Introduction
Following the advancement of shock treatment and the
establishment and promotion of arterial bypass, thrombolytic
therapy, extracorporeal circulation and organ transplantation in
recent years, a variety of organs can receive reperfusion after
ischemia (1-4).
In the majority of cases, ischemia/reperfusion (I/R) can restore
the function of organs and tissues and repair ischemic damage. As
blood flow is often restored following ischemia, ischemic damage
may be reduced; however, the dysfunction or structural damage
caused may be aggravated, which is known as I/R injury (5). Thus, I/R injury is a common injury in
surgical practice that affects a number of tissues and organs. It
plays a notable role in the pathophysiological evolution of serious
infections, trauma, shock, cardiopulmonary insufficiency, organ
transplantation and stroke (6).
Moreover, the intestine is an organ that is both fragile and
sensitive; thus, it remains vulnerable to the development of severe
I/R injury (7). The mechanisms
underlying intestinal I/R injury are relatively complex, involving
apoptosis, inflammation, oxidative stress, calcium overload and
leukocyte adhesion (8). The
current treatment of intestinal I/R injury depends on ischemic
preconditioning; that is, exposing the tissue to a state of
transient ischemia in advance (9).
However, due to the unpredictability of intestinal I/R, ischemic
preconditioning has encountered notable limitations in clinical
practice (10).
The intestinal mucosal barrier is a key barrier for
the body's defense against intestinal I/R injury. Under stress, the
intestinal mucosal barrier is damaged and the permeability
increases, which further aggravates the primary disease and causes
multiple organ failure (11).
Therefore, studying the regulation mechanism underlying the
intestinal epithelial barrier under stress conditions will aid in
the development of early prevention methods, and reduce the
mortality of critically ill patients (12).
TGF-β activated kinase 1 (TAK1), an important
intermediate in a variety of innate immune signaling pathways (such
as Toll-like receptor signaling), has been revealed to play a
notable role in maintaining intestinal homeostasis (13). Results of a previous study revealed
that TAK1 mediates lipopolysaccharide-induced NF-κB signal
activation, and increases the permeability of intestinal tight
junctions (14). In addition,
results of a further study demonstrated that intestinal I/R injury
promotes the release of IL-17A (also known as IL-17) (15), and TAK1 has been identified as a
key signaling molecule in the regulatory function of IL-17
(16,17). Moreover, results of a previous
study demonstrated that TAK1 activates the expression of downstream
MAPK signaling proteins in acute kidney injury (18).
To the best of our knowledge, the role of TAK1 in
intestinal I/R injury is yet to be fully elucidated. Caco-2 cells
are a type of human clonal adenocarcinoma cells, whose structure
and function are similar to differentiated small intestinal
epithelial cells, and are recognized for use in in vitro
research on the intestine (19).
Therefore, using Caco-2 cells, the present study aimed to explore
the effects of IL-17 release in intestinal I/R injury, and
investigate the underlying regulatory mechanisms. The findings of
the present study provide a theoretical basis for further study of
IL-17, and contribute to further understanding the mechanisms
underlying intestinal I/R injury.
Materials and methods
Cell culture and grouping
Caco-2 cells were obtained from Procell Life Science
& Technology Co., Ltd. Caco-2 cells were cultured in DMEM
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 20% FBS
(Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin, and maintained in an incubator containing
5% CO2 at 37˚C. Cells in the normoxic incubator were
regarded as the control group.
Caco-2 cells (3x105/well) were seeded
into six-well plates 1 day before transfection until ~80%
confluence was reached. Cells were subsequently transfected with 1
µg pcDNA3.1-TAK1 vector (ov-TAK1), empty vector (ov-NC) (each,
Hanbio Biotechnology Co., Ltd.), short hairpin (sh)RNAs against
TAK1 (shRNA-TAK1; 5'-CCCGTGTGAACCATCCTAATA-3') or a nonspecific
sequence as negative control (shRNA-NC;
5'-CAACAAGATGAAGAGCACCAA-3') (each, Shanghai GenePharma Co., Ltd.)
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) at 37˚C, according to the manufacturer's
protocol. Following transfection for 24 h, the cells were used for
subsequent experiments. These cells were regarded as the
shRNA-NC/TAK1 or ov-NC/TAK1 groups.
Anisomycin, a type of JNK agonist, was purchased
from GlpBio Technology and diluted to 4 µM. IL-17A neutralizing
antibody (cat. no. AF-317) and IgG (cat. no. AB-108-C) were
purchased from R&D Systems, Inc. and diluted to 2 µg/ml. Caco-2
cells treated with anisomycin or the aforementioned antibodies for
1 h at 37˚C were used as the anisomycin/IgG/IL-17 antibody
group.
To establish the hypoxia/reoxygenation (H/R) model,
which mimicked the I/R model in vitro (20), the aforementioned groups of Caco-2
cells were transferred into an incubator containing 94%
N2, 5% CO2 and 1% O2 at
37°C to simulate hypoxia for 12 h, and reoxygenated
under normoxic conditions at 37˚C for a further 6 h. Cells that had
undergone hypoxia and reoxygenation were labelled as the H/R (+)
group.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted from Caco-2 cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), and subsequently reverse transcribed into cDNA using a
universal reverse transcription kit (Beijing Baiao Laibo Technology
Co., Ltd.) according to the manufacturer's instructions. The mRNA
expression levels were measured using QuantiTect SYBR®
Green PCR kit (Qiagen, Inc.) on a StepOnePlus™ Real-time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The
thermocycling conditions were as follows: Initial denaturation at
95˚C for 3 min; followed by 40 cycles of denaturation at 95˚C for
30 sec, annealing at 60˚C for 30 sec and extension at 72˚C for 30
sec. GAPDH was used as the internal reference and the relative mRNA
expression levels were calculated using the 2-ΔΔCq
method (21). The sequences of
primer are listed in Table I.
 | Table IPrimer sequences used for reverse
transcription-quantitative PCR analysis. |
Table I
Primer sequences used for reverse
transcription-quantitative PCR analysis.
