons of multiple groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
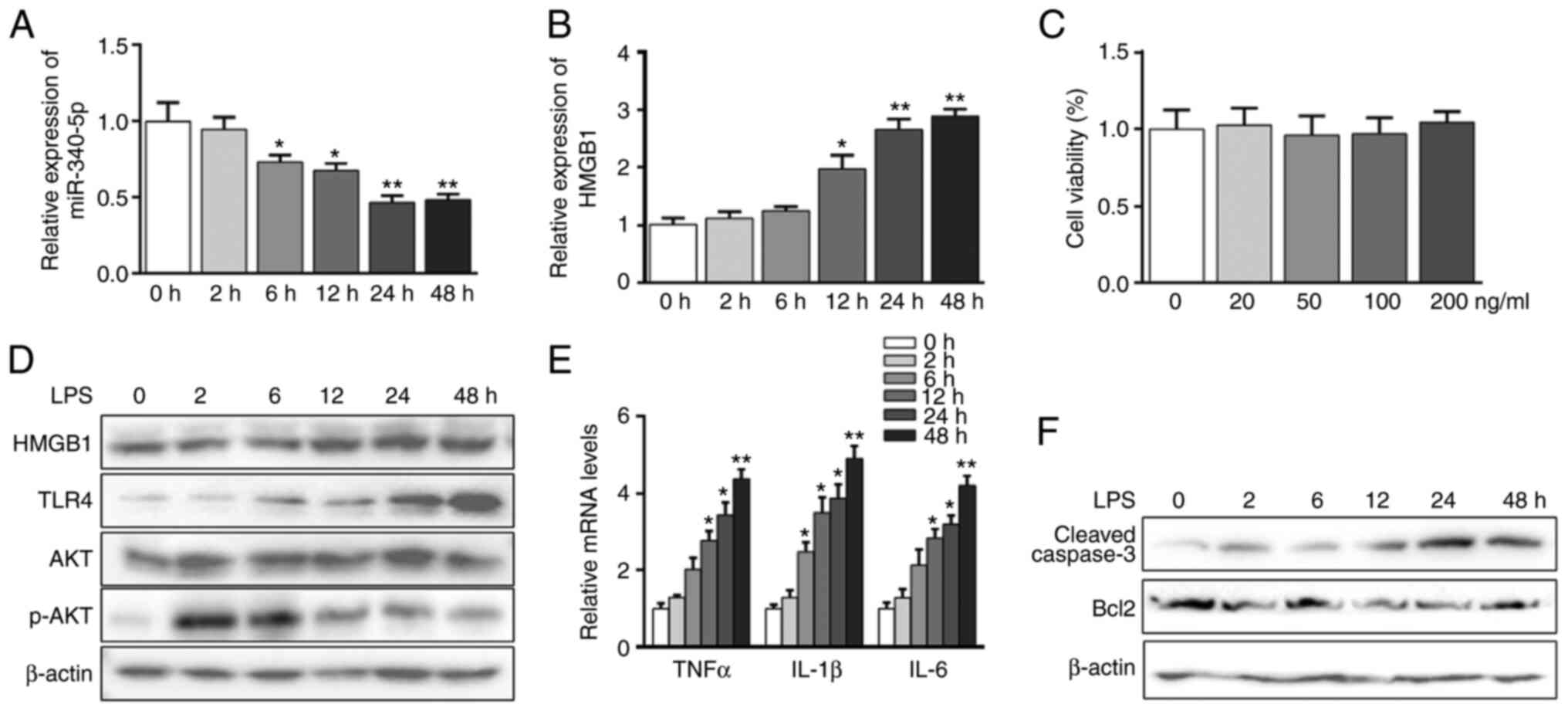
LPS-induced acute inflammation and
apoptosis parallels miR-340-5p suppression and HMGB1 elevation in
pancreatic acinar cells
The pancreatic acinar cell model induced by LPS was
used for the inflammation study (35-38).
It was found that LPS inhibited the expression of miR-340-5p and
upregulated HMGB1 mRNA expression in a time-dependent manner
(Fig. 1A and B). The cytotoxic effects of LPS were
evaluated using a CCK-8 assay, which showed no obvious cell death
at up to 200 ng/ml LPS treatment for 48 h (Fig. 1C). Western blot analysis confirmed
that HMGB1 protein expression was increased in the pancreatic
acinar cells in a time-dependent manner following LPS treatment
(Fig. 1D). Additionally, LPS
increased the expression level of TLR4, as well as enhanced those
of TNFα, IL-1β and IL-6 in a time-dependent manner (Fig. 1D and E). p-AKT expression showed a significant
increase when LPS stimulated pancreatic acinar cells within 2 h,
and then this slowly decreased (Figs.
1D and S2A). Moreover, LPS
treatment significantly enhanced the expression of cleaved-caspase3
and downregulated Bcl2 expression over time (Fig. 1F).
