Introduction
Primary myelofibrosis (PMF) is a classic
myeloproliferative neoplasm (MPN), characterized by the appearance
of diffuse fibrous tissue in the bone marrow, increased numbers of
myeloid cells, extramedullary hematopoiesis, organomegaly,
pancytopenia and an altered cytokine expression profile (1-4).
PMF comprises chronic and acute PMF (CPMF and APMF, respectively),
according to the type of onset.
The majority of PMF cases are hard to identify and
diagnose during the asymptomatic state (5). PMF-related mortality is often caused
by cardiac failure, infection, hemorrhage or acute leukemia
transformation (6). Acute leukemia
transformation occurs in ~20% of PMF patients within 10 years of
diagnosis (3). PMF is difficult to
cure, and the only known treatment method is allogeneic stem cell
transplantation (7). PMF is a
severe disease that poses a serious threat to human health. The
median survival time of PMF is 3.5-5.5 years from diagnosis
(8,9), and the 10-year survival rate for
familial PMF cases is only 30% (10). PMF predominantly occurs in patients
aged >50 years. However, half of the children with PMF are
diagnosed at <3 years old (1).
Familial PMF cases account for ~7.6% of chronic myeloproliferative
disorders (11). The first-degree
relatives of patients with PMF have a 5-7-fold increased risk of
developing MPN, as compared with other more distant relatives
(10). Janus kinase 2 (JAK2)
belongs to the identified Janus family of non-receptor tyrosine
kinases that are important for the transduction of
cytokine-mediated signals in several cell types (2). The JAK2 V617F somatic mutation arises
from a single base G-T transversion in the pseudokinase domain of
JAK2. The somatic mutation is observed in 50% of cases with PMF.
This mutation results in a valine-to-phenylalanine substitution at
codon 617(12), leading to
constitutive kinase activation (13). The JAK2 V617F mutation stimulates
various signaling pathways downstream of JAK2, thus leading to
cytokine-independent cell survival and proliferation (14). A previous study showed that the rate
of splenomegaly in JAK2 V617F-mutated PMF patients was higher than
that in JAK2-negative PMF patients, whereas the incidence of
leukemia transformation was lower in JAK2 V617F-mutated PMF than in
JAK2-negative patients (15).
Since the reports of familial PMF are infrequent,
the present case report describes two familial cases (two sisters)
with PMF, both of whom carried a JAK2 V617F mutation. Both sisters
provided written informed consent for participation in the present
study and publication of the case report. The karyotype of the
older sister was 47, XX, +8, and that of the younger sister 46, XX.
The clinical data and Dynamic International Prognostic Scoring
System (DIPSS) plus (16) risk
stratification score of the two sisters are shown in Table I. The patients were followed up
until the older sister succumbed to the disease 16 months after the
initial diagnosis. The study protocol was approved by the Ethics
Committee of Yuyao People's Hospital. Additionally, a meta-analysis
of candidate genes in PMF was also performed, in order to examine
their molecular links with the pathophysiology of the disease.
 | Table IClinical data and DIPSS-plus risk
stratification of the two sisters. |
Table I
Clinical data and DIPSS-plus risk
stratification of the two sisters.
| Parameters | Older sister | Younger sister |
|---|
| Time of initial
diagnosis, date | 19.10.2011 | 27.8.2008 |
| Age at initial
diagnosis, years | 35 | 30 |
| White blood cell
count, x109/l | 20.7 | 2.7 |
| Neutrophil count,
x109/l | 8.07 | 1.7 |
| Peripheral blood
primordial cells, % | 1 | 0 |
| Middle and late
myelocytes, % | 11 | 8 |
| Naive nucleated red
blood cells, % | 5 | 5 |
| Lymphocyte count,
x109/l | 1.45 | 0.19 |
| Eosinophil count,
x109/l | 5.38 | 0.05 |
| Basophil count,
x109/l | 1.24 | 0.14 |
| Monocyte count,
x109/l | 2.07 | 0.41 |
| Platelet count,
x109/l | 31 | 148 |
| Hemoglobin,
g/l | 110 | 132 |
| RBC transfusion
dependence | No RBC
transfusion-dependence occurred following hemolysis after
exacerbation | Never
transfused |
| B-ultrasound
findings | Diffuse liver
disease, increased liver capacity, anterior-posterior diameter of
the left lobe was 78 mm, oblique diameter of the left lobe was 161
mm, the spleen was significantly enlarged, the thickness of the
spleen was 72 mm; the lower edge was ~32 mm below the umbilical
level, the right side of the umbilicus was ~40 mm. Portal
hypertension: The diameter of the portal vein was 14 mm, the flow
rate 12.1 cm/sec, and the diameter of the splenic vein 9 mm. | Diffuse liver
disease, normal liver size, markedly enlarged spleen, splenomegaly
located 4 finger widths below the umbilicus, 65-mm thick, 175-mm
long, laminar hypoechoic area (spleen infarction) in the spleen,
