Introduction
As a systemic inflammatory disorder, rheumatoid
arthritis (RA) represents one of the most prevalent autoimmune
diseases worldwide (1). The immune
system in RA can significantly damage the joints and other tissues,
ultimately leading to irreversible joint deformity (1-3).
Although advances in understanding the pathogenesis of RA have
promoted the development of novel therapeutics against this
disease, the detailed cause of RA remains unknown and the prognosis
is uncertain (4). A number of
previous studies have demonstrated that the development of RA
involves the activation of a wide range of immune cells and
macrophages (5,6). After their activation, these cells
can promote RA development by overexpressing a series of important
factors, such as major histocompatibility complex class Ⅱ,
proinflammatory cytokines and growth factors (7,8).
Recently, targeted intervention of the inflammatory process by
disease-modifying antirheumatic drugs (DMARDs) has been used as a
promising strategy for the treatment of RA (9-11).
However, the risk of subsequent infections was an unavoidable major
concern associated with the long-term use of DMARDs (12).
Bruton's tyrosine kinase (BTK), as one of the
members of Tec family of non-receptor tyrosine kinases, has been
found to serve a key role in the regulation of B cells and
macrophage kinase (13,14). BTK in macrophages has been
recommended as a promising therapeutic target for RA, as it plays a
significant role in the polarization of proinflammatory macrophages
and the production of proinflammatory cytokines (15-17).
A number of previous studies have confirmed that knockdown of BTK
expression could effectively ameliorate arthritis by significantly
reducing the levels of autoantibodies and cytokines (18,19).
However, unavoidable immune suppression was diagnosed during the
use of BTK inhibitors, mainly due to the off-target effects
(20). Therefore, a novel strategy
to specifically deliver BTK inhibitors is urgently needed to
effectively treat RA.
In addition to the BTK, activated macrophages in the
inflamed joints of patients with RA can also produce a plethora of
cytokines, such as TNF-α, which serves as the main driver of the
vicious cycle of inflammation and tissue damage (21,22).
It has been demonstrated that inhibition of TNF-α could
significantly alleviate the symptoms of RA and delay its
progression (23,24).
However, the detailed role of BTK and TNF-α in the
regulation of RA and the underlying mechanisms have yet to be
extensively investigated. Therefore, in the present study, the
potential effects of BTK and TNF-α in the regulation of RA were
examined in macrophages in inflamed RA joints. Additionally, the
underlying mechanisms through which BTK and TNF-α can regulate RA
were also investigated.
Materials and methods
Materials
The short interfering (si)RNAs for BTK and TNF-α
(siBTK with sequence of 5'-AAUCCAGCGCUUUCUCAGCd TdT-3' and siTNF-α
with sequence of 5'-AAGAGAACCUGG GAGUAGAUAAGGU-3', respectively)
were purchased from Shanghai GenePharma Co., Ltd. The negative
control siRNA (siNC, with sequence of 5'-ACGUGACACGUUCGGAGAAd
TdT-3') was obtained from the Suzhou Ribo Life Science Co., Ltd.
MTT was purchased from MilliporeSigma. DMEM, FBS and
penicillin-streptomycin were all from Gibco; Thermo Fisher
Scientific, Inc.
Cell culture and transfection
The lipopolysaccharide-induced inflammatory mouse
macrophage cell line RAW 264.7 and the normal mouse macrophage cell
line Ana-1 were obtained from the American Type Culture Collection.
The two cells were cultured in DMEM supplemented with 10% fetal
bovine serum, 100 U/ml penicillin and 100 µg/ml streptomycin at
37˚C in a 5% CO2/95% air humidified environment
incubator (Thermo HERA cell; Thermo Fisher Scientific Inc.).
Transfection of RAW 264.7 cells with various genes
were performed using Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.). In brief, the RAW 264.7 cells were
seeded into each well of a six-well plate at the density of
1x106 cells per well. After that, the cells were
respectively incubated with siBTK and siTNF-α at 37˚C in a 5%
CO2/95% air humidified environment incubator according
to the manufacturer's instructions. After 72 h of transfection, the
aforementioned cells were harvested for further experiments.
Development of collagen-induced
arthritis (CIA) mouse model
A total of 50 DBA/1 mice aged 6-8 weeks (20±2 g)
were purchased from Shanghai SIPR-BK Laboratory Animal Co., Ltd.
and raised at 25±1˚C with humidity of 50-60%, 12-h light/dark cycle
and free access to food and water. Thereafter, RA mouse models were
developed based on the previously reported CIA approach (25). In brief, DBA/1 mice were
intradermally injected at the base of the tail with bovine type II
collagen (Chondrex, Inc.) at a dose of 5 mg/kg emulsified by 100 µl
Complete Freund's Adjuvant (Chondrex, Inc.) on day 0. After being
maintained for 21 days, the mice were again treated with emulsified
bovine type II collagen (5 mg/kg). Then, the CIA scores (26) of the mice were carefully recorded
by the following criteria: 0, normal; 1, one hind paw or fore paw
joint affected or minimal diffuse erythema and swelling; 2, two
hind or fore paw joints affected or mild diffuse erythema and
swelling; 3, three hind or fore paw joints affected or moderate
diffuse erythema and swelling; 4, marked diffuse erythema and
swelling or four digit joints affected; 5, severe diffuse erythema
and severe swelling of entire paw, unable to flex digits.
Of note, all the experimental procedures were
performed according to the guidelines approved by the Chongqing
Ninth People's Hospital (approval no. CQSY201911).