| Gene | Sequence
(5'-3') |
|---|
| TAK1 forward |
ATGCGGTACTTTCCAGGAGC |
| TAK1 reverse |
CTGTCCGTTGCCTGTGGTT |
| TNF-α forward |
CACCACTTCGAAACCTGGGA |
| TNF-α reverse |
AGGAAGGCCTAAGGTCCACT |
| IL-6 forward |
CTTCGGTCCAGTTGCCTTCTC |
| IL-6 reverse |
GGCATTTGTGGTTGGGTCAG |
| IL-1β forward |
CTGAGCTCGCCAGTGAAATG |
| IL-1β reverse |
TGTCCATGGCCACAACAACT |
| IL-17A forward |
AACCGATCCACCTCACCTTG |
| IL-17A reverse |
TCTCTTGCTGGATGGGGACA |
| GAPDH forward |
GACTCATGACCACAGTCCATGC |
| GAPDH reverse |
AGAGGCAGGGATGATGTTCTG |
Western blotting
Total proteins were extracted from Caco-2 cells
using RIPA lysis buffer (Beyotime Institute of Biotechnology). The
protein samples were determined by the BCA method and separated (25
µg/lane) via SDS-PAGE on a 10 or 12% gel, and subsequently
transferred onto PVDF membranes. Following blocking with 5% skimmed
milk for 1.5 h at room temperature, the membranes were incubated
with primary antibodies against TAK1 (1:1,000; cat. no. ab109526),
JNK (1:1,000; cat. no. ab76125), phosphorylated (p)-JNK (1:1,000;
cat. no. ab124956), inducible nitric oxide synthase (iNOS; 1:1,000;
cat. no. ab178945), cytochrome C oxidase subunit 2 (Cox2; 1:1,000;
cat. no. ab179800), cleaved caspase 3 (1:500; cat. no. ab2302),
cleaved caspase 9 (1:200; cat. no. ab2324), occludin (1:1,000; cat.
no. ab216327), claudin 1 (1:2,000; cat. no. ab211737), ZO-1 tight
junction protein (ZO-1; 1:1,000; cat. no. ab216880), IL-17A (1:500;
cat. no. ab79056) or GAPDH (1:1,000; cat. no. ab8245) (all Abcam)
at 4˚C overnight. Following primary incubation, the membranes were
washed thrice with TBS-0.01% Tween-20 for 10 min each and then
incubated with a goat anti-rabbit HRP-conjugated secondary antibody
(1:5,000; cat. no. ab97080; Abcam) for 2 h at room temperature.
Protein bands were visualized using an ECL kit (cat. no. P0018AM;
Beyotime Institute of Biotechnology), and the gray values were
measured using ImageJ software (version 1.8; National Institutes of
Health).
Cell Counting Kit-8 (CCK-8) assay
Caco-2 cells (5x103 cells/well) were
cultured in 96-well plates in the normoxic incubator for 24 h, and
10 µl CCK-8 reagent (Beyotime Institute of Biotechnology) was added
to each well. Cells were subsequently incubated at 37˚C for a
further 2 h. The absorbance of each well was measured using a
microplate reader (Molecular Devices, LLC) at a wavelength of 450
nm.
TUNEL assay
Caco-2 cells (2x104 cells/well) were
seeded into a 24-well plate and the TUNEL assay was performed using
a TUNEL kit (cat. no. C1086; Beyotime Institute of Biotechnology),
according to the manufacturer's instructions. Briefly, cells were
fixed with 4% paraformaldehyde for 30 min at room temperature, and
subsequently incubated with PBS containing 0.3% Triton X-100 for 5
min at room temperature. Following the addition of TUNEL working
fluid, cells were incubated for a further 1 h at 37˚C in the dark.
The nuclei was counterstained with DAPI for 10 min at room
temperature. The results were observed at five random fields of
view using a fluorescence microscope (magnification, x200; Olympus
Corporation).
Transepithelial electrical resistance
(TEER) assay
The TEER assay was used to determine the levels of
barrier function. Caco-2 cells (3x104) were seeded into
the upper chamber of 24-well Transwell system plates (pore size,
0.4 µm; Corning, Inc.) and incubated with DMEM for 20 days at 37˚C.
TEER was measured daily using an epithelial volt ohmmeter. The
calculation used was as follows: TEER=(R1-R2)
x M. R1 represents the background resistance,
R2 represents the collagen layer and membrane insert
resistance and M represents the insert membrane area.
Statistical analysis
All experiments were performed at least three times.
Data are presented as the mean ± standard deviation, and
statistical analysis was performed using GraphPad Prism version 8.0
(GraphPad Software, Inc.). Comparisons between multiple groups was
performed using one-way ANOVA followed by a Tukey's post hoc test,
and unpaired Student's t-tests were used for comparisons between
two groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
TAK1 inhibits JNK phosphorylation, and
TAK1 knockdown improves cell viability and alleviates the
inflammation of H/R-induced Caco-2 cells
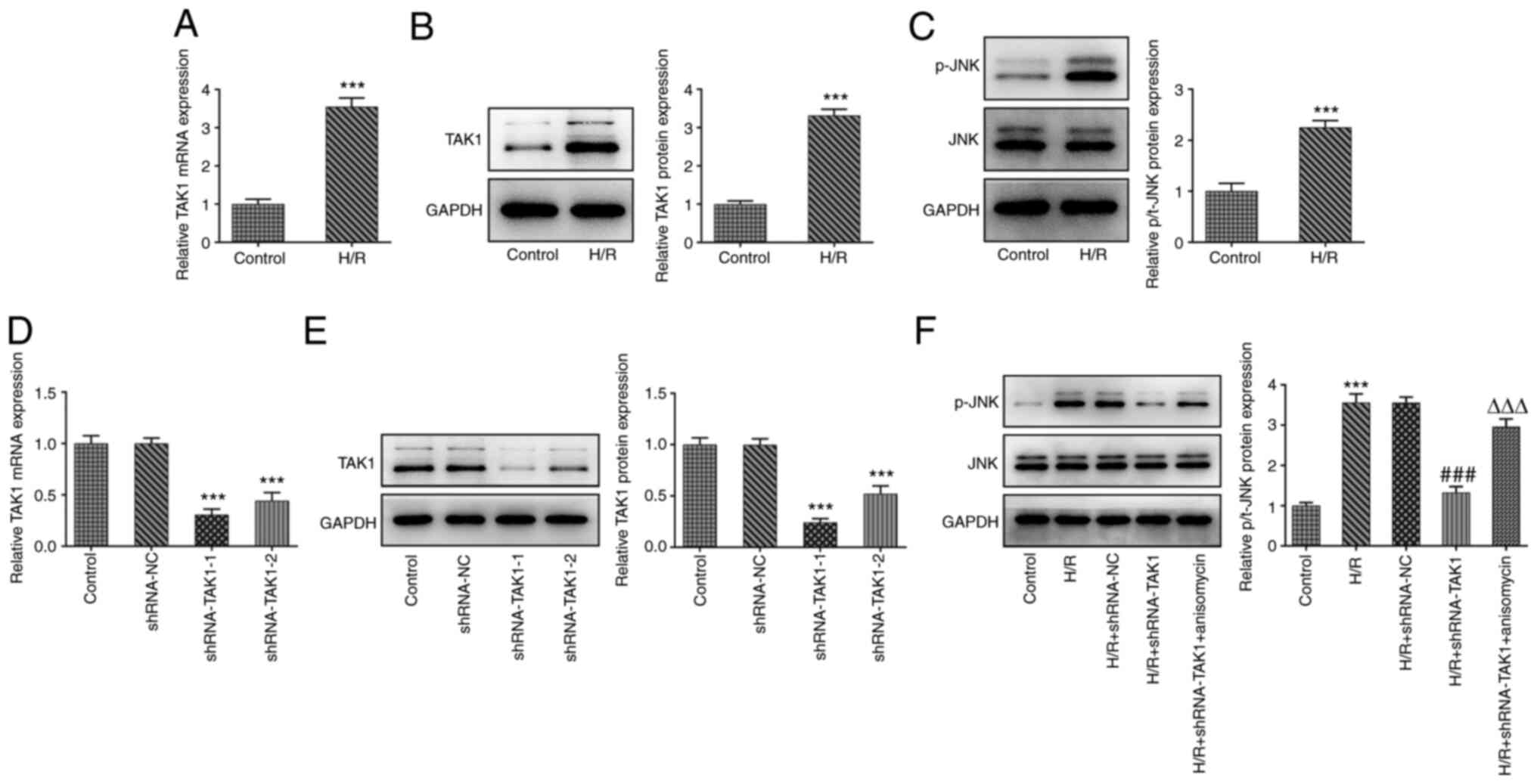
The expression levels of TAK1 in the control and H/R
groups were determined using RT-qPCR and western blotting. The
results indicated that TAK1 expression was significantly
upregulated in the H/R group compared with the control group
(Fig. 1A and B). Moreover, the expression levels of
p-JNK and JNK in the control and H/R groups were also assessed
using western blotting. The expression level of p-JNK was
significantly upregulated in the H/R group compared with the
control group, whereas no marked difference was observed for the
expression of JNK, suggesting that H/R may promote JNK
phosphorylation (Fig. 1C).