portal hypertension: portal vein diameter was 14 mm, flow rate 13.3
cm/sec, and splenic vein diameter 11 mm. |
| JAK2 mutation
ratio | 40% | 100% |
| BCR/ABL fusion gene
P210 and P190 | Negative | Negative |
| Karyotype (R band)
4 | 47, XX, +8[15] | 46, XX[15] |
| IPSS (risk
group) | 1 point,
(moderate-risk group 1) | 0 points (low-risk
group) |
| DIPSS | 1 point
(moderate-risk group 1) | 0 points (low-risk
group) |
| DIPSS plus | 3 points
(moderate-risk group 2) | 0 points (low-risk
group) |
| Prognosis, overall
survival | 16 months | >11
yearsa |
Materials and methods
Collection and analysis of
samples
Clinical and laboratory features of PMF, such as
peripheral blood counts and smear analysis, bone marrow morphology,
karyotype and high-molecular risk mutations were used to diagnose
or monitor the disease progression (17). In the present study, patients were
made to fast overnight and 5-8 ml of antecubital venous blood was
drawn. Routine blood tests were performed, which included detection
of 24 important indicators using a Sysmex XN-3000 hematology
analyzer (Sysmex Corporation) and EDTA anticoagulated blood. A
mutation at codon 617 of the JAK2 gene (JAK2 V617F) mutation was
detected by allele-specific PCR (18), which was performed using DNA
isolated from peripheral white blood cells using the QIAamp DNA
Blood Mini kit (Qiagen) following the manufacturer's protocol. RNA
was extracted using an RNeasy Mini kit (Qiagen), quantitative PCR
for identifying BCR-ABL fusions was performed using specific
primers and the PCR products were electrophoresed in agarose gels
(19). All of the PCR reactions
were performed by Adicon Clinical Laboratories, Ltd, and they were
performed as described in the reference. Subsequently, the blood
was centrifuged at 1,500-1,800 x g for 8 min at room temperature to
isolate the serum. The serum was tested for hepatitis B surface
antigen (HBsAg) using ELISA (cat. no: DECO0844; DECO), and
classified as HBsAg positive or negative (20).
Liver function parameters were assessed using a
Beckman AU5800 automated biochemistry analyzer (Beckman Coulter,
Inc.) using 3 ml coagulated peripheral blood after centrifugation
at 1,800 x g for 10 min at room temperature. Liver fibrosis stage
was evaluated semi-quantitatively according to the METAVIR scoring
system (21).
Bone marrow aspiration was performed following a
standard operating procedure by a trained clinician (22). The posterior iliac crest is usually
the preferred location of biopsy (22). Films of aspirated marrow and, when
appropriate, films of crushed particles were prepared and labeled,
as previously described (22). Once
thoroughly dry, films were fixed in fresh methanol in ethanol at
37˚C for at least 30 min and stained with Romanowsky stains at 37˚C
for at least 8 min. A cover slip was applied, and the bone marrow
films were assessed and reported in a systematic manner. The films
were first examined under a low power (x10 objective) to assess the
number of fragments, the cellularity, the number of megakaryocytes
and to detect any low incidence abnormal cells. Then the films were
examined in detail using a x50 objective to systematically assess
the cellularity and contents of fragments, megakaryocyte number,
morphology and cytological features of other lineages. Finally,
fine cytological details were assessed using an oil immersion x100
objective, as previously reported (22). The bone marrow findings were
interpreted, taking into account the clinical and hematological
features of the specific patients. Chromosome preparations were
processed for R banding, as previously described (23). At least 10 metaphases, and typically
more, were fully karyotyped for karyotype analysis. The ultrasound
examination was performed to analyze liver morphology and
echogenicity as well as spleen size, which was carried out as
described by Khan et al (24). Abdominal computed tomography was
used to define abdominal involvement in PMF, such as the
splenomegaly and the possible low-density lesions on lumbar. All
data were analyzed by an experienced pathologist.
Literature search
A literature search for PMF-related studies between
January 2002 and February 2016 was performed using PubMed, CNKI and
Wanfang literature databases. No restrictions on language were
applied. Keywords and Medical Subject Headings, including
‘idiopathic myelofibrosis’, ‘IMF’, ‘primary myelofibrosis’, ‘PMF’
and ‘mutation’, were used, where IMF stands for idiopathic
myelofibrosis.
Inclusion criteria
Studies were included in the present meta-analysis
if they met all of the following inclusion criteria: i) They
assessed patients with PMF; ii) the subjects in every study
provided the number of cases and controls; iii) they assessed the
JAK2 mutation; and iv) the publications appeared in a peer-reviewed
journal.