Measurement of cell proliferation
The inhibitory effects of siBTK and siTNF-α on the
proliferation of RAW 264.7 cells were determined using an MTT
assay. Briefly, stably transfected RAW 264.7 cells were seeded into
96-well plates at a density of 5x103 cells per well.
After an overnight incubation at 37˚C, the old medium was replaced
with fresh medium. After incubation for 24, 36 or 48 h, 20 µl of
MTT solution was added into each well followed by further
incubation for 4 h. Then, 150 µl of dimethyl sulfoxide was added
and the absorbance of each well was measured at 490 nm via a
microplate reader (Multiskan MK3; Thermo Fisher Scientific,
Inc.).
Measurement of cell colony-forming
ability
The effects of siBTK and siTNF-α on the
proliferation and clonogenic potential of RAW 264.7 cells were
evaluated using a cell colony formation assay. A total of
1x105 transfected RAW 264.7 cells were seeded into each
well of a 24-well plate and incubated overnight. Then, the old
medium in the plates was replaced with fresh medium followed by
another 24 h of incubation. Subsequently, the cells were fixed with
4% paraformaldehyde for 30 min at room temperature and cell colony
(>50 cells) were quantified by evaluation of absorbance at 570
nm in a plate reader (Thermo Multiskan MK3; Thermo Fisher
Scientific, Inc.) after staining with 1% crystal violet solution
for 30 min at room temperature. The qualitative analysis was
evaluated under a phase-contrast microscope (Leica Microsystems
GmbH).
Reverse transcription-quantitative
(RT-q)PCR assay
Total RNA from the cells (RAW 264.7 cells or Ana-1
cells) or inflammation tissues was isolated using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and the miScript Reverse Transcription kit (Qiagen NV) was
used to generate the cDNA according to the manufacturer's
instructions. Subsequently, the detection of BTK and TNF-α was
carried out using the miScript SYBR-Green PCR kit (Qiagen NV)
followed by analysis using the ABI 7500 PCR analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.). Of great importance,
the PCR cycles were: Pretreatment for 10 min at 95˚C, 96˚C for 15
sec, 64˚C for 45 sec (45 cycles), 96˚C for 15 sec, 64˚C for 1 min,
95˚C for 15 sec, a final extension at 75˚C for 10 min and held at
4˚C. The relative gene expression of BTK and TNF-α was calculated
using the 2-∆∆Cq method (27) and normalized with β-actin. The
primer sequence, designed by Invitrogen (Thermo Fisher Scientific,
Inc.), used for BTK was F, 5'-TGTTGAAACAGT GGTTCCTGA-3' and R,
5'-TGCTCCATTTCACTGGAC TCT-3'; TNF-α was F,
5'-CAGCCTCTTCTCCTTCCTGA-3' and R, 5'-GGAAGACCCCTCCCAGATAGA-3' and
β-actin was F, 5'-ATGGGCCAGAAAGATGCCTATGT-3' and R,
5'-ATGCCAGGGAACATAGTTGAGCC-3'.
Western blot analysis
The cells (RAW 264.7 cells or Ana-1 cells) were
collected and lysed using RIPA lysis buffer (Beyotime Institute of
Biotechnology). Subsequently, protein concentration was determined
using the BCA detection method. Subsequently, the protein samples
(BTK 81.3 kDa, TNF-α 17.4 kDa, p-ERK 45 kDa, p-JNK 51 kDa, p-P38 43
kDa, iRhom2 97 kDa, BAFF 17 kDa and β-actin 42 kDa all at 100 µg)
were separated by 10% SDS-PAGE and electrophoretically transferred
to PVDF membranes. The membranes were blocked in 5% skimmed milk
for 1 h at room temperature and were then incubated with primary
antibodies against BTK and TNF-α (cat. nos. ab208937 and ab1793,
Abcam; 1:1,500) were added. After an overnight incubation under
4˚C, horseradish peroxidase-conjugated IgG (cat. no. ab10183,
Abcam; 1:3,000) was added at 37˚C. After 1 h, the signals of BTK
and TNF-α were visualized using the ECL kit (Merck KGaA) with
β-actin serving as the internal control.
Treatment of CIA mouse models in
vivo
The developed CIA mouse models were randomly divided
into five groups (n=10 per group) as follows: i) Control, ii) siNC,
iii) siBTK, iv) siTNF-α and v) siBTK + siTNF-α. The siRNA dose was
2 mg/kg and mice treated with saline served as the control group.
Then, the CIA scores were carefully recorded at the indicated time
points (0, 2, 4, 6, 8, 10, 12 and 14 days). At the end of the
experimental period, all the mice were euthanized by cervical
dislocation.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 5 (GraphPad Software, Inc.). P<0.05 was considered to
indicate a statistically significant difference. All experiments
were performed in triplicates and the data are presented as the
mean ± SD. Unpaired Student's t-test was used for comparisons
between two groups and one-way ANOVA with Bonferroni tests was used
for multiple-group analysis.