Subsequently, two shRNAs targeting TAK1 were transfected into
Caco-2 cells, and the expression levels of TAK1 were subsequently
assessed using RT-qPCR and western blotting. The expression levels
of TAK1 were reduced to a greater extent following transfection
with shRNA-TAK1-1 compared with shRNA-TAK1-2; thus, cells
transfected with shRNA-TAK1-1 were used in subsequent experiments
(Fig. 1D and E).
In order to study the regulatory effects of TAK1 on
the MAPK signaling protein JNK, a JNK agonist was used to treat the
cells, and Caco-2 cells were divided into five groups: i) Control;
ii) H/R; iii) H/R + shRNA-NC; iv) H/R + shRNA-TAK1; and v) H/R +
shRNA-TAK1 + anisomycin. Firstly, the effects of TAK1 on JNK
expression were evaluated using western blotting, and the results
revealed that TAK1 knockdown induced the significant upregulation
of p-JNK expression following H/R. Moreover, anisomycin treatment
significantly reversed the effects of TAK1 knockdown on p-JNK
expression (Fig. 1F).
Cell viability was subsequently measured in the
aforementioned groups using a CCK-8 assay. The results demonstrated
that TAK1 knockdown significantly increased cell viability
following H/R compared with the H/R + shRNA-NC group, while
anisomycin reduced the level of cell viability after knockdown of
TAK1 (Fig. 2A).
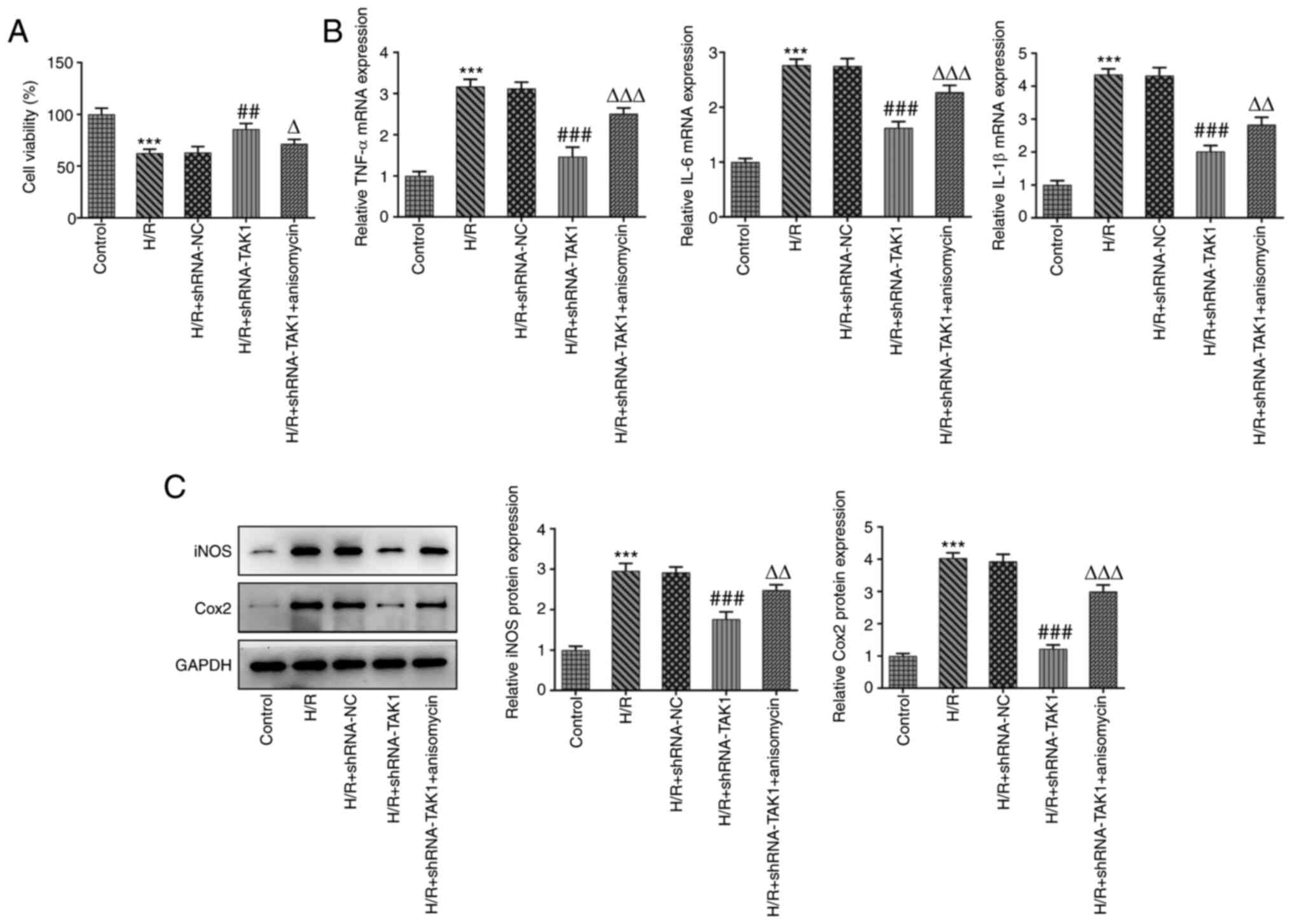 | Figure 2TAK1 knockdown improves cell viability
and alleviates the inflammation of H/R-induced Caco-2 cells. (A)
Cell viability in the five groups were measured using a Cell
Counting Kit-8 assay. (B) Expression levels of TNF-α, IL-6 and
IL-1β in the five groups were determined using reverse
transcription-quantitative PCR. (C) Expression levels of iNOS and
Cox2 were determined western blotting. ***P<0.001 vs.