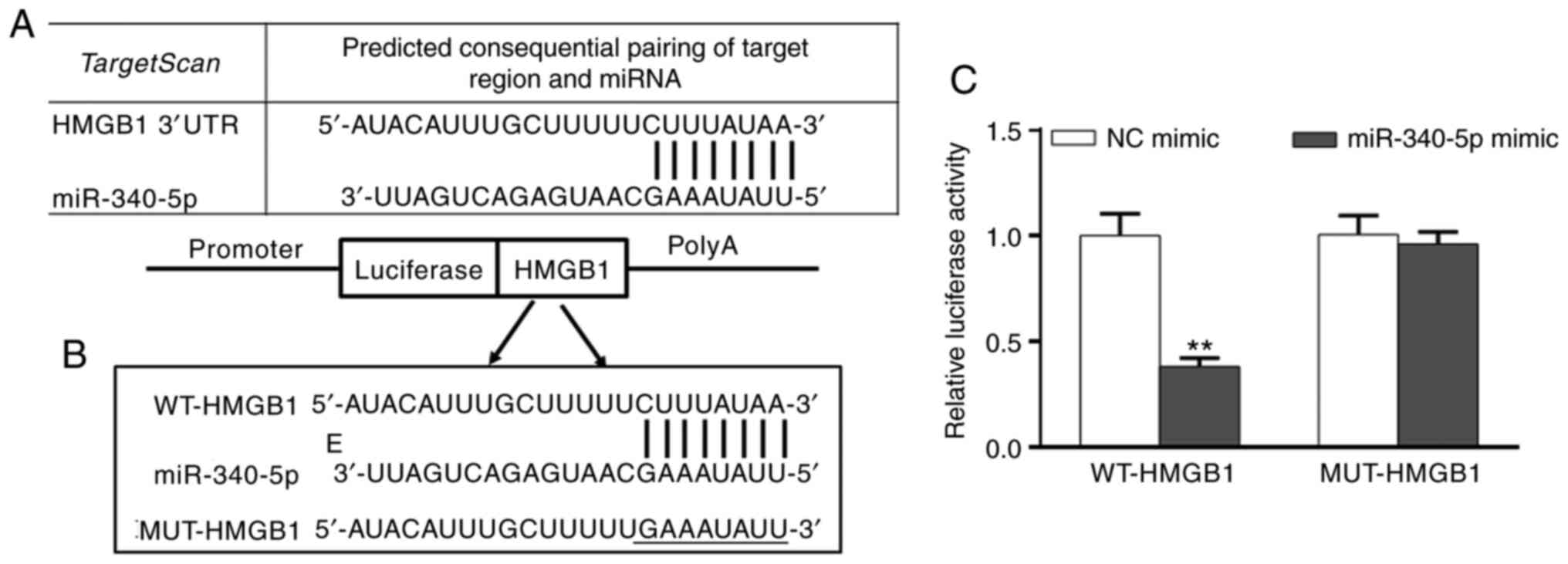
miR-340-5p inhibits HMGB1
expression
A query of the microRNA.org
online target gene database predicted that there was a targeted
binding site for miR-340-5p on the 3'UTR of HMGB1 mRNA (Fig. 2A and B). Furthermore, transfection of
miR-340-5p mimics significantly reduced the relative luciferase
activity of HMGB1 compared with NC mimic, suggesting that
miR-340-5p was capable of inhibiting the expression of HMGB1
(Fig. 2C).
miR-340-5p inhibits inflammation and
apoptosis in pancreatic acinar cells following LPS treatment
To further investigate the role of miR-340-5p in the
current model, it was determined whether miR-340-5p exerted
anti-inflammatory and anti-apoptotic effects in pancreatic acinar
cells treated with LPS. Cells were transfected with miR-340-5p
mimics or NC mimics, subjected to LPS 24 h and then the
aforementioned endpoints were examined. miR-340-5p expression was
upregulated in cells transfected with miR-340-5p mimics (Fig. S2C). Transfection with miR-340-5p
mimics led to decreased HMGB1, TLR4 and AKT upregulation following
LPS (Fig. 3A and B), as well as enhanced p-AKT expression
(Fig. 3B). In addition,
LPS-induced expression of the pro-inflammatory cytokines TNF-α,
IL-1β and IL-6 were significantly decreased compared with NC mimics
(Fig. 3C). The LPS-induced
expression levels of cleaved-caspase3 were notably inhibited, while
anti-apoptotic Bcl2 expression was markedly enhanced in cells
transfected with miR-340-5p mimics compared with NC mimics
(Fig. 3D).