Results
High expression of BTK and TNF-α is
detected in macrophages from inflamed RA joints
To investigate the possible expression of BTK and
TNF-α in macrophages from inflamed RA joints, RAW 264.7
inflammatory macrophages were used. Additionally, the levels of BTK
and TNF-α were also evaluated in normal Ana-1 macrophages and
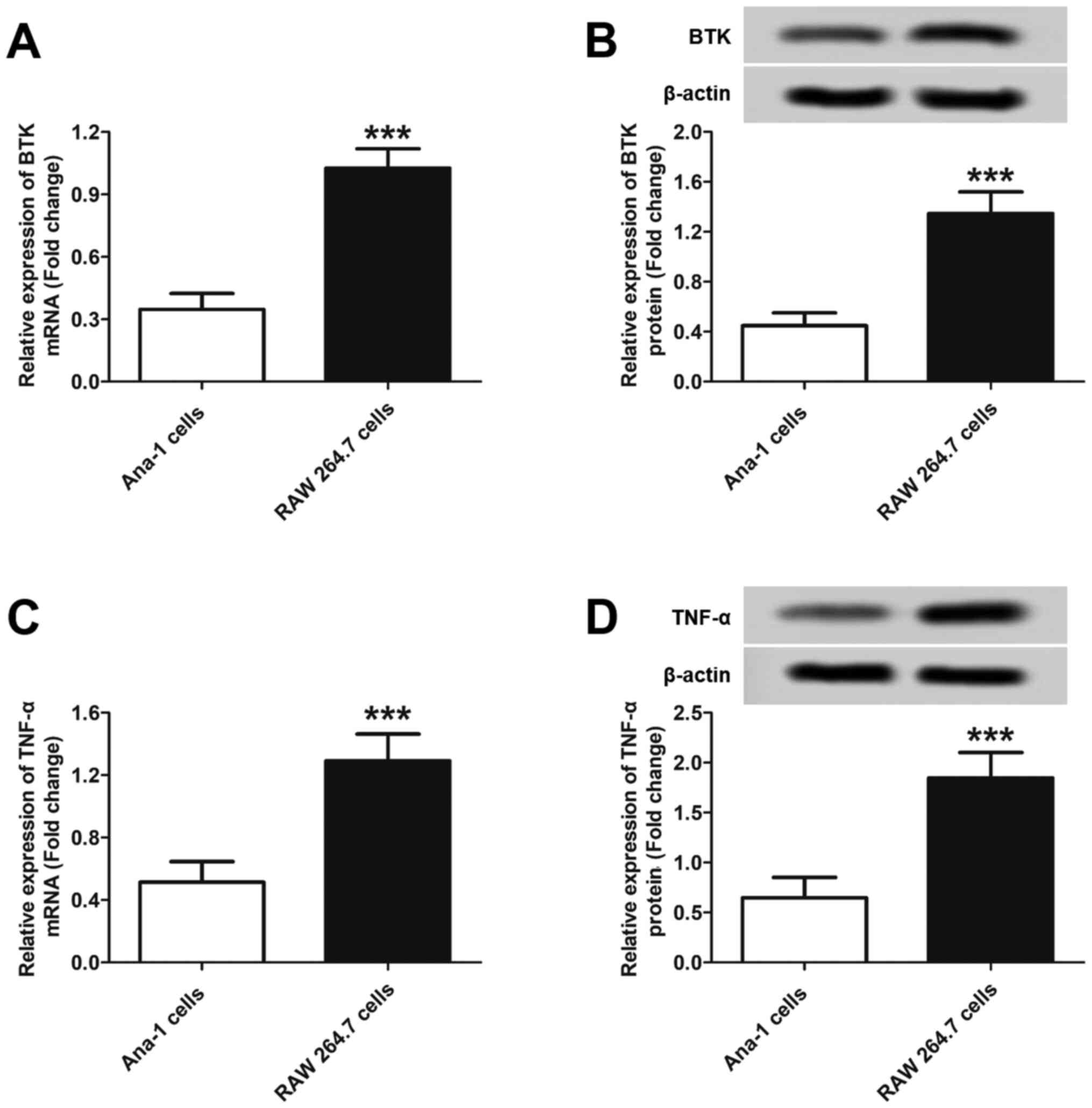
compared with the results of RAW 264.7 cells. As shown in Fig. 1A, significantly high levels of BTK
mRNA were detected in RAW 264.7 cells. However, a relative low
expression of BTK mRNA was detected in Ana-1 cells. These results
were further confirmed by western blot analysis. As shown in
Fig. 1B, the inflammatory
macrophages expressed significantly higher BTK protein levels
compared with normal macrophages. Semi-quantitative analysis
revealed that the BTK protein level in inflammatory macrophages was
3.01-fold that in normal macrophages. As regards TNF-α expression,
similar results were obtained, with higher levels of TNF-α mRNA and
protein detected in RAW 264.7 cells compared with Ana-1 cells
(Fig. 1C and D). Further semi-quantitative analysis
revealed that the TNF-α protein level in RAW 264.7 cells was
2.01-fold that in Ana-1 cells. Collectively, the aforementioned
results demonstrated that higher levels of BTK and TNF-α may be
present in macrophages in inflamed RA joints compared with those in
normal joint tissues.
