1. Introduction
Stem cells are undifferentiated cells that have the
ability to proliferate (self-renewal) both in vitro and
in vivo and differentiate into mature specialized cells
(1). Stem cells can be divided
into five groups: Totipotent stem cells (TSCs), pluripotent stem
cells (PSCs), multipotent stem cells, oligopotent stem cells (OSCs)
and unipotent stem cells (USCs) (2). TSCs have the highest differentiation
potential, able to produce an entire living organism on their own,
and most notably a zygote. PSCs can form cells in all the germ
layers, with the exception of the cells that form structures
outside the embryo, and embryonic stem cells are a prime example.
Multipotent stem cells can produce certain lineages of cells, and
the majority of adult stem cells are multipotent, including
hematopoietic stem cells (HSCs), mesenchymal stem cells (MSCs), and
other adult progenitor cells (3).
OSCs can differentiate into numerous cell types, such as bone
marrow stem cells which may develop into white blood cells but not
red blood cells. Eventually, USCs may form only one cell type, such
as skin cells.
Among multipotent stem cells, HSCs are the most
common multipotent stem cells with the ability to maintain
homeostasis by self-renewal or differentiation into all blood cell
lineages (4). The stemness of the
HSCs combines the ability of the HSCs to perpetuate its lineage, to
produce differentiated cells (such as lymphocytes and granulocytes)
and to interact with the hematopoietic microenvironment to maintain
a balance between quiescence, proliferation, and regeneration
(5). In short, the stemness of
HSCs helps maintain the homeostasis of the blood system by
balancing the proliferation and differentiation of HSCs. When the
stemness of HSCs is destroyed, abnormal production of blood cells
occurs and further abnormal blood system tumors are produced,
namely leukemia (6). Common
subtypes of leukemia include acute myelogenous leukemia (AML),
acute lymphoblastic leukemia (ALL), chronic myelogenous leukemia
(CML) and chronic lymphoblastic leukemia (CLL). Therefore, a full
understanding of the signaling pathways or regulatory factors that
are capable of regulating the stemness of HSCs may provide further
insight into HSCs and hope for a cure for leukemia.
In recent years, it has been gradually revealed that
a series of signaling pathways, such as Wnt, Notch, the TGF-β
family, Hedgehog and Hippo signaling, could affect the stemness of
HSCs, and that dysregulations of these pathways alone or
coordinated may lead to the development of leukemia (7,8).
Among them, Notch signaling, an evolutionarily conserved signaling
pathway, is essential for the establishment of the earliest
embryonic HSCs and is closely associated with the emergence,
development, and maintenance of HSCs in adulthood (9). In this signaling pathway, Notch1
receptor is most closely associated with the stemness of HSCs and
plays a key role in T-cell development and transformation (10). Abnormally activated or mutated
Notch1 receptors severely affect the balance of proliferation and
differentiation of HSCs which triggers the continuous emergence of
abnormal lymphocytes, thus leading to lymphocytic leukemia,
particularly T-cell acute lymphoblastic leukemia (T-ALL). In
addition to the Notch1 receptor itself, its post-translational
modifications, such as glycosylation, phosphorylation, and
ubiquitination, also affect the activation of the Notch1 pathway,
thereby affecting the stemness of HSCs (11). Among these post-transcriptional
modifications, particular attention has been paid to the
ubiquitination modification of Notch1, since it affects the
degradation of Notch1 receptor (12), the activation of Notch1 signaling
(11), and the process of
endocytosis that Notch1 receptor undergoes (13). Therefore, this suggests that the
key enzymes responsible for the ubiquitination modification of
Notch1 may also, directly or indirectly, affect the stemness of
HSCs and the development of leukemia (14-16).
In the present review, the structure of the Notch signaling pathway
was firstly summarized in detail and the effects of the Notch1
receptor on HSC origin, proliferation, differentiation and
associated T-ALL, were described. Subsequently, the ubiquitination
modification of Notch1 receptor and its effects on HSCs were
elucidated. Finally, the clinical application of HSCs, as well as
the potential therapeutic targets and prognostic indicators of
Notch were reviewed.
2. Overview of the Notch pathway
Notch signaling is a major mediator in determining
cell fate during development, and it regulates a variety of cell
functions, including differentiation, proliferation, and
homeostasis (17). Evidence
suggests that the Notch signaling pathway has markedly opposite
functions in tumor development, possibly acting as an oncogene or a
tumor suppressor (18). In the
process of hematopoiesis, Notch signaling controls the fate of
hematopoietic progenitor stem cells by inhibiting certain
differentiation steps and inducing self-renewal or lymphatic
lineage differentiation (19).
Notch receptor, Notch ligand and DNA binding
sequence CSL [CBF1/SU(H)/LAG-1] are the three main components of
the canonical Notch signaling pathway (20). There are four transmembrane Notch
receptors (Notch1, Notch2, Notch3, and Notch4) and five typical
transmembrane ligands [Delta-like (DLL) 1, DLL 3, DLL 4, Jagged1,
and Jagged 2] in mammals (21).
The extracellular region of the Notch receptor (NECD) contains
29-36 epidermal growth factor (EGF)-like repeats, three LIN12/Notch
repeats and a heterodimerization (HD) domain. Notch intracellular
domains (NICD) include a RAM domain, seven cdc10/ankyrin repeats,
two nuclear localization sequences, a transactivation domain (TAD)
(Notch1 and Notch2), and a C-terminal PEST motif (22) (Fig.
1A). Notch ligand is a type I transmembrane protein that
contains extracellular EGF-like repeats, a Delta, Serrate and LAG-2
domain and a Delta and OSM-11-like protein domain, which together
are responsible for Notch receptor interactions (21).
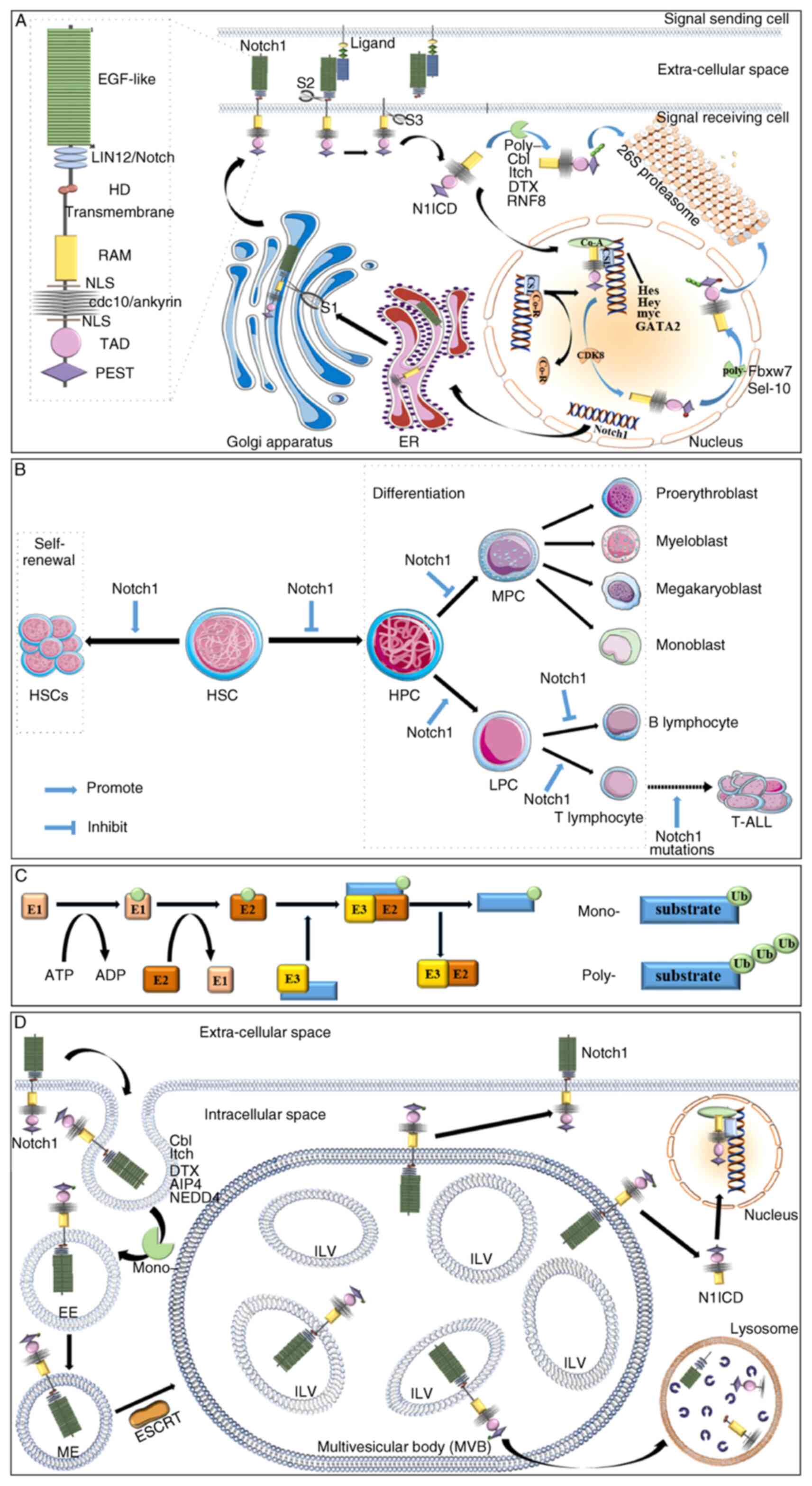 | Figure 1Notch1 and HSCs. (A) Notch1 receptor
structure and Notch1 signaling. Notch1 receptors are composed of an
intracellular domain (36 EGF-like repeats, three LIN12/Notch
repeats and an HD domain), a transmembrane domain and an
extracellular domain (RAM domain, seven cdc10/ankyrin repeats, two
NLS, a TAD domain, and a PEST motif). The Notch1 receptor undergoes
three proteolysis processes to become N1ICD. Above all, the
precursor Notch1 receptor is cleaved by a furan-like convertase
(S1) in the Golgi apparatus. When bound to the ligand, TACE or Kuz
perform a second cleavage (S2). The remaining domain is cleaved by
γ-secretase (S3) to release N1ICD. The N1ICD enters the nucleus and
the target gene is detached from the Co-R. Then, N1ICD, together
with SCL and Co-A, promotes the transcription of target genes.