Data extraction
For each eligible study, information was collected
regarding the mutation position, names of first authors,
publication year, country of origin, age, number of cases and
controls, number of male and female patients, mutation frequencies
and mutations detection method.
Statistical analysis
The meta-analysis was performed using Review Manager
version 5.2 (Cochrane). The combined odds ratios (ORs) and the
corresponding 95% confidence intervals (CIs) were calculated in the
forest plots using the fixed effect model (moderate heterogeneity,
I2<50%) or random effects model (moderate
heterogeneity, I2≥50%). Funnel plots were used to show
potential publication bias amongst the involved studies. P<0.05
was considered to indicate a statistically significant
difference.
Case report
Case of the younger sister
According to the medical records, on August 27th,
2008 the younger sister was 30 years old and had no permanent
teeth. The patient was diagnosed with asymptomatic splenomegaly 3
months after prenatal examination. The physical examination
performed whilst at the hospital showed that her splenomegaly was
four-finger widths below the umbilicus. The auxiliary examination
indicated a hemoglobin level of 125 g/l, total leukocyte count of
2.7x109/l, platelet count of 148x109/l, 7%
lymphocytes, 15% mononuclear cells, 2% eosinophils, 5% basophils,
5% immature nucleated erythrocytes, 63% neutrophils, 8% myelocytes
and metamyelocytes. The hepatitis B surface antibody was
electropositive. The B-ultrasonic test showed megalosplenia, portal
hypertension, splenic infarction and diffuse liver lesions. The
four indices of liver fibrosis showed that procollagen peptides III
and IV, type IV collagen, hyaluronic acid and laminin determination
in the serum were all normal. The routine examination of her bone
marrow on both sides of the posterior superior iliac crest revealed
‘dry tap’, which is described as a failure to obtain bone marrow
upon attempted marrow aspiration (25). In addition, it was hard to insert
the biopsy needle, which was referred to as ‘bone hard’. Routine
inspection and biopsy of the bone marrow confirmed the
transformation of fibrosis. The karyotype of the patient was 46, XX
(Fig. 1A), and the JAK2 genotype in
the bone marrow was 100% of the heterozygous mutation JAK2 V617F,
with no evidence of aberrant BCR-ABL fusion gene.
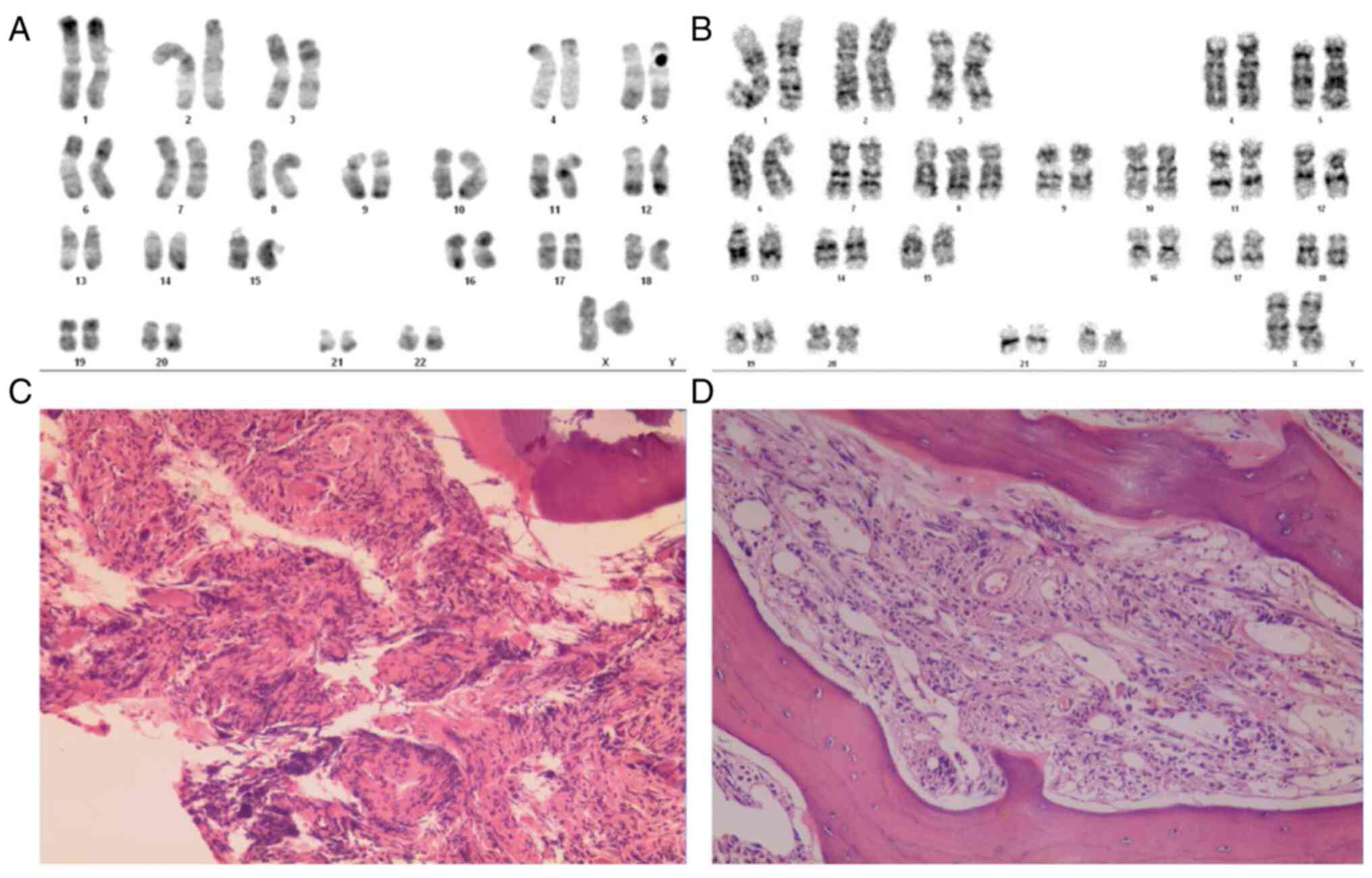 | Figure 1Karyotypes and bone marrow biopsy of
the sisters. (A) Karyotype of the younger sister was 46, XX (Giemsa
stain; magnification, x1,000). (B) Karyotype of the older sister
was 47, XX, +8 (Giemsa stain; magnification, x1,000). (C) Bone
marrow hematopoietic tissue biopsy of the younger sister (H&E
staining; magnification, x100). (D) Bone marrow hematopoietic