Aberrant expression of BTK and TNF-α
promotes the proliferation and clonogenic potential of inflammatory
macrophages
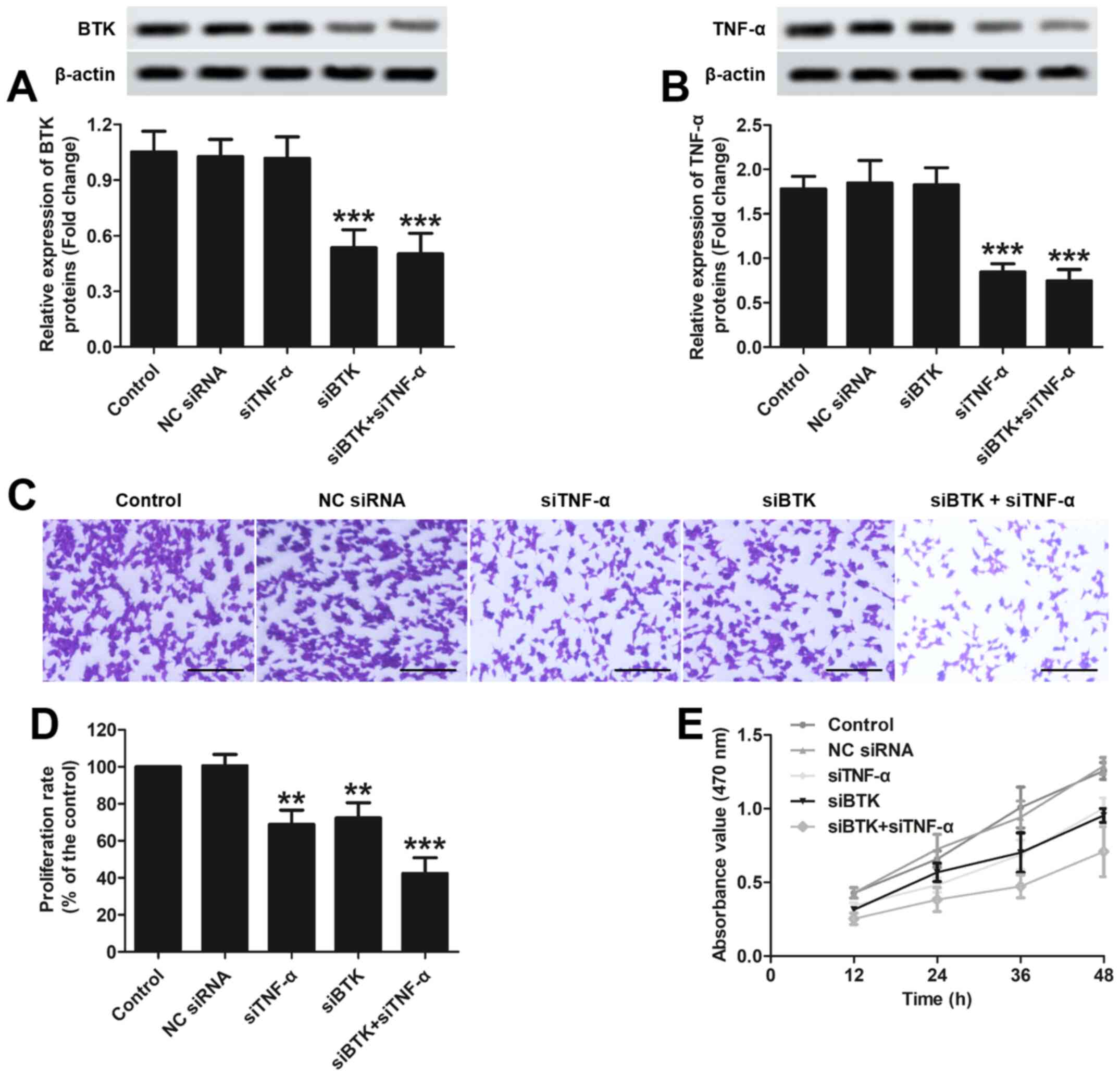
The transfection efficacy in RAW 264.7 cells was
first evaluated via detection of BTK and TNF-α protein levels using
western blotting. As shown in Fig.
2A, cells transfected with siBTK exhibited significantly lower
expression of the BTK protein compared with the control group or
cells transfected with siNC. However, the cells transfected with
siTNF-α exhibited a similar expression level of the BTK protein as
the control group or cells transfected with siNC. As regards TNF-α
expression, it was demonstrated that the cells transfected with
siTNF-α exhibited lower expression of the TNF-α protein compared
with the control group or cells transfected with siNC (Fig. 2B). However, the cells transfected
with siBTK exhibited slightly lower expression of TNF-α compared
with the control group or cells transfected with siNC, thus
indicating that silencing of BTK expression exerted a negative
regulatory effect on the expression of TNF-α.
Subsequently, the inhibitory effects of siBTK and
siTNF-α on the colony-forming ability of RAW 264.7 cells was
determined using colony formation assay. As shown in Fig. 2C, the cells in the of siBTK and
siTNF-α groups displayed a similar extent of crystal violet
staining, which was signally lower compared with that in the
control and siNC groups. Moreover, the cells simultaneously
transfected with siBTK and siTNF-α exhibited the lowest extent of
crystal violet staining. These results were further confirmed by
semi-quantitative analysis (Fig.
2D). Furthermore, the inhibitory effects of siBTK and siTNF-α
on the proliferation of RAW 264.7 cells were also determined using
MTT assay. As demonstrated in Fig.
2E, similar cell proliferation rates were observed in cells
transfected with siBTK and siTNF-α, and they were markedly lower
compared with those in the control and siNC groups. Furthermore,
the proliferation rate of the cells simultaneously transfected with
siBTK and siTNF-α was the lowest among all groups. Taken together,
these results indicated that BTK and TNF-α play a major role in the
proliferation and clonogenic potential of inflammatory macrophages
and may exert a synergistic effects.