N1ICD in the nucleus and cytoplasm is degraded by 26S proteasome
through a process of poly-ubiquitination modification. However,
N1ICD in the nucleus is phosphorylated by CDK8 before the
ubiquitination modification. (B) Effects of Notch1 signaling on
self-renewal (proliferation) and differentiation of HSCs. In
general, Notch1 promotes the proliferation of HSCs and inhibits
their differentiation. However, when hematopoietic stem cells begin
to differentiate, Notch1 promotes hematopoietic stem cells to
differentiate into T lymphocyte lines rather than myeloid lines. In
addition, Notch1 signaling drives T-cell development at the expense
of the development of B cells. In the end, the most important
carcinogenic pathway in T-ALL is the activation mutation of Notch1
signaling. (C) Processes and types of ubiquitination modification.
The ubiquitin molecule is added to the substrate by the action of
E1, E2 and E3 in turn. Ubiquitination modification mainly involves
mono-ubiquitination modification and poly-ubiquitination
modification. (D) The process of endocytosis. When no ligand binds,
Notch1 undergoes endocytosis. Notch1 is mono-ubiquitinated before
EE is formed. Then, EE will gradually mature into ME. Subsequently,
the multiple MEs are fused into MVEs with the assistance of ESCRT.
The position of Notch1 on the MEVs determines its fate. If Notch1
is present on the limiting membrane of MVBs, it may be recycled to
the cell membrane for utilization. If Notch1 on the MVB-limiting
membrane is cleaved and N1ICD is released, Notch1 signaling will be
activated. However, the residual Notch1 in ILVs is transported to
lysosomes for degradation. EGF-like, 36 epidermal growth factor
(EGF)-like repeats; LIN12/Notch1, three LIN12/Notch repeats; HD,
heterodimerization domain; transmembrane, transmembrane domain;
RAM, RAM domain; NLS, nuclear localization sequences;
cdc10/ankyrin, seven cdc10/ankyrin repeats; TAD, transactivation
domain; PEST, PEST motif; ER, endoplasmic reticulum; S1, first
proteolytic cleavage; S2, second proteolytic cleavage; S3, third
proteolytic cleavage; N1ICD, Notch1 intracellular domains; Co-A,
co-activators; Co-R, co-inhibitors; CSL, DNA-binding protein
CSL/RBPJκ; HSC, hematopoietic stem cell; HPC, hematopoietic
progenitor cell; MPC, myeloid progenitor cell; LPC, lymphoid
progenitor cell; T-ALL, T-cell acute lymphoblastic leukemia; E1,
ubiquitin-activating enzyme; E2, ubiquitin-conjugating enzyme; E3,
ubiquitin ligase; mono-, mono-ubiquitination; poly-,
poly-ubiquitination; Ub, ubiquitin molecule; EE, early endosome;
ME, maturing endosome; ESCRT, endosomal sorting complexes required
for transport; ILV, interluminal vesicle. |
The Notch signaling pathway is activated when a
ligand on a cell membrane binds to the Notch receptor on an
adjacent cell. The Notch receptor passes through three different
proteolytic cleavages (Fig. 1A).
First, a single polypeptide precursor protein is cleaved in the
Golgi by a furin-like convertase to produce a mature Notch receptor
(S1) (21). When the mature Notch
receptor binds to the ligand, a second cleavage (S2) is performed
by TACE or Kuz of the A disintegrin and metalloprotease
metalloproteinase family to release extracellular fragments
(20). The remaining transmembrane
and intracellular domains are cleaved by γ-secretase for a third
time (S3), releasing the soluble NICD and transferring to the
nucleus (20). Then, NICD binds to
the DNA-binding protein CSL/RBPJκ in the nucleus, activating genes
which belong to the basic helix-loop-helix (bHLH)
family (20). The general
consensus is that CSL/RBPJκ persistently binds to the promoter of
the targeted gene. In the absence of NICD, CSL/RBPJκ binds with
co-inhibitors (SMRT, histone deacetylase, etc.) to inhibit gene
transcription. Conversely, when NICD enters the nucleus, it
recruits co-activators such as MAML to promote the transcription of
target genes (23). In mammals,
genes known as the Hes family (Hes1,5,7) and the
Hey family (Hey1,2,L) are the major components of the
bHLH family (24-26).
Hes1 is important in the development of the nervous system,
sensory organs (eye, inner ear), pancreas, endocrine cells, and
lymphocytes (24). Hes7 is
essential for somitogenesis (25).
By contrast, the Hey family play a key role in the
cardiovascular system (26). In
addition to the bHLH family, several other genes have also
been identified as Notch targets, including
p27Kip1 (27),
cyclin D1 (28), myc
(29), and Deltex1
(30).
The main substrates that undergo ubiquitination
modification in this signaling pathway are Notch receptor, Notch
ligand and γ-secretase (31).
Among them, the ubiquitination modification of Notch receptor,
particularly Notch1 receptor, may mostly affect the signaling
strength of this pathway (31).
The Notch1 receptor firstly undergoes either mono-ubiquitination or
poly-ubiquitination, thereafter the Notch1 receptor is degraded or
the Notch1 signaling is activated, thereby affecting the Notch1
signaling and the expression of downstream genes. The detailed
process is subsequently described.
Since Notch1 receptor-mediated Notch1 signaling
plays an irreplaceable role in the blood system, The Cancer Genome
Atlas database (https://portal.gdc.cancer.gov/exploration?filters=%7B%22op%22%3A%22and%22%2C%22content%22%3A%5B%7B%22op%22%3A%22in%22%2C%22content%22%3A%7B%22field%22%3A%22cases.available_variation_ta%22%2C%22value%22%3A%5B%22ssm%22%2C%22cnv%22%5D%7D%7D%2C%7B%22op%22%3A%22in%22%2C%22content%22%3A%7B%22field%22%3A%22cases.project.project_id%22%2C%22value%22%3A%5B%22TARGET-ALL-P3%22%2C%22TCGA-DLBC%22%2C%22TCGA-LAML%22%5D%7D%7D%2C%7B%22op%22%3A%22in%22%2C%22content%22%3A%7B%22field%22%3A%22genes.gene_id%22%2C%22value%22%3A%5B%22ENSG00000148400%22%5D%7D%7D%5D%7D&searchTableTab=cases)
and the International Cancer Genome Consortium database (https://dcc.icgc.org/genes/ENSG00000148400/mutations?donors=%7B%22from%22:1%7D&filters=%7B%22donor%22:%7B%22primarySite%22:%7B%22is%22:%5B%22Blood%22%5D%7D%7D%7D)
were consulted, respectively, and data on the expression and
mutations of Notch1 were obtained by searching for
Notch1 mutations and selecting all hematology-related
malignancies in the database, including various types of lymphomas
and leukemias. The Notch1 mutation rate was 32/241 (13.28%)
in germinal B-cell derived lymphomas, 64/510 (12.55%) in CLL, 3/50
(6.00%) in T- and NK-cell lymphomas, 11/205 (5.37%) in AML and
1/136 (0.74%) in chronic myeloid disorders. Through further
integrating these data into disease categories, it was identified
that Notch1 was mutated with a frequency of 9.72% in
hematological malignancies. Furthermore, the mutation frequency of
Notch1 in lymphomas was 12.03 and 8.93% in leukemias.
3. Notch1 and HSCs
Origin of HSCs
HSCs are the cornerstone of the mammalian blood
system (32). These stem cells
self-renew to maintain a stable pool of HSCs, which are able to
differentiate into myeloid, lymphatic and erythroid cells as
required, thus maintaining blood cell homeostasis (32). The Notch signaling pathway,
particularly the Notch1 receptor, plays a key role in maintaining
undifferentiated HSCs and inducing self-renewal (9). Thus, Notch1 is biologically important
in HSCs.
During human embryonic development, two distinct
sites are involved in hematopoiesis: The extraembryonic yolk sac
(YS) and the aorta-gonadal-mesonephric (AGM) region within the
embryo (33). The hematopoiesis
starts in the YS blood islands, travels to the fetal liver, and
finally locates to the bone marrow during embryogenesis.
Additionally, adult blood cells, including lymphocytes and
hematopoietic progenitor cells (HPCs), are generated in the
para-aortic splanchnopleure (P-SP) region, which then develops into
AGM for long-term hematopoiesis before the HSCs reach the fetal
liver (34). The first long-term
regenerated HSCs are detected in the AGM region. By positively
regulating Notch1 through the transcription factor SOX17, Saito
et al (35) revealed that
Notch1 intracellular domain (N1ICD) or its downstream target
protein Hes1 transduced HSCs to maintain the ability of multipotent
colony formation in AGM. By contrast, Notch1 and Hes1
gene knockout resulted in a decrease in the ability to form
multipotent colonies. These results indicate that Notch1 and Hes1
are critical for maintaining the undifferentiated state of HSCs
(35). Thus, Notch1 is critical to
the production of HSCs during embryogenesis.
With regard to the origin of HSCs, it is generally
considered that embryonic HSCs and progenitor cells are derived
from the hematopoietic endothelium, and thus the transformation of
hematopoietic endothelial cells into HSCs and progenitor cells
(EHT) is required. Zhang et al (36) demonstrated that inhibition of
Notch1 signaling can promote EHT by G protein-coupled receptor 183.
In mouse embryos with Notch1 deletion mutations, distinct
hematopoietic endothelial cells were identified, but they did not
develop into HSCs (37).
Differences in the ligands that activate Notch1 may contribute to
the paradoxical nature of the results. Through analysis of
experimental data, Gama-Norton et al (38) revealed that 89% of endothelial
cells co-expressed Jagged1 and DLL4 ligands, and only a few
endothelial cells expressed Jagged1 ligands alone (3.8%) or DLL4
ligands alone (4.6%) or neither (2.5%). The balance of the
DLL4-Notch1 and Jagged1-Notch1 signaling pathways may ensure the
correct establishment of endothelial and hematopoietic cell fates
in AGM. Furthermore, they suggested that the deletion of Jagged1
ligand leads to increased Notch activity in the aortic endothelium
of AGM through the microarray analysis of AGM subpopulations,
thereby improving the fate of endothelial cells at the expense of
HSC formation. Conversely, when lacking the Jagged1-Notch1
signaling and experiencing high DLL4-Notch1 signaling, endothelial
cells select the endothelial protocol, thus preventing the
formation of HSCs. It was hypothesized that precursor hematopoietic
cells responding to Jagged1 would attenuate the DLL4-Notch1
signaling, replacing it with an effective low Notch1 signaling,
which is necessary and sufficient for activation of hematopoietic
genes such as GATA2 (38).
In addition to GATA2, Fox2 from the Fox gene
family induced by N1ICD also plays a role in hematopoietic
endothelium. Data from a study by Jang et al (39) established a pathway that binds
Notch signaling to its downstream Fox2 in hematopoietic
endothelial cells, thereby promoting hematopoietic development.
Collectively, these studies suggested that Notch1 has an
indispensable role prior to HSC production (37).