control; ##P<0.01, ###P<0.001 vs. H/R +
shRNA-NC; ΔP<0.05, ΔΔP<0.01,
ΔΔΔP<0.001 vs. H/R + shRNA-TAK1. TAK1, TGF-β
activated kinase 1; H/R, hypoxia/reoxygenation; shRNA, short
hairpin RNA; NC, negative control; iNOS, inducible nitric oxide
synthase; Cox2, cytochrome C oxidase subunit 2. |
Furthermore, the expression levels of inflammatory
factors were measured in the aforementioned groups using RT-qPCR
and western blotting. Results of the present study demonstrated
that TAK1 knockdown significantly reduced the expression levels of
TNF-α, IL-6 and IL-1β compared with the H/R + shRNA-NC group, while
treatment with anisomycin significantly reversed this reduction in
expression levels (Fig. 2B).
Results of western blotting demonstrated that TAK1 knockdown
significantly reduced the expression levels of iNOS and Cox2 to
varying degrees compared with the H/R + shRNA-NC group, while
treatment with anisomycin returned the expression levels to a level
similar to those observed under H/R conditions (Fig. 2C). These results suggested that
TAK1 knockdown may improve cell viability and alleviate
inflammation in Caco-2 cells subjected to H/R via inhibiting MAPK
signal activation.
TAK1 knockdown alleviates the
apoptosis and barrier dysfunction of H/R-induced Caco-2 cells by
inhibiting MAPK signal activation
The effects of TAK1 knockdown on apoptosis and
barrier dysfunction were evaluated. Apoptosis was assessed using
TUNEL assays and western blotting. Results of the present study
demonstrated that TAK1 knockdown led to a significant reduction in
the intensity of green fluorescence emitted by apoptotic cells
compared with the H/R + shRNA-NC group, indicating that TAK1
knockdown inhibited apoptosis. Moreover, treatment with anisomycin
significantly increased the levels of apoptosis in the TAK1
knockdown cells (Fig. 3A). Results
of the western blotting also revealed that the expression levels of
cleaved caspase 9 and cleaved caspase 3 were significantly reduced
following TAK1 knockdown compared with the H/R + shRNA-NC groups,
whereas treatment with anisomycin increased the expression levels
of both cleaved caspase 9 and cleaved caspase 3 (Fig. 3B). Moreover, cell barrier function
was assessed using TEER and western blotting. Results of the TEER
assay indicated that TAK1 knockdown significantly reversed the
H/R-mediated reduction in TEER value, while treatment of TAK1
knockdown cells with anisomycin reduced the TEER value (Fig. 3C). Furthermore, the expression
levels of occludin, claudin-1 and ZO-1 were notably increased by
TAK1 knockdown compared with those of the H/R + shRNA-NC group, and
anisomycin treatment significantly reduced the expression levels of
the aforementioned proteins (Fig.
3D).
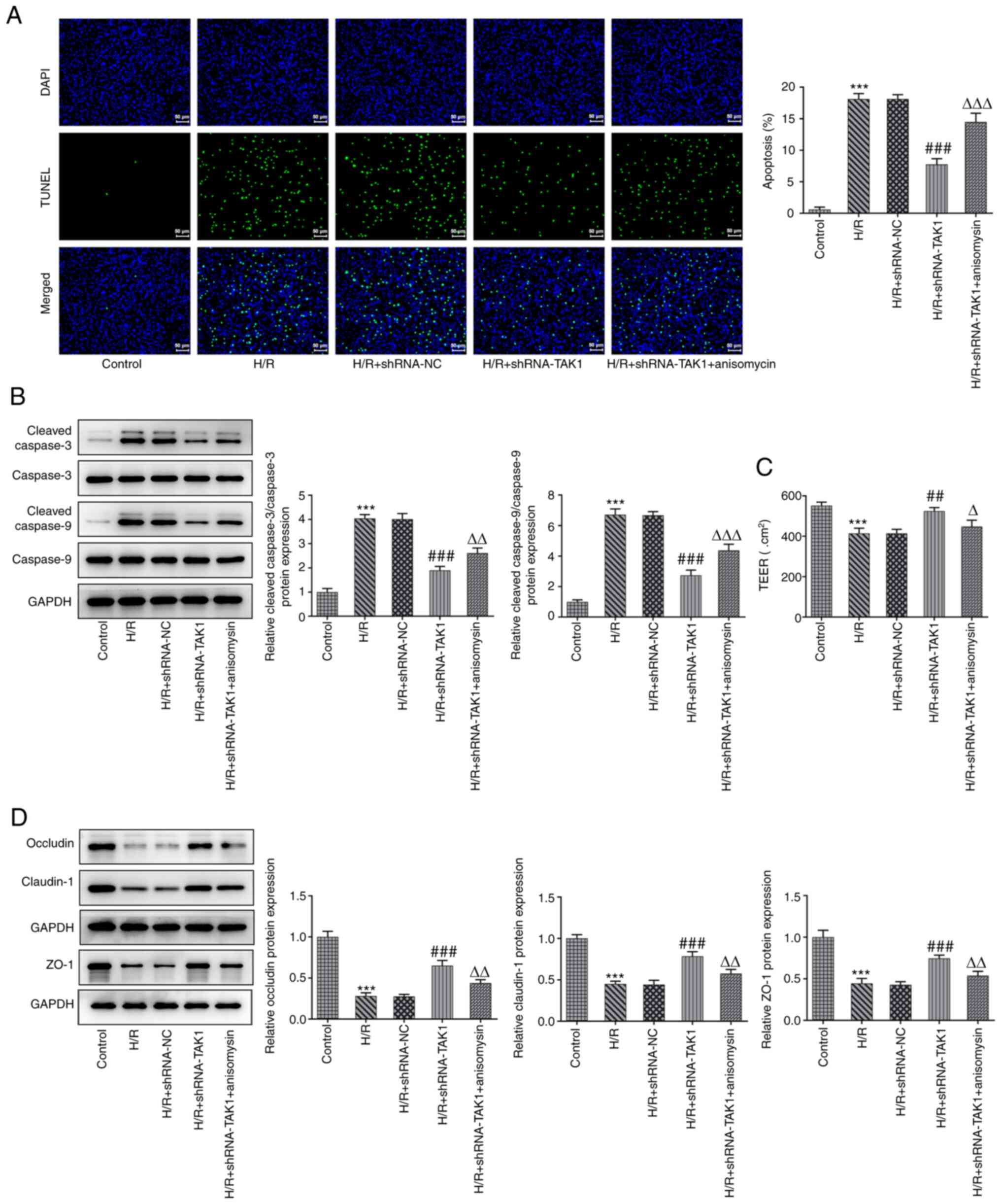 | Figure 3TAK1 knockdown alleviates the
apoptosis and barrier dysfunction of H/R-induced Caco-2 cells via
by inhibiting MAPK signal activation. (A) Apoptosis was assessed