BTK primarily regulates the ERK/JNK
pathway whereas the TNF-α modulates the inactive rhomboid protein 2
(iRhom2)/B-cell-activating factor (BAFF) pathway
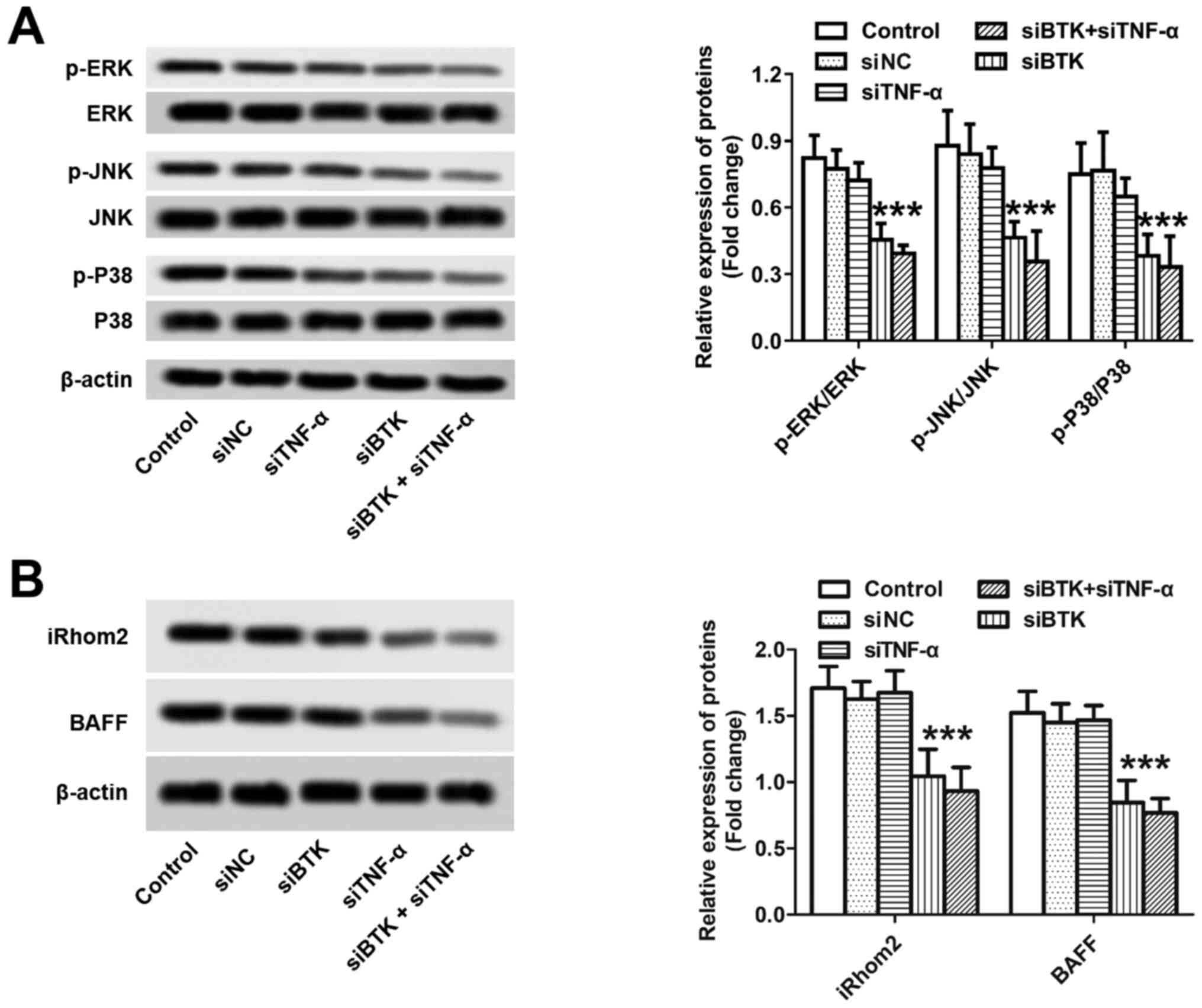
Subsequently, the underlying mechanisms through
which BTK and TNF-α can promote RA were investigated at the
cellular level. As demonstrated in Fig. 3A, silencing of BTK in inflammatory
macrophages substantially downregulated the protein expression of
ERK, p38 and JNK. However, no significant difference was observed
among the control, siNC and siTNF-α groups, thereby suggesting that
TNF-α did not affect the ERK/JNK pathway. The mechanisms underlying
the action of TNF-α were determined as well. As shown in Fig. 3B, the cells transected with
siTNF-α, but not siBTK, displayed the lowest protein expression of
iRhom2 and BAFF among all the groups. By contrast, there was no
significant difference observed among the control, siNC and siBTK
groups, thereby indicating that BTK did not exert a regulatory
effect on the iRhom2/BAFF pathway. Taken together, these results
demonstrated that BTK and TNF-α can induce RA mainly through
activation of the ERK/JNK and the iRhom2/BAFF pathways,
respectively.