Proliferation and differentiation of
HSCs
Through downstream proteins or genes, particularly
Hes1, the Notch signaling pathway mediated by Notch1 receptor
promotes self-renewal of HSCs and inhibits their differentiation
(Fig. 1B). Using
Rag-1-/- mouse stem cells, Stier et al
(40) documented Notch1-induced
reduction of in vivo differentiation and an increased stem
cell population due to enhanced stem cell self-renewal. The
research of Shao et al (41) revealed that endothelial
Jagged1-Notch1 deficiency severely affects the development of fetal
blood vessels and impedes the proliferation and differentiation of
HSCs in vitro and in vivo. Additionally, it was
specified that Notch1-Hes1 may act on hematopoietic precursor
cells, which are produced following the fate of HSCs. Furthermore,
Hes1 not only preserved the long-term recombination activity of
HSCs in vitro, but also accumulated side population cells
in vivo (42). These
results suggested that Hes1 inhibits HSC differentiation. However,
once HSCs enter the differentiation stage, Notch1 signaling
promotes HSC differentiation, with a preference for the lymphatic
rather than the myeloid line (40)
(Fig. 1B). A previous study
conducted by Henning et al (43) suggested that Notch1 signaling
mediates this process via a p53-dependent pathway. Collectively,
the main effect of Notch1 signaling on HSCs is to promote its
proliferation and inhibit its differentiation.
HSCs differentiate into T cells
When HSCs first enter the thymus and become early
T-cell precursor (ETP) cells, they receive high levels of Notch
signaling regulation (44). On the
one hand, excessive Notch1 signaling drives premature commitment of
T cells, leading to loss of ETP cells and the fate of replacement
cells (44). By contrast, complete
loss of Notch1 signaling impairs ETP cell proliferation and leads
to loss of ETP cells (44). Thus,
maintaining a good balance of Notch signaling can maintain the
stemness of HSCs.
In both mouse and human, Notch1 activation is the
primary driver of inducing T-cell development in hematopoietic stem
progenitor cells (Fig. 1B). The
role of Notch1 in lymphogenesis has been well studied, and in
particular the most prominent characteristic function of Notch1
signaling is maturation of T cells and lineage commitment in the
mouse thymus (45). It has been
demonstrated that the expression of Notch1 transgene in HSCs
leads to thymus-independent development of
CD4+CD8+T cells (45). In addition, the study of Gerhardt
et al (46) revealed that
TAD in Notch1 drives T-cell development at the expense of common
precursor development of B cells.
Notably, downstream target genes of Notch1, such as
Hes and myc, are the driving force in the
differentiation of HSCs into T cells. Hes1 has been revealed
to be expressed in both the thymus and thymus stroma, and its
expression in the thymus was regulated by Notch signaling (47). More than 90% of
Hes1-/- mice lacked thymus glands, suggesting
that Hes1 is critical for the in vivo proliferation
of early T-cell precursors (48).
In a recent study, De Decker et al (49) identified that Hes1 and Hes4 were
upregulated in a Notch-dependent manner during early T-cell
development and Hes1 acted as a differentiation inhibitor since it
maintained quiescent stem cell characteristics in CD34+
HPCs. However, Hes4 promoted the initiation of early T-cell
development. Importantly, knockout of Hes1 or Hes4
significantly reduced human T-cell development. As for myc,
in a well-established model of HSC T-lymphocyte differentiation
in vitro, Haque et al (50) determined that Notch1 and 4 directly
promoted myc expression. It was further demonstrated that
overexpression of myc promoted T-cell differentiation, while
dominant-negative myc delayed T-cell differentiation. These
results confirmed that myc is an important mediator of Notch
signaling in the differentiation of HSCs into T lymphocytes. The
Notch1-mediated emergence of these two different effects on HSC
differentiation into T cells may be attributed to the Notch ligand.
OP9-cell co-culture experiments revealed that Jagged2 induced
T-line differentiation and inhibited B cell and bone marrow
development, as did DLL ligands (51). However, the results of Van de Walle
et al (51) revealed a
unique role of Jagged1 in preventing induction of differentiation
of HSCs in T lines.
T-ALL
T-ALL is an aggressive hematologic tumor in which
the malignant transformation of HSCs and HPCs lead to the
development of T cells (52).
Although T-ALL accounts for only 25% of ALL cases in adults and 15%
in children, they have a higher risk of central nervous system
recurrence in the presence of mutations activated by the Notch1
signaling pathway (53).
Constitutive activation of Notch1 signaling is the most important
oncogenic pathway in T-cell transformation, and >65% of T-ALL
patients have Notch1 activation mutations (52). In addition, Ma et al
(53) concluded that Notch1
signaling promotes cell regeneration in human T-ALL. Most of the
abnormal activation of Notch1 observed in T-ALL is due to mutations
in its HD domain and/or PEST domain (54). Of the 15 T-ALL patients studied by
Bhanushali et al (54), 6
(40%) patients had at least one Notch1 mutation, with 2/15
(13%) occurring in the HD domain and 4/15 (27%) in the PEST domain.
In addition, mutations are considered to occur in 4 out of 10 (40%)
adult patients; in the pediatric cohort, two out of five (40%) had
both mutations in the PEST domain (54). Mutations in the HD domain of Notch1
receptor render it more susceptible to protein cleavage and then
release of N1ICD, while mutations in the PEST domain of Notch1
receptor inhibit proteasomal degradation of N1ICD by F-box and WD
repeat domain containing 7 (Fbxw7), which is a ubiquitin ligase,
thus prolonging its half-life in T-All cells. In addition, deletion
or inactivation mutations of Fbxw7 are frequently observed
in T-ALL. In addition, Ding et al (55) revealed that fetal-derived T-cell
precursor stem cells may play a role as leukemia initiation cells.
This may be due to their discovery of overexpression of N1ICD in
P-SP and YS cells. P-SP cells overexpressing N1ICD rapidly
developed T-ALL, while YS cells exhibited no leukemia proliferation
following N1ICD induction. To date, Notch1 mutations have
also been reported in CLL (56).
Di Ianni et al (56)
reported Notch1 mutation in HSCs of CLL patients, and
aberrant activation of Notch1 in HSCs of CLL patients without
Notch1 mutation.
4. Notch1 and ubiquitination
Ubiquitination
Ubiquitination is a common and important
post-translational modification process that plays a key role in
protein homeostasis (57). It is
mainly achieved through labeling the ubiquitin (an 8.6 kDa
regulatory protein) to the substrate, which is then degraded in the
26S proteasome to release the ubiquitin molecule (58). In addition, ubiquitination also
includes certain non-proteolytic functions, such as receptor
internalization (59),
multiprotein complex assembly (60), inflammatory signaling (61), DNA damage repair (62), cell death (63), metabolism (64) and signaling activation (65,66).
Ubiquitination involves three different biochemical
steps: activation, conjugation, and ligation, which are catalyzed
by three types of ubiquitination enzymes: Ubiquitin-activating
enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin
ligase (E3), respectively (67)
(Fig. 1C). Initially, E1 causes
the C-terminal adenylation of ubiquitin (Ub) to catalyze the
activation of ubiquitin in an ATP-dependent manner (68). The mature ubiquitin is then
transferred to cysteine at the active site of the E2 binding enzyme
via trans-thiesterification (58).
Finally, E3 and E2 jointly catalyze the formation of isopeptide
bonds between Ub and the substrate protein (69). Once attached to the target protein,
ubiquitin can be ubiquitinated on any of its lysine residues (K6,
K11, K27, K29, K33, K48, K63) or on its N-terminal methionine (M1).
The human proteome contains two E1s, ~50 E2s, and 600 E3s (70). Since largely determining substrate
specificity, E3 plays a key role in the entire process of
ubiquitination modification. E3s could be roughly divided into
three families: the HECT family, the RING family, and the RBR
family (64).
The E1-E2-E3 cascade is capable of producing several
types of Ub modifications, resulting in the different fates of
substrates (3). In general, two
types of ubiquitination modification are prevalent in cells:
Mono-ubiquitination and poly-ubiquitination (Fig. 1C). On the one hand,
mono-ubiquitination is the addition of a single Ub molecule to the
substrate. Poly-ubiquitination, by contrast, is the addition of Ub
chains to one or more lysine residues of the substrate (3). In most cases, membrane-bound proteins
are mono-ubiquitinated, which contributes to their endocytosis and
lysosomal degradation (31,71).
In addition, mono-ubiquitination is also involved in meiosis and
chromatin remodeling. However, poly-ubiquitination plays a role in
the ubiquitin-proteasome system (UPS), DNA repair, and immune
signaling transduction (72).
Ubiquitination modification of Notch1
receptor
Recent studies have suggested that the
ubiquitination modification of Notch1 receptor plays an
irreplaceable role in the regulation of Notch signaling (73-81).
The ubiquitinated Notch1 receptor has three distinct fates:
Transferring to the 26S proteasome, promoting N1ICD-mediated
signaling activation, and the endocytosis of Notch1 receptor. The
fate of the Notch1 receptor that transfers to the 26S proteasome is
degradation. However, the entry of Notch1 receptors into the
process of endocytosis has three different outcomes: Cycling back
to the cell membrane, becoming N1ICD and functioning in the nucleus
or being degraded in lysosomes.
Above all, the most important function of
ubiquitinated Notch1 is degradation in the 26S proteasome (Fig. 1A). The E3s that mediate this
process are mainly Sel-10, Fbxw7 and RNF8. When N1ICD enters the
nucleus, it forms complexes with MAML and CSL. Among them, MAML can
recruit CDK8 to phosphorylate the PEST domain of N1ICD.
Subsequently, Fbxw7, an E3, modifies the phosphorylated N1ICD for
poly-ubiquitination and then enters into the 26S proteasome for
degradation (12). Similarly, Wu
et al (82) demonstrated
that human Sel-10 (hSel-10) and Sel-10 bind N1ICD proteins in a
region-specific manner and that the interaction between Sel-10 and
N1ICD is phosphorylation-dependent. In vitro ubiquitination
modification experiments also revealed that Sel-10 and hSel-10
mediated ubiquitination modification of N1ICD, which were
subsequently degraded by the 26S proteasome in cells. As for RNF8,
it acts as a negative regulator of Notch signaling through
ubiquitination modification of N1ICD, leading to its degradation,
thereby regulating Notch1 signaling and cell fate determination in
lumen progenitor cells of the breast (83).
Another essential role of the ubiquitination
modification of Notch1 is the activation of Notch signaling.