using a TUNEL assay (magnification, x200). (B) Expression levels of
cleaved caspase 9 and cleaved caspase 3 were assessed using western
blotting. (C) Cell barrier function was assessed using a
transepithelial electrical resistance assay. (D) Expression levels
of occludin, claudin-1 and ZO-1 were assessed using western blot
analysis. ***P<0.001 vs. control;
##P<0.01, ###P<0.001 vs. H/R +
shRNA-NC; ΔP<0.05, ΔΔP<0.01,
ΔΔΔP<0.001 vs. H/R + shRNA-TAK1. ZO-1, ZO-1 tight
junction protein; TAK1, TGF-ß activated kinase 1; H/R,
hypoxia/reoxygenation; shRNA, short hairpin RNA; NC, negative
control. |
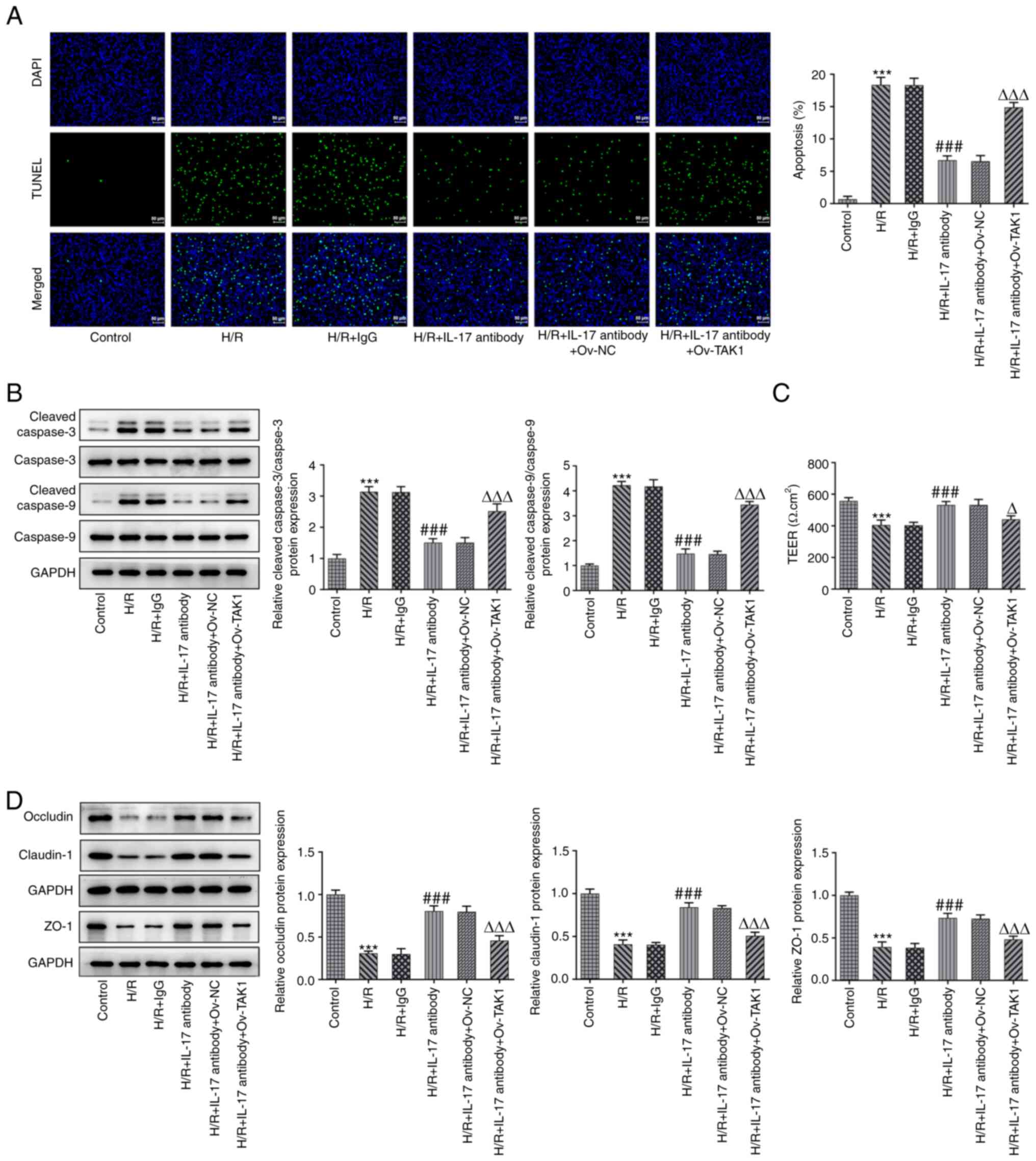
IL-17 antibody improves cell viability
and alleviates the inflammation of H/R-induced Caco-2 cells via a
reduction in TAK1 expression
Results of the present study demonstrated that TAK1
knockdown alleviated cell inflammation, apoptosis and dysfunction
caused by intestinal I/R by regulating MAPK signaling; however,
whether TAK1 participates in the IL-17 signaling pathway remains to
be fully elucidated. Thus, the expression levels of IL-17 in the
H/R group were initially determined using western blotting and
RT-qPCR. Results of the present study revealed that IL-17 was
highly expressed in cells that had undergone H/R. Subsequently, the
expression levels of TAK1 in the H/R and H/R + IL-17 antibody
groups were assessed using western blotting and RT-qPCR. TAK1
expression was significantly increased in the H/R group compared
with the control group (Fig. 4A
and B), and TAK1 expression was
markedly reduced in the H/R + IL-17 antibody group compared with
the H/R group (Fig. 4C and
D). These results indicated that
the IL-17 antibody may reduce the expression of TAK1 by
neutralizing secreted IL-17. Following the verification of high
TAK1 expression levels in the transfected cells using western
blotting and RT-qPCR (Fig. 4E and
F), cells were divided into six
groups: i) Control; ii) H/R; iii) H/R + IgG; iv) H/R + IL-17
antibody; v) H/R + IL-17 antibody + ov-NC; and vi) H/R + IL-17
antibody + ov-TAK1. Cell viability in the aforementioned six groups
was measured using a CCK-8 assay. The results demonstrated that the
addition of the IL-17 antibody significantly increased the levels
of cell viability compared with H/R + IgG, while the viability was
significantly reduced in the H/R + IL-17 + ov-TAK1 group (Fig. 4G). In addition, the mRNA expression
levels of TNF-α, IL-6a and IL-1β, and the protein expression levels
of iNOS and Cox2 were determined using RT-qPCR and western
blotting, respectively. Results of the present study demonstrated
that treatment with H/R + IL-17 antibody significantly reduced the
levels of inflammatory factors and inflammation-associated enzymes
compared with the H/R + IgG group, whereas transfection with
ov-TAK1 (H/R + IL-17 + ov-TAK1) significantly increased the IL-17
antibody-mediated reduced expression levels (Fig. 4H and I).