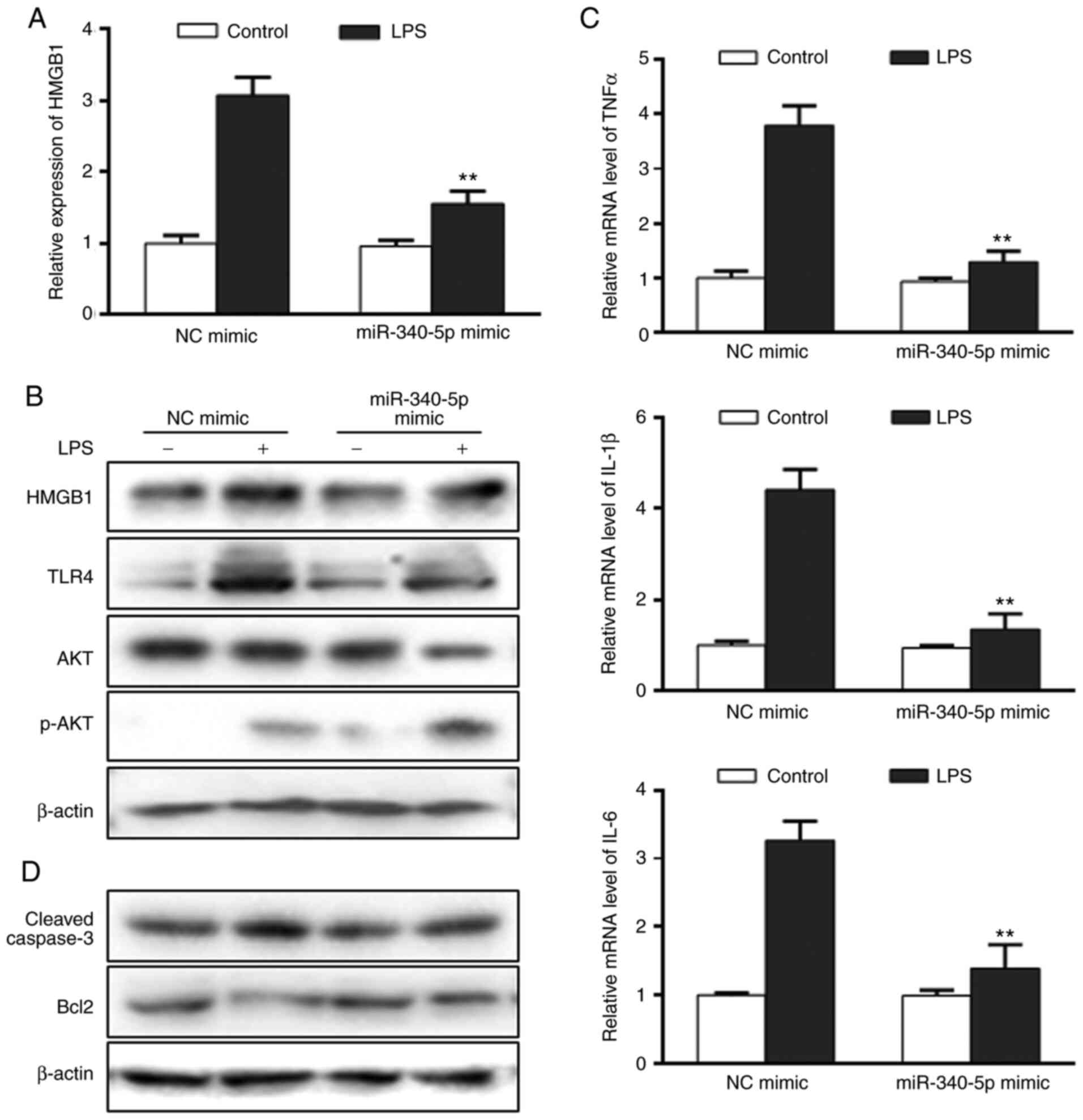 | Figure 3miR-340-5p overexpression can inhibit
HMGB1 expression, as well as inflammation and apoptosis in
pancreatic acinar cells with LPS. (A) HMGB1 expression in
pancreatic acinar cells transfected with miR-340-5p mimics was
assayed via RT-qPCR. (B) Expression levels of HMGB1, TLR4, AKT and
p-AKT in pancreatic acinar cells with miR-340-5p mimic were assayed
via western blotting. (C) mRNA expression levels of TNFα, IL-1β and
IL-6 in pancreatic acinar cells transfected with miR-340-5p mimics
were examined via RT-qPCR. (D) Expression level of cleaved-caspase3
and Bcl2 in pancreatic acinar cells with miR-340-5p mimic were
assayed via western blotting. Data are expressed as the mean ± SD.
**P<0.01 vs. control. miR-340-5p, microRNA-340-5p;
NC, negative control; RT-qPCR, reverse transcription-quantitative
PCR; TLR, Toll-like receptor; HMGB1, high mobility group box 1; p-,
phosphorylated; LPS, lipopolysaccharide. |
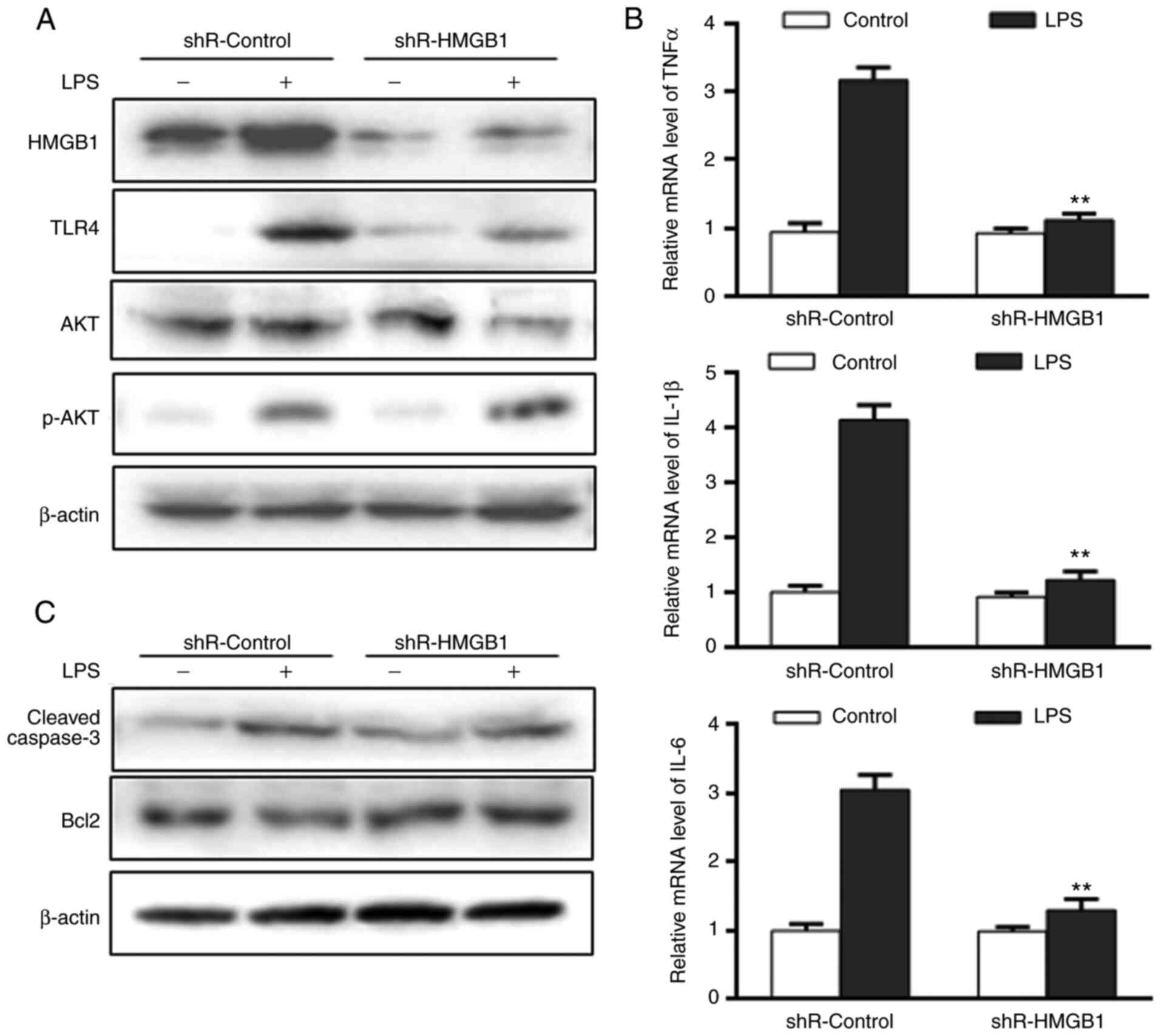
miR-340-5p could inhibit inflammation
and apoptosis via HMGB1 targeting in pancreatic acinar cells
following LPS treatment
To further examine the role of HMGB1, it was
determined whether the miR-340-5p-mediated anti-inflammatory and
anti-apoptotic effects in pancreatic acinar cells following LPS
treatment were mediated by HMGB1. Cells were transfected with
either non-specific shRNA (shR-control) or shR-HMGB1, subjected to
LPS for 24 h and then the same endpoints were examined. The
expression level of HMGB1 was significantly decreased in cells
transfected with shR-HMGB1 (Figs.
4A and S2B). HMGB1 knockdown
caused marked decreases in TLR4 and AKT upregulation following LPS
(Fig. 4A), and enhanced p-AKT
expression (Fig. 4A).
Additionally, cells transfected with shR-HMGB1 had significantly
lower expression levels of pro-inflammatory cytokines (Fig. 4B), decreased cleaved-caspase3 and
increased Bcl2 compared with shR-control (Fig. 4C) following LPS treatment.
Discussion
A critical component in the pathophysiology of AP is
inflammation (39,40). The present study demonstrated the