tissue biopsy of the older sister (H&E staining; magnification,
x100). Atypical nuclear morphology was identified and staining of
the reticular protein from a bone marrow biopsy suggested that
there was increased collagen compared to levels typically seen in
individuals without myelofibrosis. H&E, hematoxylin and
eosin. |
The patient was administered 0.5 g hydroxycarbamide
tablets orally once a day, on days 1-3 following diagnosis. Due to
2nd degree gastrointestinal reactions and chest discomfort, the
medication was withdrawn. Subsequently, she was treated with an
interferon injection only once for treatment of 3rd-degree bone
marrow depression, and subsequently administered Calcitriol soft
capsules orally for 3 months 0.25 µg, 3 times a day. Previous
studies have shown that calcitriol can be used for the treatment of
bone marrow fibrosis, although its effect is relatively weak
compared with that of JAK2 inhibitors (26,27).
Her spleen was palpable 2-3 finger widths below the umbilicus, and
it was softer than before. Since then, she has received no
treatments, is under continuous review, and her erythrocyte and
erythrocyte platelet counts were maintained at normal levels.
Case of the older sister
The older sister was 35 years old and had no
permanent teeth. She had repeatedly presented with bruise blocks in
the skin for >10 years. According to our medical records on
October 27, 2011, she presented with splenomegaly for >1 month.
Physical examination performed whilst at the hospital revealed
slight scattered mucocutaneous hemorrhage in all 4 limbs. Auxiliary
examination showed that her hemogram results comprised of
hemoglobin levels of 110 g/l, total leukocyte count of
20.7x109/l, platelet count of 31x109/l, 26%
mononuclear cells, 6% eosinophils, 10% basophils, 5% immature
nucleated erythrocytes, 39% neutrophils and 11% myelocytes and
metamyelocytes. Lactate dehydrogenase was 508 U/l. The hepatitis B
surface, hepatitis B e, and hepatitis B core antibodies were all
electropositive. The four indices of liver fibrosis showed a serum
procollagen peptide level of 34.2 µg/l and a serum type IV collagen
of 36.75 µg/l. Computed tomography revealed megalosplenia, portal
hypertension and the formation of collateral circulation. The 5
different locations of routine examination of the bone marrow on
both sides of the posterior superior iliac crest showed 3 instances
of ‘dry tap’ (as mentioned above) and another 2 instances of
‘analogous to diluted marrow’. Bone marrow biopsy revealed that the
bone marrow was ‘quite hard’, making it difficult to insert the
biopsy needle. Bone marrow fibrosis confirmed transformation of
fibrosis with scattered atypical cells. The karyotype of the older
sister was 47, XX, +8 (Fig. 1B).
The JAK2 genotype in the bone marrow showed a 40% heterozygous
mutation (JAK2 V617F).