Pettersson et al (11)
revealed that MDM2 also regulates Notch signaling through direct
interaction with N1ICD, leading to ubiquitination modification of
N1ICD. However, this type of ubiquitination modification does not
result in the degradation of N1ICD, but triggers the activation of
the Notch signaling pathway. In addition, MDM2 also interacts with
Notch regulator NUMB and induces its ubiquitination modification
and degradation (11).
With the exception of the Notch1 proteasomal
degradation and signaling activation, ubiquitination modification
also regulates the endocytosis of Notch1 (Fig. 1D). In the absence of a ligand,
Notch1 is continuously internalized and then degraded in lysosomes
(84), circulating back to the
plasma membrane (85,86) or activating Notch signaling. This
mechanism is a way to maintain Notch1 function and ultimately
regulate Notch signaling strength by targeting Notch1 levels on the
cell surface. Notch1 begins the process by its internalization in
the early endosome (EE) vesicles and then fuses with the EE. The EE
then matures and merges into a maturing endosome (ME). Finally,
multiple MEs fuse to form multivesicular bodies (MVBs). In this
step, Notch1 has the three distinct aforementioned fates,
specifically, it either returns to the membrane by circulating
endosomes, remains in MVBs, or activates the Notch1 signaling.
These different fates depend on the position of Notch1 in MVBs. If
Notch1 is present on the limiting membrane of MVBs, it can be
recycled, and when the part of Notch1 present on the MVB-limiting
membrane is cleaved to release N1ICD, Notch1 signaling can be
activated (31). However, Notch1
remaining in MVB interluminal vesicles (ILVs) can be further
degraded by lysosomes. MVB formation is controlled by endosomal
sorting complexes required for transport (ESCRT), a sequentially
acting macromolecular protein complex that ultimately allows ILV
formation (87,88). Mono-ubiquitination modification of
Notch1 has been revealed to be necessary for effective recruitment
to the endosomal membrane by the ESCRT machinery components and
formation of ILV (89). If the
ESCRT mechanical component is not functional, the
mono-ubiquitinated Notch1 accumulates on the limiting membrane of
MVBs, resulting in aberrant signaling activation. This suggests
that mono-ubiquitination modification may be directly or indirectly
involved in Notch endocytosis regulation and vesicular transport.
The E3s that mediate this process include Su(dx)/Itch/AIP4, Cbl,
NEDD4, and Deltex (DTX). The results of their effects depend on the
cell contexts, as well as their abundance. In addition,
Su(dx)/Itch/AIP4, DTX, and NEDD4 may also enable Notch1 to be
labeled by poly-ubiquitination modification and then degraded into
proteasome (73-81)
(Table I).
 | Table IE3s of Notch1 receptor. |
Table I
E3s of Notch1 receptor.
| E3 | Substrate | Species | E3-type | Ubiquitination | Effect | (Refs.) |
|---|
| Sel-10 | N1ICD |
Caenorhabditis elegans | RING | poly- | Proteasome
degradation | (73) |
| hSel-10 | N1ICD | Human | RING | poly- | Proteasome
degradation | (82) |
| Fbxw7 | N1ICD | Mammal | RING | poly- | Proteasome
degradation | (12) |
| RNF8 | N1ICD | Mammal | RING | poly- | Proteasome
degradation | (83) |
| MDM2 | N1ICD | Mammal | RING | mono- | Signaling
activation | (11) |
| Su(dx) | Notch1 |
Drosophila | HECT | mono- | Endocytosis | (74,75) |
| | | | | poly- | Proteasome
degradation | |
| Itch | Notch1 | Mammal | HECT | mono- | Endocytosis | (76,77) |
| | | | | poly- | Proteasome
degradation | |
| AIP4 | Notch1 | Human | HECT | mono- | Endocytosis | (85) |
| | | | | poly- | Proteasome
degradation | |
| NEDD4 | Notch1 |
Drosophila, | HECT | mono- | Endocytosis | (77,78) |
| | | mammal | | poly- | Proteasome
degradation | |
| DTX | Notch1 |
Melanogaster, mammal | RING | mono- | Endocytosis
(upgrade signaling) | (13,79,80) |
| | | | | poly- | Proteasome
degradation | |
| Cbl | Notch1 | Vertebrate | RING | mono- | Lysosomal
degradation | (16,81) |
| | | | | poly- | Proteasome
degradation | |
Effects on HSCs
Regulation of N1ICD through ubiquitination
modification is absolutely critical for proper Notch signaling, as
maintaining Notch signaling over long periods of time can lead to
severe diseases. For example, either a deletion of the
Notch1 gene, leading to a deletion of the PEST domain of
Notch, or a mutation in the Fbxw7 gene, encoding an inactive
or absent enzyme is associated with T-ALL (88). In the present study, three E3s were
focused on, all of which affect the stemness of HSCs through Notch1
receptor.
Cell cycle quiescence maintains the stemness of HSCs
by protecting them from differentiation or senescence (90). Fbxw7 can induce the degradation of
positive regulators such as myc and Notch1 in the cell cycle.
Iriuchishima et al (91)
revealed that Fbxw7 maintained HSCs and inhibited leukemia by
mediating ubiquitin-dependent degradation of myc and Notch1.
Thompson et al (92) also
demonstrated that the Fbxw7-/- severely affected
the maintenance of HPCs in the bone marrow, and the cell autonomy
defect of stem cell self-renewal led to the defect of HSC silencing
and self-renewal, which was attributed to the loss of the function
of Fbxw7 deletion to ubiquitination modification and
degradation of Notch1 or myc. Therefore, Fbxw7 serves as a key
fail-safe device to prevent premature loss of HSCs and the
development of T-ALL (93).
In addition, Cbl is a new negative regulator of HSC
development and functional characteristics. Rathinam et al
(94) determined that HSCs of
Cbl-/- mice had increased pool capacity,
increased proliferation, and increased long-term regeneration.
Furthermore, Zhu et al (16) revealed that flavone promoted
Cbl-induced ubiquitination modification and degradation of N1ICD,
resulting in resistance to T-ALL.
Ultimately, HSCs in Itch-/- mice
exhibited increased frequency, ability, and long-term regenerative
activity. Rathinam et al (95) demonstrated that Itch-deficient HSCs
exhibited accelerated proliferation rates and sustained progenitor
cell properties due to increased accumulation of Notch1 activation,
as well as increased Notch1 signaling by the transcription factor.
Therefore, E3 ubiquitin ligase Itch negatively regulates the
development and function of HSCs.
5. Clinical application
Multipotent stem cells, particularly HSCs and MSCs,
are widely used in clinical practice due to their characteristics
of self-renewal, multidirectional differentiation as well as
numerous others. For example, MSCs have paracrine,
anti-inflammatory, and immunomodulatory effects in addition to
their role in tissue regeneration (96). MSC-derived chambers or substances
(including exosomes, microvesicles, and microRNA) can serve as
practical tools for diagnosing, following up, managing, and
monitoring disease. In addition, Tehrani et al (97) suggested that MSCs could serve as a
vehicle for gene-directed enzyme prodrug therapy, in which suicide
genes are directed to tumor cells, attributing to their remarkable
homing properties to the tumor sites. Mirzaei et al
(98) considered that MSCs could
carry 5-fluorouracil, suicide genes such as pigment
epithelium-derived factor, INF-α, INF-β and
INF-γ to melanoma sites to inhibit tumor growth. More
specifically, interferon-γ-induced protein 10 kDa (IP-10) secreted
by human adipose-derived MSCs may be involved in this process
(99). In addition, it has been
gradually determined that this method of gene therapy can also be
applied to the treatment of osteoarthritis (100), cardiovascular disease (101) as well as other diseases, in
recent years. However, MSCs also secrete certain growth factors,
chemokines, and cytokines, which increase the burden of tumors, and
this may be the most important unresolved issue with this treatment
approach (102).
Since the Notch1 signaling pathway and its UPS
mainly affect the stemness of HSCs, attention should be paid to the
progression of HSCs in the treatment of leukemia, so as to provide
a better direction for the treatment of leukemia. HSC-related
therapies are gradually applied in the treatment of leukemia.
Transplantation of HSCs from bone marrow, peripheral blood or cord
blood is currently one of the most popular stem cell therapies in
blood system diseases (103). HSC
transplantation (HSCT) is a therapeutic method in which patients
receive a massive high-dose of radiotherapy or chemotherapy
(usually a lethal dose of radiotherapy or chemotherapy),
occasionally combined with other immunosuppressive drugs to remove
tumor cells and aberrant clonal cells in the body, and then
reinfuse the HSCs collected from the patients themselves or other
individuals to reconstruct normal hematopoietic and immune
functions (104). HSCT is widely
used in the treatment of hematological malignancies, such as acute
leukemia, CML, lymphoma, multiple myeloma (MM), myelodysplastic
syndrome (MDS), and certain hematological non-malignant tumors,
such as severe aplastic anemia (AA) and thalassemia (105). The process may be autologous
(using the patient's own cells), allogeneic (stem cells from a
donor), or syngeneic (from identical twins) (2). For numerous types of leukemia,
allogeneic HSCT (allo-HSCT) is a more suitable standard cell
treatment option than autologous HSCT (auto-HSCT) (106-110)
(Table II).
| Table IIHSCT. |
Table II
HSCT.
| Type | Recurrence rate at
100 days following HSCT | 5-Year survival
rate | Main risk factor
for late mortality | Indications for
malignant tumors | Indications for
other diseases | (Refs.) |
|---|
| Auto-HSCT | 57% | 88% | Relapse | MM, NHL, HL, AML,
ALL, neuroblastoma, ovarian cancer, germ-cell tumors, etc. | Autoimmune
disorders, amyloidosis, etc. | (106-109) |
| Allo-HSCT | 46% | 83% | Chronic GVHD | AML, ALL, CML NHL,
HL, CLL, MM, MDS, myeloproliferative disorders, etc. | AA, PNH, Fanconi's
anemia, sickle cell anemia, Wiskott-Aldrich syndrome, etc. | (107,108,110) |
In view of the fact that HSCs are derived from the
patients themselves during the process of auto-HSCT, there will be
no graft rejection and graft-vs.-host disease (GVHD) and there are
few transplantation complications. The low transplant-related
mortality and favorable quality of life following transplantation
are due to the no limitation of donor constraints. However, given
the lack of graft antitumor effect and the possibility of residual
tumor cells in the graft, the recurrence rate is high. Auto-HSCT
has become a routine treatment option for patients with lymphoma
(111), certain low-risk acute
leukemias (112), highly
invasive, relapsed/refractory non-Hodgkin's lymphoma (NHL)
(113) and MM (114). For example, the clinical efficacy
of auto-HSCT for AML has gradually improved. A group of European
researchers retrospectively analyzed the survival outcomes of 809
patients with AML in their first complete response and identified
that the 2-year leukemia-free survival rate and overall survival
rate were 51 and 65%, respectively, and the non-recurrence
mortality rate was only 3.7% (115). Taking it a step further, Passweg
(116) revealed that the 3-year
overall survival rate of AML was 34 (21-56)%
following chemotherapy, but 75 (60-95)%
following consolidation with auto-HSCT. In fact, a large number of
studies have revealed that auto-HSCT is associated with lower
recurrence rates and an acceptable non-recurrent mortality rate in
AML patients compared with chemotherapy alone (115). In addition, in certain AML
patients, auto-HSCT was comparable to allo-HSCT in overall survival
(116).