Simultaneously silencing BTK and TNF-α
in inflammatory macrophages significantly relieves RA symptoms in
vivo
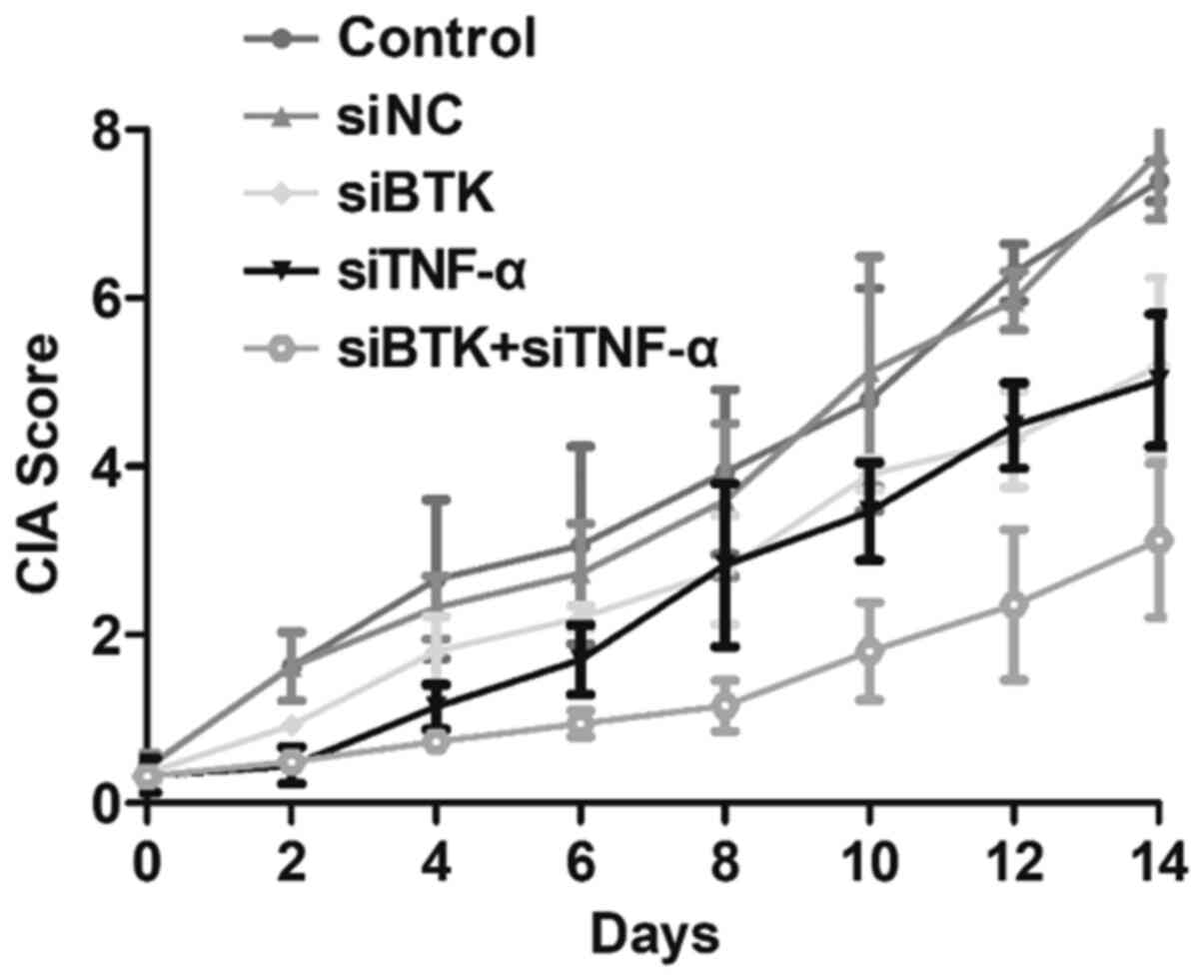
The effects of siBTK and siTNF-α on RA were
evaluated in vivo using the developed CIA mouse models. For
the experiments, the CIA mice were randomly grouped (n=10 per
group) and treated with saline (control group), siNC, siBTK,
siTNF-α or siBTK + siTNF-α. Thereafter, the CIA scores of the
treated RA mice were carefully observed and recorded every 2 days.
As shown in Fig. 4, the CIA scores
in the control group rapidly increased. However, after treatment
with siBTK or siTNF-α, the CIA scores of RA mice were significantly
reduced. More importantly, the mice treated with siBTK + siTNF-α
displayed the lowest CIA scores among all the groups. These results
indicated that simultaneously silencing BTK and TNF-α markedly
relieves the RA symptoms in vivo.
Discussion
A number of previous studies have demonstrated that
macrophages, fibroblast-like synoviocytes and dendritic cells,
among others, may play pivotal roles in the development of RA by
inducing extensive destruction of articular cartilage (28-30).
Among these cells, activated macrophages constitute the most
prominent cell population in the inflamed RA joints, and markedly
affect joint inflammation through regulation of the mRNA expression
of various factors (31). Among
these, BTK may serve as a potential therapeutic target for RA
(19). In addition, certain
cytokines produced by the macrophages, such as TNF-α, play a
critical role in driving inflammation during RA (22). In the present study, it was
demonstrated that BTK was highly expressed at the mRNA and protein
level in the inflamed RA joints compared with normal joint tissues.
Similar results were obtained for the expression of TNF-α. These
results confirmed the role of BTK and TNF-α in the development of
RA.
The MAPK pathway, comprising the ERK, JNK and p38
proteins, is one of the most important signal transduction cascades
implicated in the activation of macrophages in RA (32,33).
It has been demonstrated that persistent activation of the ERK/JNK
pathway may be involved in the development of autoimmune and
inflammatory diseases (34).
Therefore, the ERK/JNK pathway may serve as an important molecular
target for the control of inflammatory diseases. In the present
study, it was demonstrated that the activity of the ERK/JNK pathway
in the inflammatory macrophages was significantly inhibited by
silencing the expression of BTK. Additionally, the downregulation
of ERK/JNK pathway-related proteins finally resulted in decreased
proliferation of inflammatory macrophages and contributed to the
significant improvement of RA symptoms in vivo.
A number of previous studies have demonstrated that
TNF-α, iRhom2 and BAFF can play a significant role in the
development of RA, and a substantial decrease in TNF-α, iRhom2 and
BAFF levels may notably improve the symptoms of RA (35,36).
Of note, the expression of BAFF has been reported to be regulated
by TNF-α, while the production of TNF-α could be effectively
controlled by the activity of iRhom2 in synovial macrophages
(36-38).