The patient was then treated with an injection of
150 µg pegylated interferon (hypodermic injection administered once
a week for 4 weeks). Her spleen extended to 5 cm below the
umbilicus during treatment. A routine blood test revealed no
decline in the leukocyte count. However, the platelet count dropped
to 26x109/l, and treatment was discontinued due to
3rd-degree gastrointestinal reactions. The patient was later
transferred to another hospital was administered Ganoderma lucidum
(10 capsules, orally, three times per day) and Calcitriol soft
(0.25 µg, orally, three times per day) capsules. Her spleen was
palpable at 3 cm below the umbilicus, the side had dwindled and it
was softer than before. The leukocyte count increased slowly and
the platelet count was maintained at 30-40x109/l. The
results of the February 2012 follow-up showed a leukocyte count of
59.8x109/l, 10% myelocytes and metamyelocytes, 63%
neutrophils, 7% lymphocytes, 9% mononuclear cells, 6% eosinophils,
5% basophils, 4% immature nucleated erythrocytes, hemoglobin levels
of 86 g/l and a platelet count of 40x109/l. She was then
treated with hydroxyurea 0.5 g (orally, once per day) and did not
experience any notable toxicity. Unfortunately, she succumbed to
intracranial hemorrhage in February 2013.
Summary of the two cases
The clinical manifestations of the sisters in the