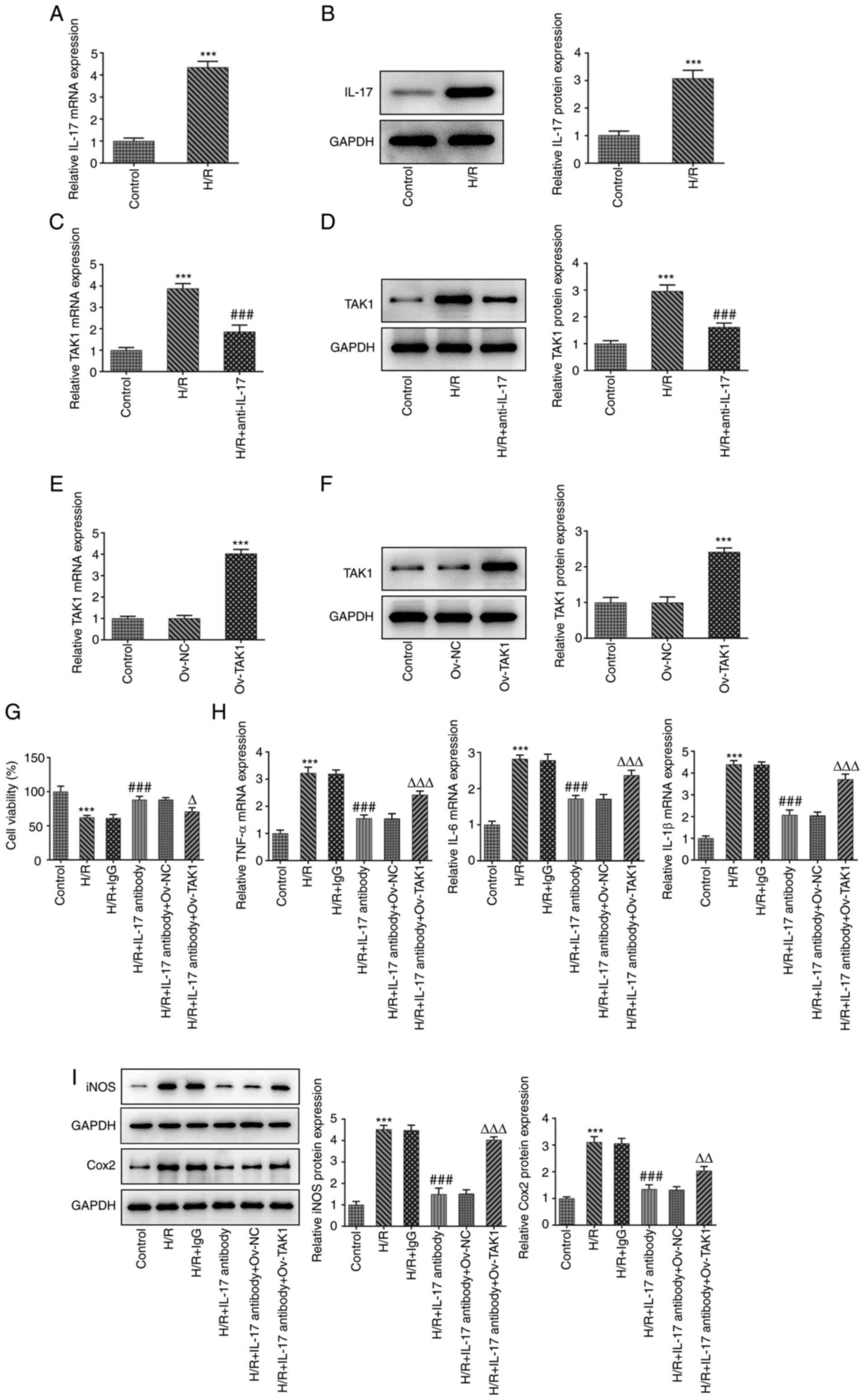 | Figure 4IL-17 antibody improves cell viability
and alleviates the inflammation of H/R-induced Caco-2 cells through
a reduction of TAK1 expression. Expression levels of IL-17 in the
H/R group were determined using (A) RT-qPCR and (B) western
blotting. Expression levels of TAK1 were determined using (C)
RT-qPCR and (D) western blotting. Expression levels of TAK1 in the
transfected cells were determined using (E) RT-qPCR and (F) western
blotting. (G) Cell viability in the six groups was measured using a
Cell Counting-Kit 8 assay. (H) mRNA expression levels of TNF-α,
IL-6 and IL-1β were determined using RT-qPCR. (I) Protein
expression levels of iNOS and Cox2 were determined using western
blot analysis. ***P<0.001 vs. control/Ov-NC;
###P<0.001 vs. H/R + IgG; ΔP<0.05,
ΔΔP<0.01, ΔΔΔP<0.001 vs. H/R + IL-17
antibody + Ov-NC. H/R, hypoxia/reoxygenation; RT-qPCR, reverse
transcription-quantitative PCR; TAK1, TGF-ß activated kinase 1;
iNOS, inducible nitric oxide synthase; Cox2, cytochrome C oxidase
subunit 2; Ov, overexpression; NC, negative control. |
IL-17A antibody alleviates the
apoptosis and barrier dysfunction of H/R-induced Caco-2 cells via a
reduction in TAK1 expression
As the previously described methods focused on
determining the effects of TAK1 knockdown on apoptosis and barrier
function, the effects of the IL-17 antibody on apoptosis and
barrier function were also investigated. The results of the TUNEL
assay demonstrated that treatment with the IL-17 antibody
significantly reduced the fluorescence emitted by apoptotic cells
compared with the H/R + IgG group, whereas transfection with
ov-TAK1 increased this fluorescence compared with the H/R + IL-17
antibody + ov-NC group (Fig. 5A).