Based on these results, it was suggested that a positive feedback
process exists in the iRhom2/TNF-α/BAFF pathway, and reducing the
expression of these genes may be a promising strategy for the
treatment of RA. The present study demonstrated that cells
transfected with siTNF-α, but not siBTK, exhibited the lowest
expression of iRhom2 and BAFF proteins among all groups. By
contrast, there were no significant differences observed among
cells in the control, NC siRNA and siBTK groups, thereby indicating
that BTK did not exert regulatory effects on the iRhom2/BAFF
pathway.
In conclusion, the present study demonstrated that
BTK and TNF-α are expressed at higher levels in inflamed RA joints
compared with normal joint tissues, whereas the aberrant expression
of BTK and TNF-α contributed to high proliferation rate and
clonogenic potential of inflammatory macrophages. Importantly, the
activation of inflammatory macrophages was significantly inhibited
by BTK and/or TNF-α silencing. Moreover, the most prominent
inhibitory effects on inflammatory macrophages were observed after
simultaneous silencing of the expression of both BTK and TNF-α.
Subsequent mechanistic studies demonstrated that BTK primarily
regulated the ERK/JNK pathway, whereas TNF-α modulated the
iRhom2/BAFF pathway. In summary, the present study demonstrated
that simultaneously silencing both BTK and TNF-α can significantly
alleviate the symptoms associated with RA and provide a promising
strategy for treatment of RA in the clinical setting. However, due
to the limited human and material resources, the complicated
relationship between the BTK and TNF-α was not thoroughly
investigated in the present study and needs to be studied in
future.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JD was the guarantor of integrity of the entire
study. JD prepared, edited and reviewed the manuscript. He was also
involved in the definition of intellectual content, literature
research and responsible for the study design, data acquisition and
analysis.
Ethics approval and consent to
participate
The present study was approved by the Research
Ethics Committee of Chongqing Ninth People's Hospital (approval
no.CQSY201911).
Patient consent for publication
Not applicable.
Competing interests
The author declares that they have no competing
interests.
References
|
1
|
De Cock D and Hyrich K: Malignancy and
rheumatoid arthritis: Epidemiology, risk factors and management.
Best Pract Res Clin Rheumatol. 32:869–886. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Wasserman AM: Diagnosis and management of
rheumatoid arthritis. Am Fam Physician. 84:1245–1252.
2011.PubMed/NCBI
|
|
3
|
Wasserman A: Rheumatoid arthritis: Common
questions about diagnosis and management. Am Fam Physician.
97:455–462. 2018.PubMed/NCBI
|
|
4
|
McInnes IB and Schett G: The pathogenesis
of rheumatoid arthritis. N Engl J Med. 365:2205–2219.
2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Croia C, Bursi R, Sutera D, Petrelli F,
Alunno A and Puxeddu I: One year in review 2019: Pathogenesis of
rheumatoid arthritis. Clin Exp Rheumatol. 37:347–357.
2019.PubMed/NCBI
|
|
6
|
Udalova IA, Mantovani A and Feldmann M:
Macrophage heterogeneity in the context of rheumatoid arthritis.
Nat Rev Rheumatol. 12:472–485. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Rana AK, Li Y, Dang Q and Yang F:
Monocytes in rheumatoid arthritis: Circulating precursors of
macrophages and osteoclasts and, their heterogeneity and plasticity
role in RA pathogenesis. Int Immunopharmacol. 65:348–359.
2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Huang QQ, Birkett R, Doyle R, Shi B,
Roberts EL, Mao Q and Pope RM: The role of macrophages in the
response to TNF inhibition in experimental arthritis. J Immunol.
200:130–138. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
de La Forest Divonne M, Gottenberg JE and
Salliot C: Safety of biologic DMARDs in RA patients in real life: A
systematic literature review and meta-analyses of biologic
registers. Joint Bone Spine. 84:133–140. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Massalska M, Maslinski W and Ciechomska M:
Small molecule inhibitors in the treatment of rheumatoid arthritis
and beyond: Latest updates and potential strategy for fighting
COVID-19. Cells. 9(1876)2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ternant D, Bejan-Angoulvant T, Passot C,
Mulleman D and Paintaud G: Clinical pharmacokinetics and
pharmacodynamics of monoclonal antibodies approved to treat
rheumatoid arthritis. Clin Pharmacokinet. 54:1107–1123.
2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Accortt NA, Bonafede MM, Collier DH, Iles
J and Curtis JR: Risk of subsequent infection among patients
receiving tumor necrosis factor inhibitors and other
disease-modifying antirheumatic drugs. Arthritis Rheumatol.
68:67–76. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Lv J, Wu J, He F, Qu Y, Zhang Q and Yu C:
Development of Bruton's tyrosine kinase inhibitors for rheumatoid
arthritis. Curr Med Chem. 25:5847–5859. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Hendriks RW, Yuvaraj S and Kil LP:
Targeting Bruton's tyrosine kinase in B cell malignancies. Nat Rev
Cancer. 14:219–232. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
15
|
Norman P: Investigational Bruton's
tyrosine kinase inhibitors for the treatment of rheumatoid
arthritis. Expert Opin Investig Drugs. 25:891–899. 2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Hsu J, Gu Y, Tan SL, Narula S, DeMartino
JA and Liao C: Bruton's Tyrosine Kinase mediates platelet
receptor-induced generation of microparticles: A potential
mechanism for amplification of inflammatory responses in rheumatoid
arthritis synovial joints. Immunol Lett. 150:97–104.