case report were similar, suggesting that the same type of
complications (bleeding) may occur in familial PMF patients, which
may result in progression of the disease. A previous study showed
that the clinical manifestations of PMF include progressive anemia,
massive splenomegaly, cachexia and extramedullary hematopoiesis
(28). Both cases here exhibited
bone marrow hematopoietic tissue hyperplasia, an increased
proportion of granulocyte erythrocytes (particularly myeloid and
bone marrow cells), increased megakaryocytes, aggregation into
small clusters and a large number of fibroblasts in the
interstitium (Fig. 1C and D).
Both sisters harbored a JAK2 mutation, but their
all-round healthy parents did not. PMF was shown to be a disease
with autosomal recessive inheritance (10), suggesting that the JAK2 mutation in
these cases was acquired and occurred as a secondary genetic event
in familial chronic MPD (CMPD). Furthermore, the older sister had a
trisomy 8 (47, XX, +8) karyotype and 40% JAK2 V617F mutation in the
bone marrow, and her outcome was more severe than that of the
younger sister, who had a normal karyotype and 100% JAK2 V617F
mutation in the bone marrow. Research has shown that patients with
trisomy 8 mosaicism are at a higher risk of developing leukemia
(29), and karyotype abnormalities
are associated with lower survival in the univariate analysis