The HSCs in allo-HSCT are derived from normal
donors without tumor cell contamination. Considering the
immune-antitumor effect of the graft, it has a low recurrence rate,
a high long-term disease-free survival rate (also known as cure
rate), a wide range of indications, and is even the only cure for
certain diseases (114). However,
due to the limited sources of donors, GVHD is prone to occur with
numerous transplant complications, leading to high graft-related
mortality. Therefore, patients need to be treated with
immunosuppressants for a long period of time, and the quality of
life of long-term survivors may be poor. Patients at moderate or
high risk for acute leukemia (117), AML (118), MDS (119), severe AA (120), and thalassemia (121) are suitable for allo-HSCT
(122). To date, allo-HSCT
remains the only radical treatment for CML. Allo-HSCT is exhibiting
better results in the treatment of CML due to improved HLA
gene matching techniques, the use of tyrosine kinase inhibitors,
advances in postoperative immune status and fusion gene monitoring
and improvements in postoperative complications, particularly GVHD
(123). Similar to CML, allo-HSCT
is effective in alleviating highly complex and severe AML. However,
relapse is a major cause of treatment failure for AML patients
undergoing allo-HSCT. Therefore, an effective and safe approach is
required, in the future, to improve survival following remission of
AML (117). In addition, a female
patient with adult T-cell leukemia/lymphoma involving bone, skin,
and skeletal muscle exhibited a favorable post-transplant course
after receiving cyclophosphamide following allo-HSCT from her son
in a clinical case report (124).
There has been no progression of disease for more than two years,
suggesting that this approach offers a well-tolerated and
potentially curable treatment for this hard-to-treat disease
(124). Given the high toxicity
of this treatment, graft-anti-leukemia response and the high
recurrence and mortality rate, novel post-transplantation
maintenance regimens need to be studied.
In a series of studies, it was revealed that the
Notch1/Fbxw7 mutations in T-ALL patients may be useful
biomarkers for predicting the prognosis of T-ALL. In a survey of 50
patients with T-ALL in southern India, there were 20 out of the 50
(40%) patients with Notch1/Fbxw7 mutations among which the
13 out of the 20 (65%) T-ALL patients with Notch1/Fbxw7
mutations exhibited favorable prednisone responses (P=0.01) and
improved clinical outcomes compared with patients without
Notch1/Fbxw7 mutations (P=0.03) (125). In the survival analysis of the
sample (n=50) studied by Valliyammai et al (126), it was determined that patients
with Notch1/Fbxw7 hotspot mutation had earlier response to
treatment and improved survival. Additionally, it was suggested
that Notch1/Fbxw7 hotspot-mutated T-ALL cases responded
better to the ALL BFM-95 protocol. Furthermore, pediatric T-ALL
patients with either double Notch1 mutations
(Notch1DoubleFbxw7WT) or mutations in
both genes (Notch1MUTFbxw7MUT),
hereafter termed as Notch1±Fbxw7Double,
had an improved outcome (127).
Jenkinson et al (127)
screened 162 pediatric T-ALL patients treated in the MRC UKALL2003
trial for Notch1/Fbxw7 gene mutations and associated
genotypes in response to treatment and long-term outcomes. Of the
162 patients, 57 (35%) patients were both Notch1 and
Fbxw7 wild-type, 62 (38%) patients had single Notch1
mutations, 5 (3%) patients had single Fbxw7 mutations, and
39 (24%) patients had Notch1±Fbxw7Double.
It was revealed that while 14
Notch1±Fbxw7Double patients were
classified as high risk, only 2 patients progressed in disease and
all survived. Collectively, these data suggested that detecting the
Fbxw7 mutations adds important prognostic value to the
separate assessment of Notch1 status, justifies individual
treatment stratification of T-ALL (128), and allows the identification of
the majority (72%) of Notch1/Fbxw7-mutated T-ALL patients
with a relatively favorable prognosis, who cannot be treated with
more classical, clinical, immunophenotypic or carcinogenic markers
(128). Conversely, loss of
Fbxw7 in primary T-ALL has also been reported to provide a
favorable prognosis for patients. Loss of Fbxw7 reduces
ubiquitination modification and degradation of glucocorticoid
receptor α, thus enhancing glucocorticoid sensitivity. This
increased sensitivity can enhance glucocorticoid response to
treatment and provide a favorable prognosis for T-ALL (129).
In addition, several studies identified Cbl and
Fbxw7 as new targets for anti-Notch1 therapy. Saito et al
(130) revealed that flavonoids
induced N1ICD degradation through the UPS by increasing Cbl in
T-ALL. Flavonoid-induced resistance to T-ALL was also revealed, and
Cbl was identified as a new N1ICD binding partner critical for
regulating its stability and carcinogenic function. In the case of
Fbxw7, oridonin has exhibited an anti-leukemia activity in
vitro and in vivo by promoting Fbxw7-mediated
ubiquitination modification and degradation of myc (131). These studies suggest that
flavonoid and oridonin are potential drugs for T-ALL.
6. Conclusions and perspectives
Notch signaling, particularly Notch1 receptor, is
the primary regulator of HSC stemness in embryos and adulthood, and
its role in inducing leukemia (e.g., T-ALL) has been detailed in a
variety of studies (52-55,132).
In general, Notch1 can promote the proliferation of HSCs and
inhibit its differentiation (133). However, due to the context
dependence of the Notch signaling pathway and activation by
different ligands, Notch1 receptor can also partially inhibit the
proliferation of HSCs and promote their differentiation.
Additionally, the lifetime and activity of the Notch1 receptor is
largely determined by the UPS that regulates Notch1 receptor
degradation, activation of Notch1 signaling, and Notch1 receptor
endocytosis and its subsequent fate. Additionally, the signaling
enhancement or mutations of the Notch1 pathway and the
dysregulations or mutations of Notch1-related UPS have been
demonstrated to be closely associated with the aberrancy of HSCs
and the occurrence of leukemia (134). Therefore, further revealing the
details of this pathway and the factors that regulate the UPS could
help improve the treatment and prognosis of leukemia.
Since the present review is limited to the effects
of Notch1 receptor and its ubiquitination modification on HSCs,
other receptors and ligands of the Notch signaling pathway, or
other regulatory modes of this pathway such as phosphorylation,
require clarification. In addition, several studies have revealed
that Notch signaling interacts closely with other signaling
pathways, such as the Wnt, hippo, TGF-β family and Hedgehog that
regulate stem cell properties, but these associations have not been
well elucidated (135-139).
Therefore, future research may also focus on the interactions or
crosstalk between these pathways.
Currently, Notch1 and Fbxw7 are mainly used as
prognostic indicators of T-ALL (140). However, studies on these two
proteins and their regulators as treatments for leukemia are
urgent. In particular, their application as therapeutic targets for
leukemia or in combination with other chemotherapeutic agents
require further study. In addition, it is necessary to examine
whether they can be used as prognostic indicators for auto-HSCT or
allo-HSCT.
Acknowledgements
Not applicable.
Funding
Funding: The present review was supported by The Fundamental
Research Funds for the Provincial Universities of Zhejiang, The
Natural Science Foundation of Zhejiang Province (grant no.
LY20C070001), The National Natural Science Foundation of China
(grant no. 31801165), and The K.C. Wong Magna Fund of Ningbo
University.
Availability of data and materials
Not applicable.
Authors' contributions
XJ, MY, YG, HZ, JW and JL made substantial
contributions to conception and design. XJ, YG, HZ, JW and JL were
involved in drafting the manuscript and revising it critically for
important intellectual content. XJ, MY, YG, HZ, JW and JL agreed to
be accountable for all aspects of the work. All authors read and
approved the final manuscript for publication. Data authentication
is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nawab K, Bhere D, Bommarito A, Mufti M and
Naeem A: Stem cell therapies: A way to promising cures. Cureus.
11(e5712)2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Zakrzewski W, Dobrzynski M, Szymonowicz M
and Rybak Z: Stem cells: Past, present, and future. Stem Cell Res
Ther. 10(68)2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wang D, Bu F and Zhang W: The role of
Ubiquitination in regulating embryonic stem cell maintenance and
cancer development. Int J Mol Sci. 20(2667)2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wang X: Stem cells in tissues, organoids,
and cancers. Cell Mol Life Sci. 76:4043–4070. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Aponte PM and Caicedo A: Stemness in
cancer: Stem cells, cancer stem cells, and their microenvironment.
Stem Cells Int. 2017(5619472)2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ruan Y, Kim HN, Ogana H and Kim YM: Wnt
signaling in leukemia and its bone marrow microenvironment. Int J
Mol Sci. 21(6247)2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Roo JJD and Staal FJT: Cell signaling
pathway reporters in adult hematopoietic stem cells. Cells.
9(2264)2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Althoff MJ, Nayak RC, Hegde S, Wellendorf
AM, Bohan B, Filippi MD, Xin M, Lu QR, Geiger H, Zheng Y, et al:
Yap1-Scribble polarization is required for hematopoietic stem cell
division and fate. Blood. 136:1824–1836. 2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Pajcini KV, Speck NA and Pear WS: Notch
signaling in mammalian hematopoietic stem cells. Leukemia.
25:1525–1532. 2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Palomero T and Ferrando A: Oncogenic
NOTCH1 control of MYC and PI3K: Challenges and opportunities for
anti-NOTCH1 therapy in T-cell acute lymphoblastic leukemias and
lymphomas. Clin Cancer Res. 14:5314–5317. 2008.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Pettersson S, Sczaniecka M, McLaren L,
Russell F, Gladstone K, Hupp T and Wallace M: Non-degradative
ubiquitination of the Notch1 receptor by the E3 ligase MDM2
activates the Notch signalling pathway. Biochem J. 450:523–536.
2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kar R, Jha SK, Ojha S, Sharma A, Dholpuria
S, Raju VSR, Prasher P, Chellappan DK, Gupta G, Kumar Singh S, et
al: The FBXW7-NOTCH interactome: A ubiquitin proteasomal
system-induced crosstalk modulating oncogenic transformation in
human tissues. Cancer Rep (Hoboken). 4(e1369)2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hori K, Sen A, Kirchhausen T and
Artavanis-Tsakonas S: Synergy between the ESCRT-III complex and
Deltex defines a ligand-independent Notch signal. J Cell Biol.