2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Whang JA and Chang BY: Bruton's tyrosine
kinase inhibitors for the treatment of rheumatoid arthritis. Drug
Discov Today. 19:1200–1204. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Di Paolo JA, Huang T, Balazs M, Barbosa J,
Barck KH, Bravo BJ, Carano RA, Darrow J, Davies DR, DeForge LE, et
al: Specific Btk inhibition suppresses B cell- and myeloid
cell-mediated arthritis. Nat Chem Biol. 7:41–50. 2011.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Wu J, Zhu Z, Yu Q and Ding C: Tyrosine
kinase inhibitors for the treatment of rheumatoid arthritis: Phase
I to Ⅱ clinical trials. Expert Opin Investig Drugs. 28:1113–1123.
2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Erickson RI, Schutt LK, Tarrant JM,
McDowell M, Liu L, Johnson AR, Lewin-Koh SC, Hedehus M, Ross J,
Carano RA, et al: Bruton's tyrosine kinase small molecule
inhibitors induce a distinct pancreatic toxicity in rats. J
Pharmacol Exp Ther. 360:226–238. 2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Gowhari Shabgah A, Shariati-Sarabi Z,
Tavakkol-Afshari J, Ghasemi A, Ghoryani M and Mohammadi M: A
significant decrease of BAFF, APRIL, and BAFF receptors following
mesenchymal stem cell transplantation in patients with refractory
rheumatoid arthritis. Gene. 732(144336)2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Li J, Hsu HC and Mountz JD: Managing
macrophages in rheumatoid arthritis by reform or removal. Curr
Rheumatol Rep. 14:445–454. 2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Moura RA, Quaresma C, Vieira AR, Gonçalves
MJ, Polido-Pereira J, Romão VC, Martins N, Canhão H and Fonseca JE:
B-cell phenotype and IgD-CD27- memory B cells are affected by
TNF-inhibitors and tocilizumab treatment in rheumatoid arthritis.
PLoS One. 12(e0182927)2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Schiff M, Combe B, Dörner T, Kremer JM,
Huizinga TW, Veenhuizen M, Gill A, Komocsar W, Berclaz PY, Ortmann
R, et al: Efficacy and safety of tabalumab, an anti-BAFF monoclonal
antibody, in patients with moderate-to-severe rheumatoid arthritis
and inadequate response to TNF inhibitors: Results of a randomised,
double-blind, placebo-controlled, phase 3 study. RMD Open.
1(e000037)2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhao G, Liu A, Zhang Y, Zuo ZQ, Cao ZT,
Zhang HB, Xu CF and Wang J: Nanoparticle-delivered siRNA targeting
Bruton's tyrosine kinase for rheumatoid arthritis therapy. Biomater
Sci. 7:4698–4707. 2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Chang BY, Huang MM, Francesco M, Chen J,
Sokolove J, Magadala P, Robinson WH and Buggy JJ: The Bruton
tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune
arthritis by inhibition of multiple effector cells. Arthritis Res
Ther. 13(R115)2011.PubMed/NCBI View
Article : Google Scholar
|
|
27
|
Cao Y, Lu G, Chen X, Chen X, Guo N and Li
W: BAFF is involved in the pathogenesis of IgA nephropathy by
activating the TRAF6/NF-κB signaling pathway in glomerular
mesangial cells. Mol Med Rep. 21:795–805. 2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
O'Neil LJ and Kaplan MJ: Neutrophils in
rheumatoid arthritis: breaking immune tolerance and fueling
disease. Trends Mol Med. 25:215–227. 2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Calabresi E, Petrelli F, Bonifacio AF,
Puxeddu I and Alunno A: One year in review 2018: Pathogenesis of
rheumatoid arthritis. Clin Exp Rheumatol. 36:175–184.
2018.PubMed/NCBI
|
|
30
|
Boissier MC, Semerano L, Challal S,
Saidenberg-Kermanac'h N and Falgarone G: Rheumatoid arthritis: From
autoimmunity to synovitis and joint destruction. J Autoimmun.
39:222–228. 2012.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Kurowska-Stolarska M and Alivernini S:
Synovial tissue macrophages: Friend or foe? RMD Open.
3(e000527)2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Thalhamer T, McGrath MA and Harnett MM:
MAPKs and their relevance to arthritis and inflammation.
Rheumatology (Oxford). 47:409–414. 2008.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Schett G, Tohidast-Akrad M, Smolen JS,
Schmid BJ, Steiner CW, Bitzan P, Zenz P, Redlich K, Xu Q and
Steiner G: Activation, differential localization, and regulation of
the stress-activated protein kinases, extracellular
signal-regulated kinase, c-JUN N-terminal kinase, and p38
mitogen-activated protein kinase, in synovial tissue and cells in
rheumatoid arthritis. Arthritis Rheum. 43:2501–2512.
2000.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Salojin KV, Owusu IB, Millerchip KA,
Potter M, Platt KA and Oravecz T: Essential role of MAPK
phosphatase-1 in the negative control of innate immune responses. J
Immunol. 176:1899–1907. 2006.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Shabgah AG, Shariati-Sarabi Z,
Tavakkol-Afshari J and Mohammadi M: The role of BAFF and APRIL in
rheumatoid arthritis. J Cell Physiol. 234:17050–17063.
2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wei F, Chang Y and Wei W: The role of BAFF
in the progression of rheumatoid arthritis. Cytokine. 76:537–544.
2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Lichtenthaler SF: iRHOM2 takes control of
rheumatoid arthritis. J Clin Invest. 123:560–562. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
38
|
Siggs OM, Xiao N, Wang Y, Shi H, Tomisato
W, Li X, Xia Y and Beutler B: iRhom2 is required for the secretion
of mouse TNFα. Blood. 119:5769–5771. 2012.PubMed/NCBI View Article : Google Scholar
|