(30). A higher mutation frequency
of JAK2 V617F in PMF is associated with an unfavorable cytogenetic
profile, and the presence of unfavorable cytogenetic abnormalities
is significantly associated with decreased survival (31). The overall survival and risk of
mortality for a patient in which trisomy 8 and JAK2 V617F mutations
exist simultaneously is currently unknown, to the best of our
knowledge. However, the findings from the cases of the two sisters
may suggest that trisomy 8 translates to an increased risk of
mortality, when compared with the JAK2 mutation.
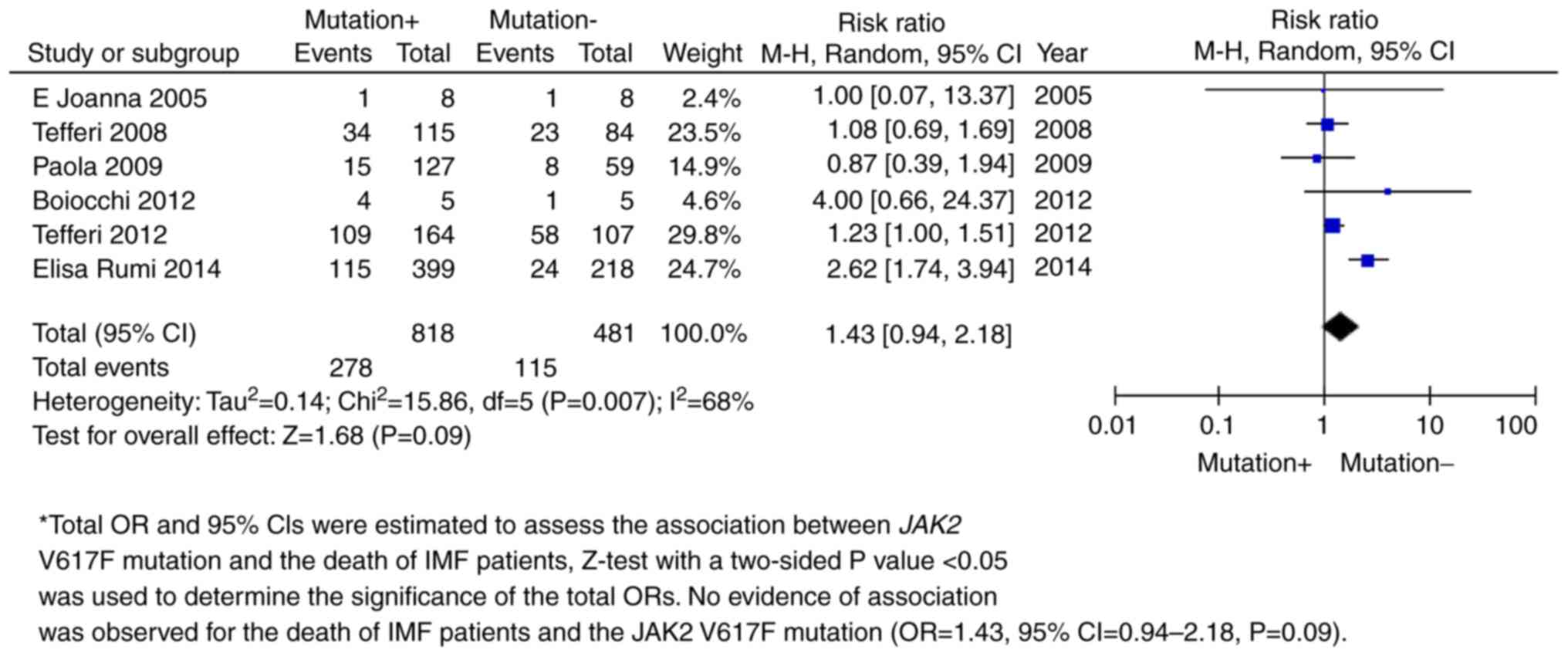
Meta-analysis
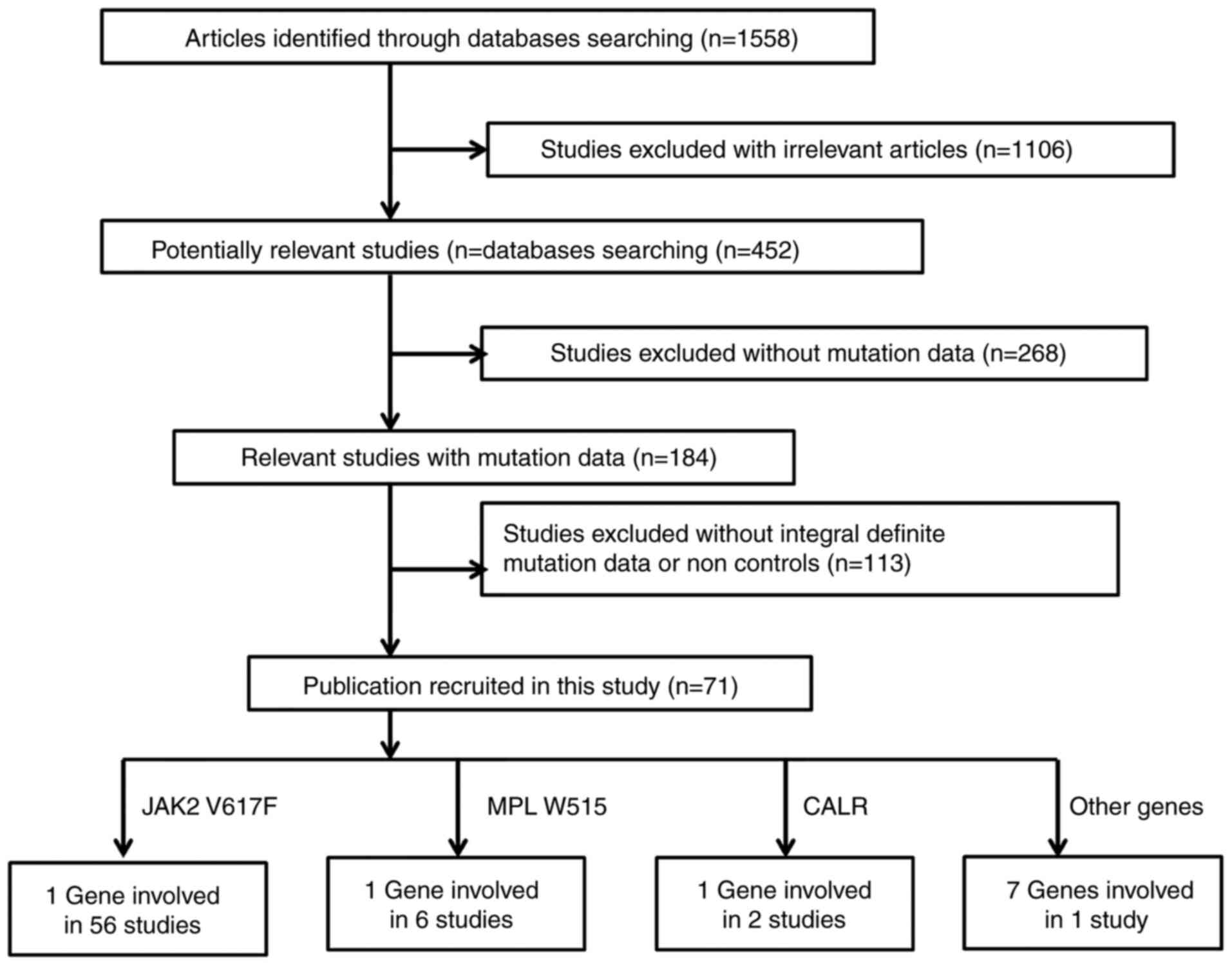
As shown in Fig. 2,
a total of 1,558 relevant publications appeared to meet the
inclusion criteria during an initial search in the PubMed, CNKI and
Wanfang literature databases. A total of 1,106 irrelevant studies,
268 studies without mutation data and 113 studies without the
controls were excluded. A total of 71 eligible studies were
included in the current meta-analysis. Fixed-effect tests were
applied for the subgroup association of JAK2 V617F by sex and
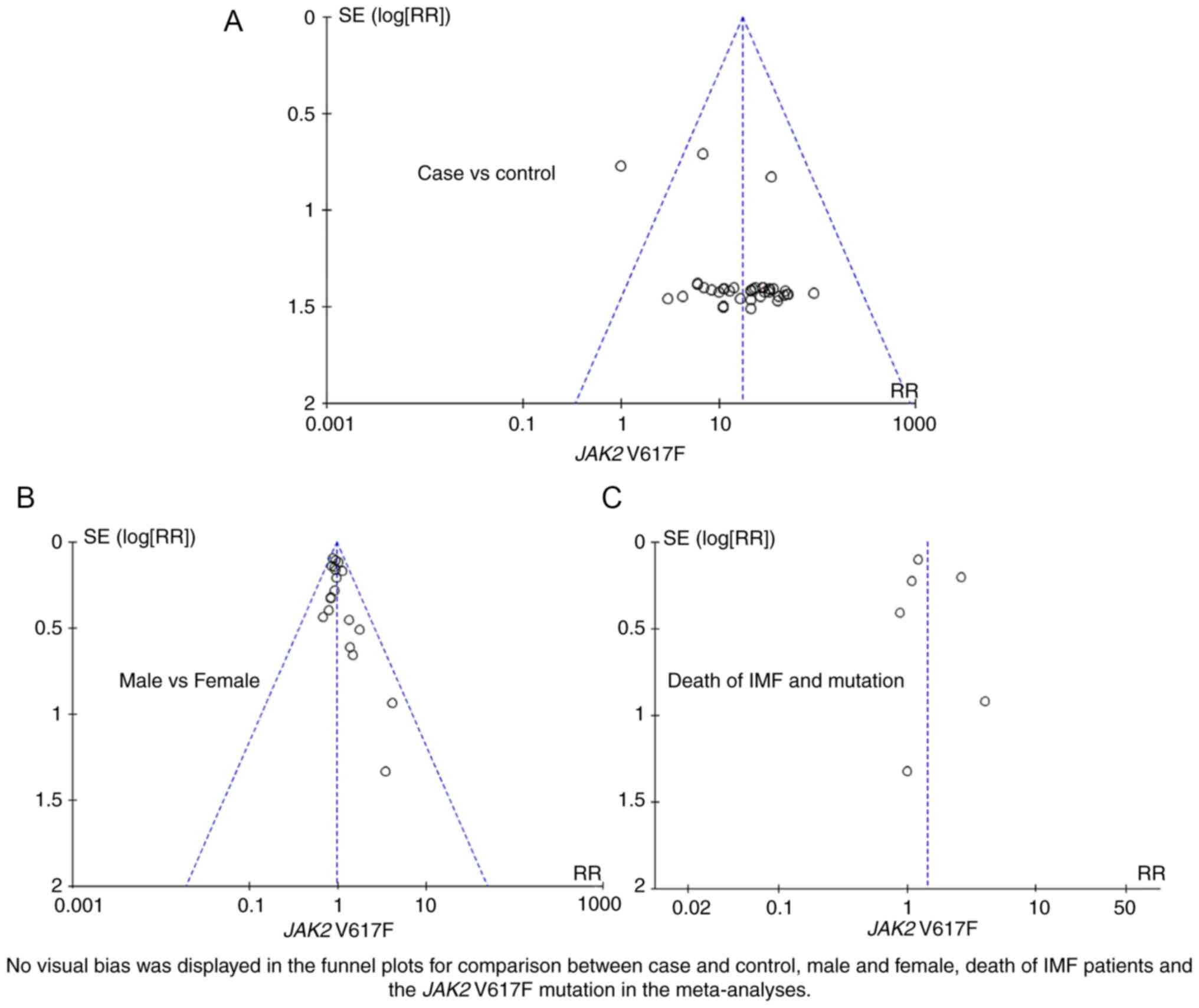
mortality of PMF (I2<1%). A funnel plot evaluated
publication bias in the meta-analysis. As shown in Fig. 3, no visual bias was present in the
current meta-analysis.