195:1005–1015. 2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
King B, Trimarchi T, Reavie L, Xu L,
Mullenders J, Ntziachristos P, Aranda-Orgilles B, Perez-Garcia A,
Shi J, Vakoc C, et al: The ubiquitin ligase FBXW7 modulates
leukemia-initiating cell activity by regulating MYC stability.
Cell. 153:1552–1566. 2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Thompson BJ, Buonamici S, Sulis ML,
Palomero T, Vilimas T, Basso G, Ferrando A and Aifantis I: The
SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell
leukemia. J Exp Med. 204:1825–1835. 2007.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhu W, Zhu Y, Xu H, Wang T, Wang J, Meng
M, Liu Y, Yan H, Yang Q and Liu P: Flavone inhibited proliferation
of T-ALL by promoting c-Cbl-induced ubiquitinylation and
degradation of Notch1. Biochem Biophys Res Commun. 522:684–689.
2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zieba JT, Chen YT, Lee BH and Bae Y: Notch
signaling in skeletal development, homeostasis and pathogenesis.
Biomolecules. 10(332)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chikara S and Reindl KM: Notch signaling:
A hero or villain in the war against cancer? Transl Lung Cancer
Res. 2:449–451. 2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Jundt F, Anagnostopoulos I, Forster R,
Mathas S, Stein H and Dorken B: Activated Notch1 signaling promotes
tumor cell proliferation and survival in Hodgkin and anaplastic
large cell lymphoma. Blood. 99:3398–3403. 2002.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Li L, Tang P, Li S, Qin X, Yang H, Wu C
and Liu Y: Notch signaling pathway networks in cancer metastasis: A
new target for cancer therapy. Med Oncol. 34(180)2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Espinoza I, Pochampally R, Xing F, Watabe
K and Miele L: Notch signaling: Targeting cancer stem cells and
epithelial-to-mesenchymal transition. Onco Targets Ther.
6:1249–1259. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Wu J and Bresnick EH: Bare rudiments of
notch signaling: How receptor levels are regulated. Trends Biochem
Sci. 32:477–485. 2007.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Meloty-Kapella L, Shergill B, Kuon J,
Botvinick E and Weinmaster G: Notch ligand endocytosis generates
mechanical pulling force dependent on dynamin, epsins, and actin.
Dev Cell. 22:1299–1312. 2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Fischer A and Gessler M: Delta-Notch-and
then? Protein interactions and proposed modes of repression by Hes
and Hey bHLH factors. Nucleic Acids Res. 35:4583–4596.
2007.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Bessho Y, Sakata R, Komatsu S, Shiota K,
Yamada S and Kageyama R: Dynamic expression and essential functions
of Hes7 in somite segmentation. Genes Dev. 15:2642–2647.
2001.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Fischer A, Schumacher N, Maier M, Sendtner
M and Gessler M: The Notch target genes Hey1 and Hey2 are required
for embryonic vascular development. Genes Dev. 18:901–911.
2004.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Dohda T, Maljukova A, Liu L, Heyman M,
Grandér D, Brodin D, Sangfelt O and Lendahl U: Notch signaling
induces SKP2 expression and promotes reduction of p27Kip1 in T-cell
acute lymphoblastic leukemia cell lines. Exp Cell Res.
313:3141–3152. 2007.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Ronchini C and Capobianco AJ: Induction of
cyclin D1 transcription and CDK2 activity by Notch(ic): Implication
for cell cycle disruption in transformation by Notch(ic). Mol Cell
Biol. 21:5925–5934. 2001.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Demarest RM, Dahmane N and Capobianco AJ:
Notch is oncogenic dominant in T-cell acute lymphoblastic leukemia.
Blood. 117:2901–2909. 2011.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhang P, Yang Y, Nolo R, Zweidler-McKay PA
and Hughes DPM: Regulation of NOTCH signaling by reciprocal
inhibition of HES1 and Deltex 1 and its role in osteosarcoma
invasiveness. Oncogene. 29:2916–2926. 2010.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Dutta D, Sharma V, Mutsuddi M and
Mukherjee A: Regulation of Notch signaling by E3 ubiquitin ligases.
FEBS J: Feb 28, 2021 (Epubs ahead of print). doi:
10.1111/febs.15792.
|
|
32
|
Seita J and Weissman IL: Hematopoietic
stem cell: Self-renewal versus differentiation. Wiley Interdiscip
Rev Syst Biol Med. 2:640–653. 2010.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Sinka L, Biasch K, Khazaal I, Peault B and
Tavian M: Angiotensin-converting enzyme (CD143) specifies emerging
lympho-hematopoietic progenitors in the human embryo. Blood.
119:3712–3723. 2012.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Suzuki T and Chiba S: Notch signaling in
hematopoietic stem cells. Int J Hematol. 82:285–294.
2005.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Saito K, Nobuhisa I, Harada K, Takahashi
S, Anani M, Lickert H, Kanai-Azuma M, Kanai Y and Taga T:
Maintenance of hematopoietic stem and progenitor cells in fetal
intra-aortic hematopoietic clusters by the Sox17-Notch1-Hes1 axis.
Exp Cell Res. 365:145–155. 2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zhang P, He Q, Chen D, Liu W, Wang L,
Zhang C, Ma D, Li W, Liu B and Liu F: G protein-coupled receptor
183 facilitates endothelial-to-hematopoietic transition via Notch1
inhibition. Cell Res. 25:1093–1107. 2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Kumano K, Chiba S, Kunisato A, Sata M,
Saito T, Nakagami-Yamaguchi E, Yamaguchi T, Masuda S, Shimizu K,
Takahashi T, et al: Notch1 but Not Notch2 is essential for
generating hematopoietic stem cells from endothelial cells.
Immunity. 18:699–711. 2003.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Gama-Norton L, Ferrando E, Ruiz-Herguido
C, Liu Z, Guiu J, Islam AB, Lee SU, Yan M, Guidos CJ, López-Bigas
N, et al: Notch signal strength controls cell fate in the
haemogenic endothelium. Nat Commun. 6(8510)2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Jang IH, Lu YF, Zhao L, Wenzel PL, Kume T,
Datta SM, Arora N, Guiu J, Lagha M, Kim PG, et al: Notch1 acts via
Foxc2 to promote definitive hematopoiesis via effects on hemogenic
endothelium. Blood. 125:1418–1426. 2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Stier S, Cheng T, Dombkowski D, Carlesso N
and Scadden DT: Notch1 activation increases hematopoietic stem cell
self-renewal in vivo and favors lymphoid over myeloid lineage
outcome. Blood. 99:2369–2378. 2002.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Shao L, Paik NY and Pajcini KV:
Hematopoietic jagged1 is required for the transition of
hematopoietic stem cells from the fetal liver to the adult bone
marrow niche. Blood. 136:10–11. 2020.
|
|
42
|
Ishiko E, Matsumura I, Ezoe S, Gale K,
Ishiko J, Satoh Y, Tanaka H, Shibayama H, Mizuki M, Era T, et al:
Notch signals inhibit the development of erythroid/megakaryocytic
cells by suppressing GATA-1 activity through the induction of HES1.
J Biol Chem. 280:4929–4939. 2005.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Henning K, Heering J, Schwanbeck R,
Schroeder T, Helmbold H, Schäfer H, Deppert W, Kim E and Just U:
Notch1 activation reduces proliferation in the multipotent
hematopoietic progenitor cell line FDCP-mix through a p53-dependent
pathway but Notch1 effects on myeloid and erythroid differentiation
are independent of p53. Cell Death Differ. 15:398–407.
2008.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Yashiro-Ohtani Y, Ohtani T and Pear WS:
Notch regulation of early thymocyte development. Semin Immunol.
22:261–269. 2010.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Allman D, Karnell FG, Punt JA, Bakkour S,
Xu L, Myung P, Koretzky GA, Pui JC, Aster JC and Pear WS:
Separation of Notch1 promoted lineage commitment and
expansion/transformation in developing T cells. J Exp Med.
194:99–106. 2001.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Gerhardt DM, Pajcini KV, D'Altri T, Tu L,
Jain R, Xu L, Chen MJ, Rentschler S, Shestova O, Wertheim GB, et
al: The Notch1 transcriptional activation domain is required for
development and reveals a novel role for Notch1 signaling in fetal
hematopoietic stem cells. Genes Dev. 28:576–593. 2014.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Deftos ML and Bevan MJ: Notch signaling in
T cell development. Curr Opin Immunol. 12:166–172. 2000.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Tomita K, Hattori M, Nakamura E, Nakanishi
S, Minato N and Kageyama R: The bHLH gene Hes1 is essential for
expansion of early T cell precursors. Genes Dev. 13:1203–1210.
1999.PubMed/NCBI View Article : Google Scholar
|
|
49
|
De Decker M, Lavaert M, Roels J, Tilleman
L, Vandekerckhove B, Leclercq G, Van Nieuwerburgh F, Van
Vlierberghe P and Taghon T: HES1 and HES4 have non-redundant roles
downstream of Notch during early human T-cell development.
Haematologica. 106:130–141. 2021.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Haque R and Song J, Haque M, Lei F, Sandhu
P, Ni B, Zheng S, Fang D, Yang JM and Song J: c-Myc-Induced
survivin is essential for promoting the Notch-dependent T cell
differentiation from hematopoietic stem cells. Genes (Basel).
8(97)2017.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Van de Walle I, De Smet G, Gärtner M, De
Smedt M, Waegemans E, Vandekerckhove B, Leclercq G, Plum J, Aster
JC, Bernstein ID, et al: Jagged2 acts as a Delta-like Notch ligand
during early hematopoietic cell fate decisions. Blood.
117:4449–4459. 2011.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Wendorff AA and Ferrando AA: Modeling
NOTCH1 driven T-cell acute lymphoblastic leukemia in mice. Bio
Protoc. 10(e3620)2020.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Ma W, Gutierrez A, Goff DJ, Geron I,
Sadarangani A, Jamieson CA, Court AC, Shih AY, Jiang Q, Wu CC, et
al: NOTCH1 signaling promotes human T-cell acute lymphoblastic
leukemia initiating cell regeneration in supportive niches. PLoS
One. 7(e39725)2012.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Bhanushali AA, Babu S, Thangapandi VR,
Pillai R, Chheda P and Das BR: Mutations in the HD and PEST domain
of Notch-1 receptor in T-cell acute lymphoblastic leukemia: Report
of novel mutations from Indian population. Oncol Res. 19:99–104.