protective effect of miR-340-5p on LPS-induced inflammation and
apoptosis in pancreatic acinar cells. miRNAs have emerged as
important post-transcriptional regulatory factors in recent years
(10). The current results
demonstrated that miR-340-5p attenuated LPS-induced upregulation of
HMGB1, decreased TLR4 activation and promoted the activation of
AKT, subsequently leading to decreased inflammatory and apoptotic
signaling. TLR4 is widely expressed in the pancreatic tissues and
plays an important role in pancreatitis (41). A previous study has shown that TLR4
participates in the early stage of SAP and may also be involved in
its progression (42). The
PI3K/AKT signaling pathway plays an important role in the
activation of pancreatic trypsinogen, a causal factor in the onset
and aggravation of AP (35).
Moreover, increasing evidence suggests that PI3K/AKT signaling is
involved in the pathogenesis of inflammatory diseases, such as AP
(43). Pharmacological activation
of the PI3K/AKT pathway may therefore represent a promising new
direction in therapeutics to limit inflammation in SAP (44), as recent studies suggest (45).
HMGB1 is an important mediator of damage-associated
molecular pattern (DAMP) signaling. DAMPs can activate pattern
recognition receptors such as TLRs and NOD-like receptors. Among
them, TLRs are considered to be ‘gateway’ proteins that initiate
the inflammatory response. TLR4 has been reported to bind HMGB1
secreted by macrophages and neutrophils to recruit myeloid
differentiation protein 88 and IL-1 receptor-related kinases,
thereby causing the downstream regulator TRAF to activate the MAPK
pathway and nuclear (46,47) transcription factors to induce the
inflammatory response. In addition to TLR4, extracellular HMGB1 can
interact with TLR2 and RAGE (48).
TLR4 activation promotes the degradation of IκB, leading to NF-κB
p65 nuclear translocation and subsequent expression of
pro-inflammatory cytokines/chemokines and other inflammatory
mediators (49-54),
ultimately leading to intestinal damage. In addition to its
deleterious effects, at low levels HMGB1 can promote tissue repair.