Moreover, the results of western blotting revealed that treatment
with the IL-17 antibody significantly decreased the expression
levels of cleaved caspase 9 and cleaved caspase 3 compared with the
H/R + IgG group, whereas transfection with ov-TAK1 reversed the
IL-17 antibody-mediated expression levels (Fig. 5B). Subsequently, the results of the
TEER assay indicated that treatment with the IL-17 antibody
significantly increased the H/R-mediated reduction in TEER value,
and transfection with ov-TAK1 significantly reversed the effects of
the IL-17 antibody on the TEER value (Fig. 5C). Furthermore, the expression
levels of occludin, claudin-1 and ZO-1 were significantly
upregulated in the H/R + IL-17 antibody group compared with the H/R
+ IgG group, while transfection with ov-TAK1 significantly reduced
these expression levels (Fig.
5D).
Discussion
Intestinal I/R injury is often a synergistic effect
of multiple factors (22).
Inflammation is a key response to pathogens and, according to a
previous report, when intestinal I/R injury occurs, the body
stimulates an immune response and simultaneously promotes the
upregulation of numerous inflammatory cytokines. Thus, an
inflammatory response is aggravated, causing damage to multiple
organs (23). Moreover, apoptosis
is also considered to be a major mechanism underlying ischemic
tissue cell death, which impacts inflammation, calcium overload and
oxidation (24). In the process of
intestinal I/R, local apoptosis occurs in the intestine, and
apoptosis may also be induced in remote organs (25). Notably, the intestinal capillary
permeability increases following I/R, which leads to intestinal
absorption dysfunction and increased mucosal permeability, allowing
macromolecular solutes to pass. Thus, damaged intestines may become
the source of numerous harmful biologically active substances
(26). Therefore, further
investigation into the mechanisms underlying intestinal I/R injury
is required for the development of novel treatment options.
Results of the present study demonstrated that
intestinal I/R causes upregulation of TAK1 expression, and TAK1
knockdown inhibits the phosphorylation of JNK. Results of a
previous study demonstrated that the expression of JNK in the MAPK
signaling family is increased in intestinal ischemic tissues
(27). Moreover, activation of
TAK1-dependent NF-κB and proto-oncogene c-Jun is associated with
the occurrence of intestinal inflammation and, in the case of
dextran sodium sulfate-induced enteritis, the expression levels of
TAK1 are significantly increased (28). These findings are consistent with
those of the present study. Furthermore, results of the present
study also demonstrated that TAK1 was involved in intestinal I/R
injury, including cell inflammation, apoptosis and barrier damage.
TAK1 knockdown alleviated the inflammation, apoptosis and barrier
dysfunction of H/R-induced Caco-2 cells by inhibiting MAPK signal
activation.
Moreover, following intestinal I/R, the release of
IL-17 is upregulated. Over the past decade, IL-17 has been revealed
to play an important role in autoimmune diseases and tumors
(29). At present, an IL-17
monoclonal antibody that has been approved for marketing is used
for the treatment of psoriasis (30). However, to the best of our
knowledge, there are few studies that describe the role of IL-17 in
the intestinal tract. Results of a previous study revealed that
IL-17 is upregulated in the gut of mice with intestinal fibrosis
(31), and the decrease of IL-17
expression levels may protect intestinal cells from inflammation
(32). Results of the present
study demonstrated that reducing IL-17 expression alleviated
inflammation, apoptosis and barrier dysfunction caused by
intestinal I/R injury. Notably, IL-17 exerted the aforementioned
effects by reducing the levels of TAK1 expression. Results of a
previous study further demonstrated that IL-17 promotes drug efflux
in patients with rheumatoid arthritis via regulation of
TAK1(33). Usually, some
researchers use recombinant IL-17 to induce inflammation to study
the role of IL-17 in cells. In the present article, cells
influenced each other through paracrine signaling, therefore, the
use of recombinant IL-17 was unnecessary. Therefore, the present
study selected to neutralize IL-17 with an antibody to explore its
effects.
In conclusion, the present study demonstrated that
intestinal I/R induces the release of IL-17 to regulate cell
viability, inflammation, apoptosis and barrier damage via TAK1/MAPK
signaling. However, the present study is limited to in vitro
research, and in vivo intestinal I/R models will be
established in the future to further explore the roles of IL-17 in
I/R injury.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LX and PH conceived the study, and designed and
participated in the experiments. LX wrote the manuscript. PH
revised the manuscript. WHZ and YH participated in the experiments
and data processing. All the authors have read and approved the
final manuscript. LX and PH confirm the authenticity of the raw
data
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jozwiak M, Bougouin W, Geri G, Grimaldi D
and Cariou A: Post-resuscitation shock: Recent advances in
pathophysiology and treatment. Ann Intensive Care.
10(170)2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Van Hoof L, Rega F, Devroe S, Degezelle K,
Pirenne J and Neyrinck A: Successful resuscitation after
hyperkalemic cardiac arrest during liver transplantation by
converting Veno-Venous bypass to veno-arterial ECMO. Perfusion.
36:766–768. 2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Warach SJ, Dula AN and Milling TJ Jr:
Tenecteplase thrombolysis for acute ischemic stroke. Stroke.
51:3440–3451. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Fernández AR, Sánchez-Tarjuelo R, Cravedi
P, Ochando J and López-Hoyos M: Review: Ischemia reperfusion
Injury-A translational perspective in organ transplantation. Int J
Mol Sci. 21(8549)2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ali M, Pham A, Wang X, Wolfram J and Pham
S: Extracellular vesicles for treatment of solid organ
ischemia-reperfusion injury. Am J Transplant. 20:3294–3307.
2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zang X, Zhou J, Zhang X, Han Y and Chen X:
Ischemia reperfusion injury: Opportunities for nanoparticles. ACS
Biomater Sci Eng. 6:6528–6539. 2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Prieto-Moure B, Cejalvo-Lapeña D,
Belda-Antolí M, Padrón-Sanz C, Lloris-Cejalvo JM and Lloris-Carsí
JM: Combination therapy of allopurinol and dantrolene and its role
in the prevention of experimental ischemia reperfusion injury of
the small intestine. J Invest Surg. 34:800–807. 2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Saleh H and El-Shorbagy HM: Mechanism
underlying methyl eugenol attenuation of intestinal
ischemia/reperfusion injury. Appl Physiol Nutr Metab. 42:1097–1105.