2010.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Ding J, Cardoso AA, Yoshimoto M and
Kobayashi M: The Earliest T-Precursors in the mouse embryo are
susceptible to leukemic transformation. Front Cell Dev Biol.
9(634151)2021.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Di Ianni M, Baldoni S, Del Papa B, Aureli
P, Dorillo E, De Falco F, Albi E, Varasano E, Di Tommaso A,
Giancola R, et al: NOTCH1 is aberrantly activated in chronic
lymphocytic leukemia hematopoietic stem cells. Front Oncol.
8(105)2018.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Spit M, Rieser E and Walczak H: Linear
ubiquitination at a glance. J Cell Sci.
132(jcs208512)2019.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Mansour MA: Ubiquitination: Friend and foe
in cancer. Int J Biochem Cell Biol. 101:80–93. 2018.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Martins S, Dohmann EMN, Cayrel A, Johnson
A, Fischer W, Pojer F, Satiat-Jeunemaître B, Jaillais Y, Chory J,
Geldner N and Vert G: Author Correction: Internalization and
vacuolar targeting of the brassinosteroid hormone receptor BRI1 are
regulated by ubiquitination. Nat Commun. 12(2982)2021.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Barbosa P, Zhaunova L, Debilio S,
Steccanella V, Kelly V, Ly T and Ohkura H: SCF-Fbxo42 promotes
synaptonemal complex assembly by downregulating PP2A-B56. J Cell
Biol. 220(e202009167)2021.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Li ZQ, Chen X and Wang Y: Small molecules
targeting ubiquitination to control inflammatory diseases. Drug
Discov Today. 26:2414–2422. 2021.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Dybas JM, Herrmann C and Weitzman MD:
Ubiquitination at the interface of tumor viruses and DNA damage
responses. Curr Opin Virol. 32:40–47. 2018.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Seo J, Kim MW, Bae KH, Lee SC, Song J and
Lee EW: The roles of ubiquitination in extrinsic cell death
pathways and its implications for therapeutics. Biochem Pharmacol.
162:21–40. 2019.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Sun T, Liu Z and Yang Q: The role of
ubiquitination and deubiquitination in cancer metabolism. Mol
Cancer. 19(146)2020.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Zuo Y, Feng Q, Jin L, Huang F, Miao Y, Liu
J, Xu Y, Chen X, Zhang H, Guo T, et al: Regulation of the linear
ubiquitination of STAT1 controls antiviral interferon signaling.
Nat Commun. 11(1146)2020.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Liu Y and Deng J:
Ubiquitinationdeubiquitination in the Hippo signaling pathway
(Review). Oncol Rep. 41:1455–1475. 2019.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Soysouvanh F, Giuliano S, Habel N,
El-Hachem N, Pisibon C, Bertolotto C and Ballotti R: An Update on
the role of ubiquitination in melanoma development and therapies. J
Clin Med. 10(113)2021.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Jeusset LM and McManus KJ: Developing
targeted therapies that exploit aberrant histone ubiquitination in
cancer. Cells. 8(165)2019.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Celebi G, Kesim H, Ozer E and Kutlu O: The
effect of dysfunctional ubiquitin enzymes in the pathogenesis of
most common diseases. Int J Mol Sci. 21(6335)2020.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Liu BQ, Jin J and Li YY: Ubiquitination
modification: Critical regulation of IRF family stability and
activity. Sci China Life Sci. 64:957–965. 2021.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Chen RH, Chen YH and Huang TY:
Ubiquitin-mediated regulation of autophagy. J Biomed Sci.
26(80)2019.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Song YQ, Wu C, Wu KJ, Han QB, Miao XM, Ma
DL and Leung CH: Ubiquitination regulators discovered by virtual
screening for the treatment of cancer. Front Cell Dev Biol.
9(665646)2021.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Gupta-Rossi N, Le Bail O, Gonen H, Brou C,
Logeat F, Six E, Ciechanover A and Israël A: Functional interaction
between SEL-10, an F-box protein, and the nuclear form of activated
Notch1 receptor. J Biol Chem. 276:34371–34378. 2001.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Fostier M, Evans DA, Artavanis-Tsakonas S
and Baron M: Genetic characterization of the Drosophila
melanogaster Suppressor of deltex gene: A regulator of notch
signaling. Genetics. 150:1477–1485. 1998.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Cornell M, Evans DA, Mann R, Fostier M,
Flasza M, Monthatong M, Artavanis-Tsakonas S and Baron M: The
Drosophila melanogaster Suppressor of deltex gene, a regulator of
the Notch receptor signaling pathway, is an E3 class ubiquitin
ligase. Genetics. 152:567–576. 1999.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Qiu L, Joazeiro C, Fang N, Wang HY, Elly
C, Altman Y, Fang D, Hunter T and Liu YC: Recognition and
ubiquitination of Notch by Itch, a hect-type E3 ubiquitin ligase. J
Biol Chem. 275:35734–35737. 2000.PubMed/NCBI View Article : Google Scholar
|
|
77
|
McGill MA and McGlade CJ: Mammalian numb
proteins promote Notch1 receptor ubiquitination and degradation of
the Notch1 intracellular domain. J Biol Chem. 278:23196–23203.
2003.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Che F, Chen J, Wan C and Huang X:
MicroRNA-27 inhibits autophagy and promotes proliferation of
multiple myeloma cells by targeting the NEDD4/Notch1 Axis. Front
Oncol. 10(571914)2020.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Baron M: Endocytic routes to Notch
activation. Semin Cell Dev Biol. 23:437–442. 2012.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Wilkin M, Tongngok P, Gensch N, Clemence
S, Motoki M, Yamada K, Hori K, Taniguchi-Kanai M, Franklin E,
Matsuno K and Baron M: Drosophila HOPS and AP-3 complex genes are
required for a Deltex-regulated activation of notch in the
endosomal trafficking pathway. Dev Cell. 15:762–772.
2008.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Jehn BM, Dittert I, Beyer S, von der Mark
K and Bielke W: c-Cbl binding and ubiquitin-dependent lysosomal
degradation of membrane-associated Notch1. J Biol Chem.
277:8033–8040. 2002.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Wu G, Lyapina S, Das I, Li J, Gurney M,
Pauley A, Chui I, Deshaies RJ and Kitajewski J: SEL-10 is an
inhibitor of notch signaling that targets notch for
ubiquitin-mediated protein degradation. Mol Cell Biol.
21:7403–7415. 2001.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Li L, Guturi KKN, Gautreau B, Patel PS,
Saad A, Morii M, Mateo F, Palomero L, Barbour H, Gomez A, et al:
Ubiquitin ligase RNF8 suppresses Notch signaling to regulate
mammary development and tumorigenesis. J Clin Invest.
128:4525–4542. 2018.PubMed/NCBI View Article : Google Scholar
|
|
84
|
McGill MA, Dho SE, Weinmaster G and
McGlade CJ: Numb regulates post-endocytic trafficking and
degradation of Notch1. J Biol Chem. 284:26427–26438.
2009.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Chastagner P, Israel A and Brou C:
AIP4/Itch regulates Notch receptor degradation in the absence of
ligand. PLoS One. 3(e2735)2008.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Sakata T, Sakaguchi H, Tsuda L,
Higashitani A, Aigaki T, Matsuno K and Hayashi S: Drosophila Nedd4
regulates endocytosis of notch and suppresses its
ligand-independent activation. Curr Biol. 14:2228–2236.
2004.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Henne WM, Buchkovich NJ and Emr SD: The
ESCRT pathway. Dev Cell. 21:77–91. 2011.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Moretti J and Brou C: Ubiquitinations in
the notch signaling pathway. Int J Mol Sci. 14:6359–6381.
2013.PubMed/NCBI View Article : Google Scholar
|
|
89
|
MacDonald C, Buchkovich NJ, Stringer DK,
Emr SD and Piper RC: Cargo ubiquitination is essential for
multivesicular body intralumenal vesicle formation. EMBO Rep.
13:331–338. 2012.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Perry JM and Li L: Self-renewal versus
transformation: Fbxw7 deletion leads to stem cell activation and
leukemogenesis. Genes Dev. 22:1107–1109. 2008.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Iriuchishima H, Takubo K, Matsuoka S,
Onoyama I, Nakayama KI, Nojima Y and Suda T: Ex vivo maintenance of
hematopoietic stem cells by quiescence induction through Fbxw7α
overexpression. Blood. 117:2373–2377. 2011.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Thompson BJ, Jankovic V, Gao J, Buonamici
S, Vest A, Lee JM, Zavadil J, Nimer SD and Aifantis I: Control of
hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7.
J Exp Med. 205:1395–1408. 2008.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Matsuoka S, Oike Y, Onoyama I, Iwama A,
Arai F, Takubo K, Mashimo Y, Oguro H, Nitta E, Ito K, et al: Fbxw7
acts as a critical fail-safe against premature loss of
hematopoietic stem cells and development of T-ALL. Genes Dev.
22:986–991. 2008.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Rathinam C, Thien CB, Langdon WY, Gu H and
Flavell RA: The E3 ubiquitin ligase c-Cbl restricts development and
functions of hematopoietic stem cells. Genes Dev. 22:992–997.
2008.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Rathinam C, Matesic LE and Flavell RA: The
E3 ligase Itch is a negative regulator of the homeostasis and
function of hematopoietic stem cells. Nat Immunol. 12:399–407.
2011.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Asgarpour K, Shojaei Z, Amiri F, Ai J,
Mahjoubin-Tehran M, Ghasemi F, ArefNezhad R, Hamblin MR and Mirzaei
H: Exosomal microRNAs derived from mesenchymal stem cells:
Cell-to-cell messages. Cell Commun Signal. 18(149)2020.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Moradian Tehrani R, Verdi J, Noureddini M,
Salehi R, Salarinia R, Mosalaei M, Simonian M, Alani B, Ghiasi MR,
Jaafari MR, et al: Mesenchymal stem cells: A new platform for
targeting suicide genes in cancer. J Cell Physiol. 233:3831–3845.
2018.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Mirzaei H, Sahebkar A, Avan A, Jaafari MR,
Salehi R, Salehi H, Baharvand H, Rezaei A, Hadjati J, Pawelek JM
and Mirzaei HR: Application of mesenchymal stem cells in melanoma:
A potential therapeutic strategy for delivery of targeted agents.
Curr Med Chem. 23:455–463. 2016.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Mirzaei H, Salehi H, Oskuee RK,
Mohammadpour A, Mirzaei HR, Sharifi MR, Salarinia R, Darani HY,
Mokhtari M, Masoudifar A, et al: The therapeutic potential of human
adipose-derived mesenchymal stem cells producing CXCL10 in a mouse
melanoma lung metastasis model. Cancer Lett. 419:30–39.