Diener et al (55) reported
that HMGB1, as an ‘alarmin’ secreted by damaged tissue, can induce
stem cells or primitive cells to migrate to the damaged area to aid
in its repair and replacement. Biscetti et al (56) also showed that HMGB1 plays a role
in protecting and repairing myocardial tissue following myocardial
infarction. However, information regarding the threshold and
mechanism via which HMGB1 promotes the pro-inflammatory and
deleterious effects, as compared with the protective effects in
tissues, remains to be determined. Recent experimental studies have
shown that early inflammatory factors reach their peak quickly
after AP model induction (e.g. LPS exposure), and then rapidly
decline to normal level, although the damage to the pancreas
persists (23,35). A good systemic indicator of organ
damage is the serum levels of HMGB1. In a mouse model of acute
necrotizing pancreatitis, serum HMGB1 increased significantly with
disease onset and correlated positively with disease severity
(57). In this regard, some
investigators have proposed using the serum HMGB1 level as a
biomarker of AP severity in patients and there are existing reports
to support its use in this manner (58). Compared with early inflammation,
HMGB1 appears late, has a long action time and forms an
inflammatory positive feedback loop. As such, it plays a key role
in regulating inflammation in the context of AP. Inhibition of
extracellular HMGB1 has also been proposed to be a therapeutic
target for AP treatment (57).
miRNAs are known to regulate a wide variety of
physiological and pathological processes, and rapidly accumulating
evidence is suggesting that they are key regulators of numerous
diseases (9,11,59).
However, regulation of target genes by miRNAs is complicated and
difficult to model and study. For example, while one single miRNA
may be capable of regulating expression of multiple genes, the
combination of several miRNAs may be necessary to fine-tune the
expression of a single gene. Most of the evidence to date suggests
that the main role of miRNA is to downregulate and, in rare cases,
upregulate target gene expression (9). miRNA sequences are complementary to
the target gene mRNA, usually inhibiting the translation of the
target mRNA and thereby acting similarly to RNA interference
(59). From a clinical
perspective, miRNA-based diagnostic and treatment methods have
exciting and have a broad potential for the future. They may serve
in a gene therapy capacity, where adenovirus vectors carrying
target miRNAs may be used to regulate the expression of target
genes (60,61). With respect to miR-340-5p, a prior
study reported that this miRNA was downregulated in hearts with
ischemia-reperfusion injury. Overexpression of miR-340-5p inhibited
ischemia-reperfusion-induced apoptosis in H9C2 cardiomyoblasts
(62). Overexpression of
miR-340-5p was also shown to significantly promote the activation
of the PI3K/AKT signaling pathway to alleviate neuronal cell damage
(63). However, to the best of our
knowledge, a role for miR-340-5p in AP has not been previous
examined. The current study is important as it identified that
miR-340-5p was capable of targeting HMGB1 and inhibiting its
expression (Fig. S1), although
the precise mechanism of action requires further investigation.
In conclusion, the presented study demonstrated that
miR-340-5p effectively attenuated the severity of LPS-induced
inflammation and apoptosis in pancreatic acinar cells, and this
effect was likely mediated via the suppression of HMGB1 and
activation of PI3K/AKT signaling. Collectively, these findings
support the notion that miR-340-5p may be a promising target for
new AP therapies, and they also support a need for additional
mechanistic studies that examine a role for HMGB1 in AP
progression.
Supplementary Material
Schematic diagram of miR-340-5p
inhibition of pancreatic acinar cell inflammation and apoptosis via
targeted inhibition of HMGB1. LPS-induced acute inflammation and
apoptosis parallels miR-340-5p (green arrows) suppression and HMGB1
elevation in pancreatic acinar cells. miR-340-5p inhibits
inflammation and apoptosis via HMGB1 targeting in pancreatic acinar
cells following LPS (red arrows) treatment. TLR, Toll-like
receptor; HMGB1 (blue arrows), high mobility group box 1; p-,
phosphorylated; LPS, lipopolysaccharide; miR-340-5p,
microRNA-340-5p; shR, short hairpin RNA.
Quantifications of the Figs. 1 and 3. (A) Pancreatic acinar cells were
treated with lipopolysaccharide for different durations, and the
ratio of p-AKT/AKT protein expression was examined.
**P<0.01 vs. 0 h. (B) HMGB1 expression was
deter-mined in pancreatic acinar cells with HMGB1 knockdown. (C)
miR-340-5p expression was measured in pancreatic acinar cells
transfected with miR-340-5p mimic. Data are expressed as the means
± SD. **P<0.01 vs. shR-control or NC mimic. shR,
short hairpin RNA; HMGB1, high mobility group box 1; p-,
phosphorylated; miR-340-5p, microRNA-340-5p; NC, negative
control.
Acknowledgements
Not applicable.
Funding
Funding: This research study was approved and financially
supported by Shaanxi Provincial Science and Technology Research
Subject of Traditional Chinese Medicine (grant no. 2019-ZZ-JC031),
Fund for Free Exploration Project of The Second Affiliated Hospital
of Xi'an Jiaotong University [grant no. 2020YJ(ZYTS)359], the
Natural Science Basic Research Plan in Shaanxi Province of China
(grant no. 2021JM-284) and the Health Research Projects of Shaanxi
Province (grant no. 2021A010).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
All authors contributed to this work. LP and YazhouG
made substantial contributions to the conception and design of the
study, and were involved in the drafting of the manuscript.