2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Tuncer FB, Durmus Kocaaslan FN, Yildirim
A, Sacak B, Arabaci Tamer S, Sahin H, Cinel L and Celebiler O:
Ischemic preconditioning and Iloprost reduces Ischemia-reperfusion
injury in Jejunal flaps: An animal model. Plast Reconstr Surg.
144:124–133. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Subramanian S, Geng H and Tan XD: Cell
death of intestinal epithelial cells in intestinal diseases. Sheng
Li Xue Bao. 72:308–324. 2020.PubMed/NCBI
|
|
11
|
Zhang X, Wu J, Liu Q, Li X, Li S, Chen J,
Hong Z, Wu X, Zhao Y and Ren J: mtDNA-STING pathway promotes
Necroptosis-dependent enterocyte injury in intestinal ischemia
reperfusion. Cell Death Dis. 11(1050)2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Liu L, Yao J, Li Z, Zu G, Feng D, Li Y,
Qasim W, Zhang S, Li T, Zeng H and Tian X: miR-381-3p knockdown
improves intestinal epithelial proliferation and barrier function
after intestinal ischemia/reperfusion injury by targeting nurr1.
Cell Death Dis. 9(411)2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Kajino-Sakamoto R, Inagaki M, Lippert E,
Akira S, Robine S, Matsumoto K, Jobin C and Ninomiya-Tsuji J:
Enterocyte-derived TAK1 signaling prevents epithelium apoptosis and
the development of ileitis and colitis. J Immunol. 181:1143–1152.
2008.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Nighot M, Rawat M, Al-Sadi R, Castillo EF,
Nighot P and Ma TY: Lipopolysaccharide-induced increase in
intestinal permeability is mediated by TAK-1 activation of IKK and
MLCK/MYLK gene. Am J Pathol. 189:797–812. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Lee HT, Kim M, Kim JY, Brown KM, Ham A,
D'Agati VD and Mori-Akiyama Y: Critical role of interleukin-17A in
murine intestinal ischemia-reperfusion injury. Am J Physiol
Gastrointest Liver Physiol. 304:G12–G25. 2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zepp J, Wu L and Li X: IL-17 receptor
signaling and T helper 17-mediated autoimmune demyelinating
disease. Trends Immunol. 32:232–239. 2011.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Huang F, Kao CY, Wachi S, Thai P, Ryu J
and Wu R: Requirement for both JAK-mediated PI3K signaling and
ACT1/TRAF6/TAK1-dependent NF-kappaB activation by IL-17A in
enhancing cytokine expression in human airway epithelial cells. J
Immunol. 179:6504–6513. 2007.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Zhou J, An C, Jin X, Hu Z, Safirstein RL
and Wang Y: TAK1 deficiency attenuates cisplatin-induced acute
kidney injury. Am J Physiol Renal Physiol. 318:F209–F215.
2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Hiebl V, Schachner D, Ladurner A, Heiss
EH, Stangl H and Dirsch VM: Caco-2 cells for measuring intestinal
cholesterol transport-possibilities and limitations. Biol Proced
Online. 22(7)2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Tang LJ, Zhou YJ, Xiong XM, Li NS, Zhang
JJ, Luo XJ and Peng J: Ubiquitin-specific protease 7 promotes
ferroptosis via activation of the p53/TfR1 pathway in the rat
hearts after ischemia/reperfusion. Free Radic Biol Med.
162:339–352. 2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Yang B, Zhang LY, Chen Y, Bai YP, Jia J,
Feng JG, Liu KX and Zhou J: Melatonin alleviates intestinal injury,
neuroinflammation and cognitive dysfunction caused by intestinal
ischemia/reperfusion. Int Immunopharmacol.
85(106596)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Liu DQ, Chen SP, Sun J, Wang XM, Chen N,
Zhou YQ, Tian YK and Ye DW: Berberine protects against
ischemia-reperfusion injury: A review of evidence from animal
models and clinical studies. Pharmacol Res.
148(104385)2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Radak D, Katsiki N, Resanovic I, Jovanovic
A, Sudar-Milovanovic E, Zafirovic S, Mousad SA and Isenovic ER:
Apoptosis and acute brain ischemia in ischemic stroke. Curr Vasc
Pharmacol. 15:115–122. 2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Feng D, Yao J, Wang G, Li Z, Zu G, Li Y,
Luo F, Ning S, Qasim W, Chen Z and Tian X: Inhibition of
p66Shc-mediated mitochondrial apoptosis via targeting
prolyl-isomerase Pin1 attenuates intestinal ischemia/reperfusion
injury in rats. Clin Sci (Lond). 131:759–773. 2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Li C, Li Q, Liu YY, Wang MX, Pan CS, Yan
L, Chen YY, Fan JY and Han JY: Protective effects of
Notoginsenoside R1 on intestinal ischemia-reperfusion injury in
rats. Am J Physiol Gastrointest Liver Physiol. 306:G111–G122.
2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Ming YC, Chao HC, Chu SM and Luo CC:
Heparin-binding epidermal growth factor-like growth factor (HB-EGF)
protected intestinal ischemia-reperfusion injury through JNK and
p38/MAPK-dependent pathway for anti-apoptosis. Pediatr Neonatol.
60:332–326. 2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Xu K, Guo Y, Ping L, Qiu Y, Liu Q, Li Z
and Wang Z: Protective effects of SIRT6 overexpression against
DSS-induced colitis in mice. Cells. 9(1513)2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
McGeachy MJ, Cua DJ and Gaffen SL: The
IL-17 family of cytokines in health and disease. Immunity.
50:892–906. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Ly K, Smith MP, Thibodeaux Q, Reddy V,
Liao W and Bhutani T: Anti IL-17 in psoriasis. Expert Rev Clin
Immunol. 15:1185–1194. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Li J, Liu L, Zhao Q and Chen M: Role of
interleukin-17 in pathogenesis of intestinal fibrosis in mice. Dig
Dis Sci. 65:1971–1979. 2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Prete R, Garcia-Gonzalez N, Di Mattia CD,
Corsetti A and Battista N: Food-borne Lactiplantibacillus plantarum
protect normal intestinal cells against inflammation by modulating
reactive oxygen species and IL-23/IL-17 axis. Sci Rep.
10(16340)2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Li Z, Niu H, Yao H, Chen M, Zhao X, Zhao
W, Luo J, Gao C, Li X and Wang C: IL-17A upregulates P-glycoprotein
expression in peripheral blood lymphocytes of patients with
rheumatoid arthritis through TAK1. Clin Exp Rheumatol. 38:299–305.
2020.PubMed/NCBI
|