2018.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Mianehsaz E, Mirzaei HR, Mahjoubin-Tehran
M, Rezaee A, Sahebnasagh R, Pourhanifeh MH, Mirzaei H and Hamblin
MR: Mesenchymal stem cell-derived exosomes: A new therapeutic
approach to osteoarthritis? Stem Cell Res Ther.
10(340)2019.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Goradel NH, Hour FG, Negahdari B,
Malekshahi ZV, Hashemzehi M, Masoudifar A and Mirzaei H: Stem cell
therapy: A new therapeutic option for cardiovascular diseases. J
Cell Biochem. 119:95–104. 2018.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Mohammadi M, Jaafari MR, Mirzaei HR and
Mirzaei H: Mesenchymal stem cell: a new horizon in cancer gene
therapy. Cancer Gene Ther. 23:285–286. 2016.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Hatzimichael E and Tuthill M:
Hematopoietic stem cell transplantation. Stem Cells Cloning.
3:105–117. 2010.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Walsh K, Margossian S and Duncan CN:
Pediatric hematopoietic cell transplantation. J Pediatr Intensive
Care. 3:91–101. 2014.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Dessie G, Derbew Molla M, Shibabaw T and
Ayelign B: Role of stem-cell transplantation in leukemia treatment.
Stem Cells Cloning. 13:67–77. 2020.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Vanderwalde AM, Sun CL, Laddaran L,
Francisco L, Armenian S, Berano-Teh J, Wong FL, Popplewell L, Somlo
G, Stein AS, et al: Conditional survival and cause-specific
mortality after autologous hematopoietic cell transplantation for
hematological malignancies. Leukemia. 27:1139–1145. 2013.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Wang Y, Chen H, Chen J, Han M, Hu J, Jiong
Hu, Huang H, Lai Y, Liu D, Liu Q, et al: The consensus on the
monitoring, treatment, and prevention of leukemia relapse after
allogeneic hematopoietic stem cell transplantation in China. Cancer
Lett. 438:63–75. 2018.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Copelan EA: Medical progress:
Hematopoietic stem-cell transplantation. N Engl J Med.
354:1813–1826. 2006.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Doubek M, Folber F, Koristek Z, Brychtova
Y, Krejci M, Tomiska M, Navratil M, Mikulasova P and Mayer J:
Autologous hematopoietic stem cell transplantation in adult acute
lymphoblastic leukemia: Still not out of fashion. Ann Hematol.
88:881–887. 2009.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Atsuta Y, Hirakawa A, Nakasone H, Kurosawa
S, Oshima K, Sakai R, Ohashi K, Takahashi S, Mori T, Ozawa Y, et
al: Late mortality and causes of death among long-term survivors
after allogeneic stem cell transplantation. Biol Blood Marrow
Transplant. 22:1702–1709. 2016.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Wang X, Xia B and Zhang YZ: Progress of
Auto-HSCT for treatment of DLBCL-review. Zhongguo Shi Yan Xue Ye
Xue Za Zhi. 26:1841–1846. 2018.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
112
|
Alimoghaddam K, Ghavamzadeh A, Jahani M,
Jalali A, Jorjani H, Iravani M, Hamidieh AA, Mousavi A, Bahar B,
Behfar M, et al: Hematopoietic stem cell transplantation in acute
promyelocytic leukemia, experience in Iran. Arch Iran Med.
14:332–334. 2011.PubMed/NCBI
|
|
113
|
Zhu J: Thoughts on autologous
hematopoietic stem cell transplantation and mobilization in Chinese
patients with non Hodgkin's lymphoma. Zhonghua Xue Ye Xue Za Zhi.
41:1–4. 2020.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
114
|
Liu JR, Li J and Huang XJ: Problems and
progress of autologous hematopoietic stem cell transplantation in
multiple myeloma. Zhonghua Xue Ye Xue Za Zhi. 42:82–86.
2021.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
115
|
Zhao Y, Chen X and Feng S: Autologous
hematopoietic stem cell transplantation in acute Myelogenous
leukemia. Biol Blood Marrow Transplant. 25:e285–e292.
2019.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Passweg JR: Autologous hematopoetic cell
transplantation: Preferred consolidation treatment in low-risk AML?
Leuk Lymphoma. 60:2341–2342. 2019.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Xuan L and Liu Q: Maintenance therapy in
acute myeloid leukemia after allogeneic hematopoietic stem cell
transplantation. J Hematol Oncol. 14(4)2021.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Yong AS, Brissot E, Rubinstein S, Savani
BN and Mohty M: Transplant to treatment-free remission: The
evolving view of ‘cure’ in chronic myeloid leukemia. Expert Rev
Hematol. 8:785–797. 2015.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Kook MH, Yhim HY, Lee NR, Song EK, Kim HS,
Yim CY and Kwak JY: Successful treatment of myelodysplastic
syndrome and Behcet colitis after allogeneic hematopoietic stem
cell transplantation. Korean J Intern Med. 29:123–125.
2014.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Chen X and Feng SZ: Advances in allogeneic
hematopoietic stem cell transplantation for severe aplastic anemia.
Zhonghua Xue Ye Xue Za Zhi. 33:963–967. 2012.PubMed/NCBI(In Chinese).
|
|
121
|
Peters C: Allogeneic hematopoietic stem
cell transplantation to cure transfusion-dependent thalassemia:
Timing Matters! Biol Blood Marrow Transplant. 24:1107–1108.
2018.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Del Galy AS, Marouf A, Raffoux E, Robin M,
Michonneau D, Sébert M, Sicre de Fontebrune F, Xhaard A, Lengline
E, Itzykson R, et al: Correction to: Allogeneic hematopoietic stem
cell transplantation in elderly patients with acute myeloid
leukemia or myelodysplastic syndromes: Myth and reality. Leukemia.
35(934)2021.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Chen XP, Cao ZR and Hu J: Advances in
allogeneic hematopoietic stem cell transplantation for chronic
Myelogenous leukemia-review. Zhongguo Shi Yan Xue Ye Xue Za Zhi.
27:1334–1338. 2019.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
124
|
Iioka F, Tanabe H, Honjo G, Misaki T and
Ohno H: Resolution of bone, cutaneous, and muscular involvement
after haploidentical hematopoietic stem cell transplantation
followed by post-transplant cyclophosphamide in adult T-cell
leukemia/lymphoma. Clin Case Rep. 8:1553–1559. 2020.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Natarajan V, Bandapalli OR, Rajkumar T,
Sagar TG and Karunakaran N: NOTCH1 and FBXW7 mutations favor better
outcome in pediatric South Indian T-cell acute lymphoblastic
leukemia. J Pediat Hematol Oncol. 37:E23–E30. 2015.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Valliyammai N, Nancy NK, Sagar TG and
Rajkumar T: Study of NOTCH1 and FBXW7 mutations and its prognostic
significance in South Indian T-cell acute lymphoblastic leukemia. J
Pediatr Hematol Oncol. 40:e1–e8. 2018.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Jenkinson S, Koo K, Mansour MR, Goulden N,
Vora A, Mitchell C, Wade R, Richards S, Hancock J, Moorman AV, et
al: Impact of NOTCH1/FBXW7 mutations on outcome in pediatric T-cell
acute lymphoblastic leukemia patients treated on the MRC UKALL 2003
trial. Leukemia. 27:41–47. 2013.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Asnafi V, Buzyn A, Le Noir S, Baleydier F,
Simon A, Beldjord K, Reman O, Witz F, Fagot T, Tavernier E, et al:
NOTCH1/FBXW7 mutation identifies a large subgroup with favorable
outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): A
Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL)
study. Blood. 113:3918–3924. 2009.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Malyukova A, Brown S, Papa R, O'Brien R,
Giles J, Trahair TN, Dalla Pozza L, Sutton R, Liu T, Haber M, et
al: FBXW7 regulates glucocorticoid response in T-cell acute
lymphoblastic leukaemia by targeting the glucocorticoid receptor
for degradation. Leukemia. 27:1053–1062. 2013.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Saito Y, Aoki Y, Muramatsu H, Makishima H,
Maciejewski JP, Imaizumi M, Rikiishi T, Sasahara Y, Kure S, Niihori
T, et al: Casitas B-cell lymphoma mutation in childhood T-cell
acute lymphoblastic leukemia. Leuk Res. 36:1009–1015.
2012.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Huang HL, Weng HY, Wang LQ, Yu CH, Huang
QJ, Zhao PP, Wen JZ, Zhou H and Qu LH: Triggering Fbw7-mediated
proteasomal degradation of c-Myc by oridonin induces cell growth
inhibition and apoptosis. Mol Cancer Ther. 11:1155–1165.
2012.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Zheng R, Li M, Wang S and Liu Y: Advances
of target therapy on NOTCH1 signaling pathway in T-cell acute
lymphoblastic leukemia. Exp Hematol Oncol. 9(31)2020.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Chiba S: Notch signaling in stem cell
systems. Stem Cells. 24:2437–2447. 2006.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Tan SH, Bertulfo FC and Sanda T:
Leukemia-initiating cells in T-cell acute lymphoblastic leukemia.
Front Oncol. 7(218)2017.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Belmonte M, Hoofd C, Weng AP and Giambra
V: Targeting leukemia stem cells: Which pathways drive self-renewal
activity in T-cell acute lymphoblastic leukemia? Curr Oncol.
23:34–41. 2016.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Gordeeva O: TGFβ family signaling pathways
in pluripotent and teratocarcinoma stem Cells' fate decisions:
Balancing between self-renewal, differentiation, and cancer. Cells.
8(1500)2019.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Ma L, Wang Y, Hui Y, Du Y, Chen Z, Feng H,
Zhang S, Li N, Song J, Fang Y, et al: WNT/NOTCH pathway is
essential for the maintenance and expansion of human MGE
progenitors. Stem Cell Reports. 12:934–949. 2019.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Rho JY, Bae GY, Chae J, Yu K, Koo DB, Lee
KK and Han YM: Transcriptional Properties of the BMP, TGF-β, RTK,
Wnt, Hh, Notch, and JAK/STAT signaling molecules in mouse embryonic
stem cells. Reproductive Dev Biol. 30:143–156. 2006.
|
|
139
|
Ding D, Lim KS and Eberhart CG: Arsenic
trioxide inhibits Hedgehog, Notch and stem cell properties in
glioblastoma neurospheres. Acta Neuropathologica Communications.
2(31)2014.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Liu H, Zhu L and Liu YH: Effect of FBXW7
and NOTCH1 mutations on prognosis of patients with adult acute T
lymphoblastic leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi.
26:1294–1300. 2018.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|