YazhouG, LW, JS and YanxiaG made substantial contributions to data
acquisition. ZN, HF and JL were responsible for the development of
the study methodology, analysis and interpretation of the data. LP
and YazhouG confirm the authenticity of the raw data. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
The use of animals and all related procedures were
in accordance with the Institutional Animal Care Committee of Xi'an
Jiaotong University. The animal experiments were approved by the
Ethics Committee of Xi'an Jiaotong University (Xi'an, China). All
efforts were made to reduce number of animals and to lower their
sufferings.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ji L, Lv JC, Song ZF, Jiang MT, Li L and
Sun B: Risk factors of infected pancreatic necrosis secondary to
severe acute pancreatitis. Hepatobiliary Pancreat Dis Int.
15:428–433. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Singh P and Garg PK: Pathophysiological
mechanisms in acute pancreatitis: Current understanding. Indian J
Gastroenterol. 35:153–166. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bhatia M, Wong FL, Cao Y, Lau HY, Huang J,
Puneet P and Chevali L: Pathophysiology of acute pancreatitis.
Pancreatology. 5:132–144. 2005.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Pandol SJ, Saluja AK, Imrie CW and Banks
PA: Acute pancreatitis: Bench to the bedside. Gastroenterology.
132:1127–1151. 2007.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Jiang J, Yamato E and Miyazaki J:
Intravenous delivery of naked plasmid DNA for in vivo cytokine
expression. Biochem Biophys Res Commun. 289:1088–1092.
2001.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Shimomura A, Shiino S, Kawauchi J,
Takizawa S, Sakamoto H, Matsuzaki J, Ono M, Takeshita F, Niida S,
Shimizu C, et al: Novel combination of serum microRNA for detecting
breast cancer in the early stage. Cancer Sci. 107:326–334.
2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Cech TR and Steitz JA: The noncoding RNA
revolution-trashing old rules to forge new ones. Cell. 157:77–94.
2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Khoshnam SE, Winlow W, Farbood Y,
Moghaddam HF and Farzaneh M: Emerging roles of microRNAs in
ischemic stroke: As possible therapeutic agents. J Stroke.
19:166–187. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Malumbres M: miRNAs and cancer: An
epigenetics view. Mol Aspects Med. 34:863–874. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Matts J, Jagadeeswaran G, Roe BA and
Sunkar R: Identification of microRNAs and their targets in
switchgrass, a model biofuel plant species. J Plant Physiol.
167:896–904. 2010.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Qu B and Shen N: miRNAs in the
pathogenesis of systemic lupus erythematosus. Int J Mol Sci.
16:9557–9572. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wang W, Dai LX, Zhang S, Yang Y, Yan N,
Fan P, Dai L, Tian HW, Cheng L, Zhang XM, et al: Regulation of
epidermal growth factor receptor signaling by plasmid-based
microRNA-7 inhibits human malignant gliomas growth and metastasis
in vivo. Neoplasma. 60:274–283. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Liu Y, Gao Y, Yang J, Shi C, Wang Y and Xu
Y: MicroRNA-381 reduces inflammation and infiltration of
macrophages in polymyositis via downregulating HMGB1. Int J Oncol.
53:1332–1342. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wei D, Miao Y, Yu L, Wang D and Wang Y:
Downregulation of microRNA-198 suppresses cell proliferation and
invasion in retinoblastoma by directly targeting PTEN. Mol Med Rep.
18:595–602. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhang J, Ning X, Cui W, Bi M, Zhang D and
Zhang J: Transforming growth factor (TGF)-β-induced microRNA-216a
promotes acute pancreatitis via Akt and TGF-β pathway in mice. Dig
Dis Sci. 60:127–135. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Engel C, Brunkhorst FM, Bone HG,
Brunkhorst R, Gerlach H, Grond S, Gruendling M, Huhle G, Jaschinski
U, John S, et al: Epidemiology of sepsis in Germany: Results from a
national prospective multicenter study. Intensive Care Med.
33:606–618. 2007.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Wu H, Li R, Pei LG, Wei ZH, Kang LN, Wang
L, Xie J and Xu B: Emerging role of high mobility group box-1 in
thrombosis-related diseases. Cell Physiol Biochem. 47:1319–1337.
2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Gao N, Yan C and Zhang G: Changes of serum
procalcitonin (PCT), C-reactive protein (CRP), interleukin-17
(IL-17), interleukin-6 (IL-6), high mobility group protein-B1
(HMGB1) and D-dimer in patients with severe acute pancreatitis
treated with continuous renal replacement therapy (CRRT) and its
clinical significance. Med Sci Monit. 24:5881–5886. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhao S, Yang J, Liu T, Zeng J, Mi L and
Xiang K: Dexamethasone inhibits NF-κBp65 and HMGB1 expression in
the pancreas of rats with severe acute pancreatitis. Mol Med Rep.
18:5345–5352. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Kang R, Zhang Q, Hou W, Yan Z, Chen R,
Bonaroti J, Bansal P, Billiar TR, Tsung A, Wang Q, et al:
Intracellular Hmgb1 inhibits inflammatory nucleosome release and
limits acute pancreatitis in mice. Gastroenterology. 146:1097–1107.
2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhang ZW, Zhang QY, Zhou MT, Liu NX, Chen
TK, Zhu YF and Wu L: Antioxidant inhibits HMGB1 expression and
reduces pancreas injury in rats with severe acute pancreatitis. Dig
Dis Sci. 55:2529–2536. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kang R, Lotze MT, Zeh HJ, Billiar TR and
Tang D: Cell death and DAMPs in acute pancreatitis. Mol Med.
20:466–477. 2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Saluja A, Dudeja V, Dawra R and Sah RP:
Early intra-acinar events in pathogenesis of pancreatitis.
Gastroenterology. 156:1979–1993. 2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Cheng L, Luo Z, Xiang K, Ren J, Huang Z,
Tang L and Tian F: Clinical significance of serum triglyceride
elevation at early stage of acute biliary pancreatitis. BMC
Gastroenterol. 15(19)2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Sit M, Aktas G, Yilmaz EE, Alcelik A,
Terzi EH and Tosun M: Effects of the inflammatory response on serum
omentin levels in early acute and chronic pancreatitis. Clin Ter.
165:e148–e152. 2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Schneider L, Jabrailova B, Strobel O,
Hackert T and Werner J: Inflammatory profiling of early
experimental necrotizing pancreatitis. Life Sci. 126:76–80.
2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Yang R, Tenhunen J and Tonnessen TI: HMGB1
and histones play a significant role in inducing systemic
inflammation and multiple organ dysfunctions in severe acute
pancreatitis. Int J Inflam. 2017(1817564)2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Habtezion A: Inflammation in acute and
chronic pancreatitis. Curr Opin Gastroenterol. 31:395–399.
2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wang S, Ni HM, Chao X, Wang H, Bridges B,
Kumer S, Schmitt T, Mareninova O, Gukovskaya A, De Lisle RC, et al:
Impaired TFEB-mediated lysosomal biogenesis promotes the
development of pancreatitis in mice and is associated with human
pancreatitis. Autophagy. 15:1954–1969. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Xin-Peng Z, Yong-Hua H, Yong L, Jing-Jing
W, Guang-Hua W, Ren-Jie W and Min Z: A high-mobility group box 1
that binds to DNA, enhances pro-inflammatory activity, and acts as
an anti-infection molecule in black rockfish, Sebastes schlegelii.
Fish Shellfish Immunol. 56:402–409. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Kayagaki N, Wong MT, Stowe IB, Ramani SR,
Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP,
Muszyński A, et al: Noncanonical inflammasome activation by
intracellular LPS independent of TLR4. Science. 341:1246–1249.
2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Keppler-Noreuil KM, Parker VE, Darling TN
and Martinez-Agosto JA: Somatic overgrowth disorders of the
PI3K/AKT/mTOR pathway & therapeutic strategies. Am J Med Genet
C Semin Med Genet. 172:402–421. 2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Estaras M, Ameur FZ, Roncero V,
Fernandez-Bermejo M, Blanco G, Lopez D, Mateos JM, Salido GM and
Gonzalez A: The melatonin receptor antagonist luzindole induces
Ca2+ mobilization, reactive oxygen species generation
and impairs trypsin secretion in mouse pancreatic acinar cells.
Biochim Biophys Acta Gen Subj. 1863(129407)2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhao Q, Zhang H, Wu J, Lv X, Jin X and Hu
J: Melatonin inhibits the endoplasmic reticulum stress-induced,
C/EBP homologous protein-mediated pathway in acute pancreatitis.
Mol Med Rep. 22:1647–1655. 2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Liu Y, Yang L, Chen KL, Zhou B, Yan H,
Zhou ZG and Li Y: Knockdown of GRP78 promotes apoptosis in
pancreatic acinar cells and attenuates the severity of cerulein and
LPS induced pancreatic inflammation. PLoS One.
9(e92389)2014.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Li Z, Xu C, Tao Y, Liang Y, Liang Q, Li J,
Li R and Ye H: Anisodamine alleviates lipopolysaccharide-induced
pancreatic acinar cell injury through NLRP3 inflammasome and NF-κB
signaling pathway. J Recept Signal Transduct Res. 40:58–66.
2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zhang H, Li YY and Wu XZ: Effect of
Tetrandrine on LPS-induced NF-kappaB activation in isolated
pancreatic acinar cells of rat. World J Gastroenterol.
12:4232–4236. 2006.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Bhatia M: Novel therapeutic targets for
acute pancreatitis and associated multiple organ dysfunction
syndrome. Curr Drug Targets Inflamm Allergy. 1:343–351.
2002.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Petrov MS, Shanbhag S, Chakraborty M,
Phillips AR and Windsor JA: Organ failure and infection of
pancreatic necrosis as determinants of mortality in patients with
acute pancreatitis. Gastroenterology. 139:813–820. 2010.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Hong YP, Yu J, Su YR, Mei FC, Li M, Zhao
KL, Zhao L, Deng WH, Chen C and Wang WX: High-fat diet aggravates
acute pancreatitis via TLR4-mediated necroptosis and inflammation
in rats. Oxid Med Cell Longev. 2020(8172714)2020.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Zhang X, Zhu C, Wu D and Jiang X: Possible
role of toll-like receptor 4 in acute pancreatitis. Pancreas.
39:819–824. 2010.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Abliz A, Deng W, Sun R, Guo W, Zhao L and
Wang W: Wortmannin, PI3K/Akt signaling pathway inhibitor,
attenuates thyroid injury associated with severe acute pancreatitis
in rats. Int J Clin Exp Pathol. 8:13821–13833. 2015.PubMed/NCBI
|
|
44
|
Xu P, Wang J, Yang ZW, Lou XL and Chen C:
Regulatory roles of the PI3K/Akt signaling pathway in rats with
severe acute pancreatitis. PLoS One. 8(e81767)2013.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Liu Y, Liao R, Qiang Z and Zhang C:
Pro-inflammatory cytokine-driven PI3K/Akt/Sp1 signalling and
H2S production facilitates the pathogenesis of severe
acute pancreatitis. Biosci Rep. 37(BSR20160483)2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Fan J, Li Y, Levy RM, Fan JJ, Hackam DJ,
Vodovotz Y, Yang H, Tracey KJ, Billiar TR and Wilson MA:
Hemorrhagic shock induces NAD(P)H oxidase activation in
neutrophils: Role of HMGB1-TLR4 signaling. J Immunol.
178:6573–6580. 2007.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Kokkola R, Andersson A, Mullins G, Ostberg
T, Treutiger CJ, Arnold B, Nawroth P, Andersson U, Harris RA and
Harris HE: RAGE is the major receptor for the proinflammatory
activity of HMGB1 in rodent macrophages. Scand J Immunol. 61:1–9.
2005.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Tian X, Sun L, Feng D, Sun Q, Dou Y, Liu
C, Zhou F, Li H, Shen H, Wang Z and Chen G: HMGB1 promotes
neurovascular remodeling via Rage in the late phase of subarachnoid
hemorrhage. Brain Res. 1670:135–145. 2017.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Akira S, Takeda K and Kaisho T: Toll-like
receptors: Critical proteins linking innate and acquired immunity.
Nat Immunol. 2:675–680. 2001.PubMed/NCBI View
Article : Google Scholar
|
|
50
|
Xu GF, Guo M, Tian ZQ, Wu GZ, Zou XP and
Zhang WJ: Increased of serum high-mobility group box chromosomal
protein 1 correlated with intestinal mucosal barrier injury in
patients with severe acute pancreatitis. World J Emerg Surg.
9(61)2014.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Kylänpää ML, Repo H and Puolakkainen PA:
Inflammation and immunosuppression in severe acute pancreatitis.
World J Gastroenterol. 16:2867–2872. 2010.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Sun J, Shi S, Wang Q, Yu K and Wang R:
Continuous hemodiafiltration therapy reduces damage of multi-organs
by ameliorating of HMGB1/TLR4/NFκB in a dog sepsis model. Int J
Clin Exp Pathol. 8:1555–1564. 2015.PubMed/NCBI
|
|
53
|
Todorova J and Pasheva E: High mobility
group B1 protein interacts with its receptor RAGE in tumor cells
but not in normal tissues. Oncol Lett. 3:214–218. 2012.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Schiraldi M, Raucci A, Muñoz LM, Livoti E,
Celona B, Venereau E, Apuzzo T, De Marchis F, Pedotti M, Bachi A,
et al: HMGB1 promotes recruitment of inflammatory cells to damaged
tissues by forming a complex with CXCL12 and signaling via CXCR4. J
Exp Med. 209:551–563. 2012.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Diener KR, Al-Dasooqi N, Lousberg EL and
Hayball JD: The multifunctional alarmin HMGB1 with roles in the
pathophysiology of sepsis and cancer. Immunol Cell Biol.
91:443–450. 2013.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Biscetti F, Ghirlanda G and Flex A:
Therapeutic potential of high mobility group box-1 in ischemic
injury and tissue regeneration. Curr Vasc Pharmacol. 9:677–681.
2011.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Kanakoudi-Tsakalidou F, Farmaki E,
Tzimouli V, Taparkou A, Paterakis G, Trachana M, Pratsidou-Gertsi
P, Nalbanti P and Papachristou F: Simultaneous changes in serum
HMGB1 and IFN-α levels and in LAIR-1 expression on plasmatoid
dendritic cells of patients with juvenile SLE. New therapeutic
options? Lupus. 23:305–312. 2014.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Gu H, Werner J, Bergmann F, Whitcomb DC,
Büchler MW and Fortunato F: Necro-inflammatory response of
pancreatic acinar cells in the pathogenesis of acute alcoholic
pancreatitis. Cell Death Dis. 4(e816)2013.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Samanta S, Balasubramanian S, Rajasingh S,
Patel U, Dhanasekaran A, Dawn B and Rajasingh J: MicroRNA: A new
therapeutic strategy for cardiovascular diseases. Trends Cardiovasc
Med. 26:407–419. 2016.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Wang AY, Ehrhardt A, Xu H and Kay MA:
Adenovirus transduction is required for the correction of diabetes
using Pdx-1 or Neurogenin-3 in the liver. Mol Ther. 15:255–263.
2007.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Wang AY, Peng PD, Ehrhardt A, Storm TA and
Kay MA: Comparison of adenoviral and adeno-associated viral vectors
for pancreatic gene delivery in vivo. Hum Gene Ther. 15:405–413.
2004.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Li D, Zhou J, Yang B and Yu Y:
MicroRNA-340-5p inhibits hypoxia/reoxygenation-induced apoptosis
and oxidative stress in cardiomyocytes by regulating the Act1/NF-κB
pathway. J Cell Biochem. 120:14618–14627. 2019.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Zheng Y, Zhao P, Lian Y, Li S, Chen Y and
Li L: MiR-340-5p alleviates oxygen-glucose
deprivation/reoxygenation-induced neuronal injury via PI3K/Akt
activation by targeting PDCD4. Neurochem Int.
134(104650)2020.PubMed/NCBI View Article : Google Scholar
|