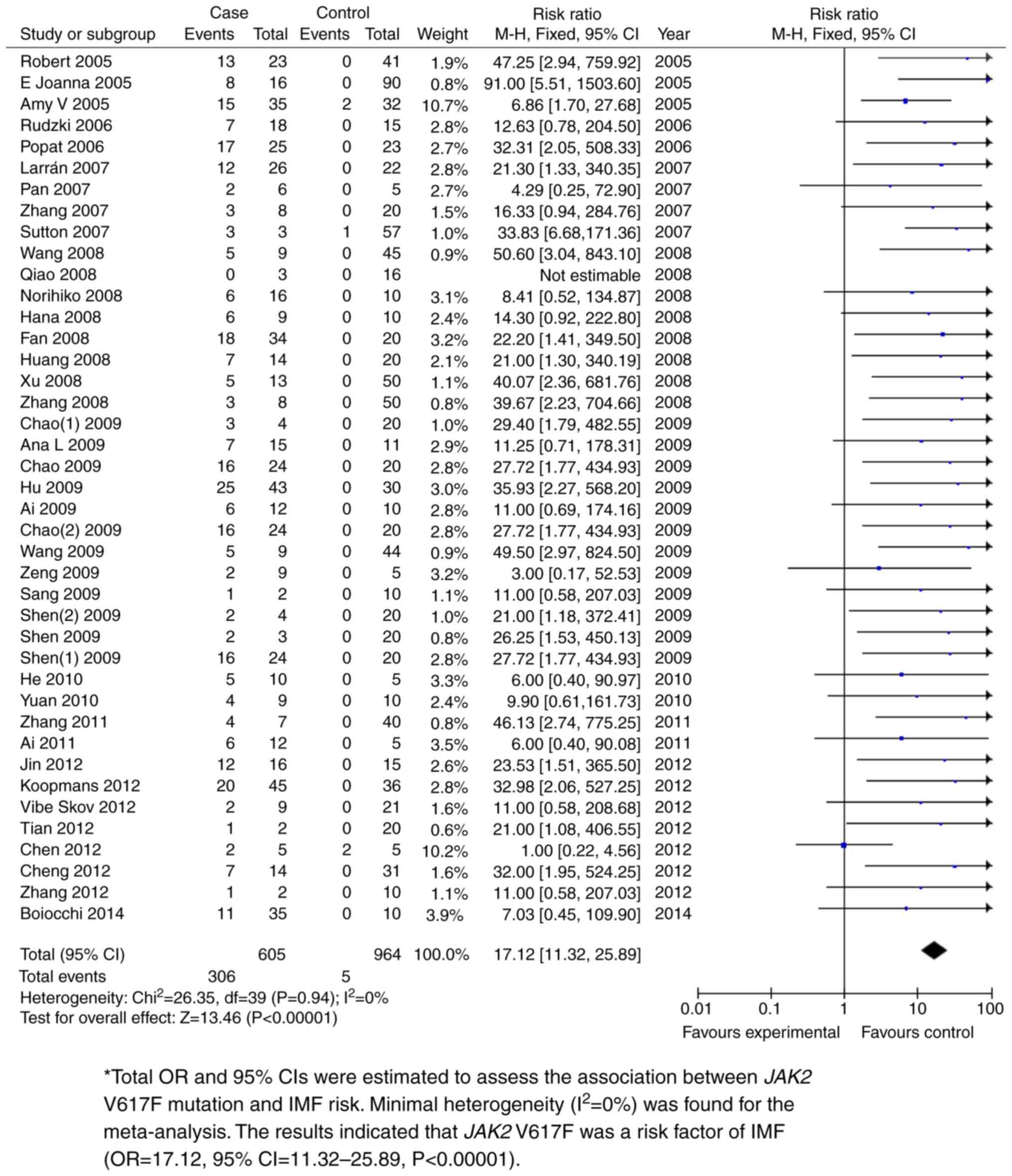
The JAK2 V617F mutation was shown to be
significantly associated with PMF. Total ORs and 95% CIs were
estimated to assess the association between JAK2 V617F mutation and
PMF risk. The fixed effects model was used for meta-analysis with
minimal heterogeneity (I2=0%). P<0.05 was used to
determine the significance of the total ORs. The results indicated
that JAK2 V617F was a risk factor for PMF (OR=17.12, 95%
CI=11.32-25.89; P<0.00001; Fig.
4 and Table II). Further
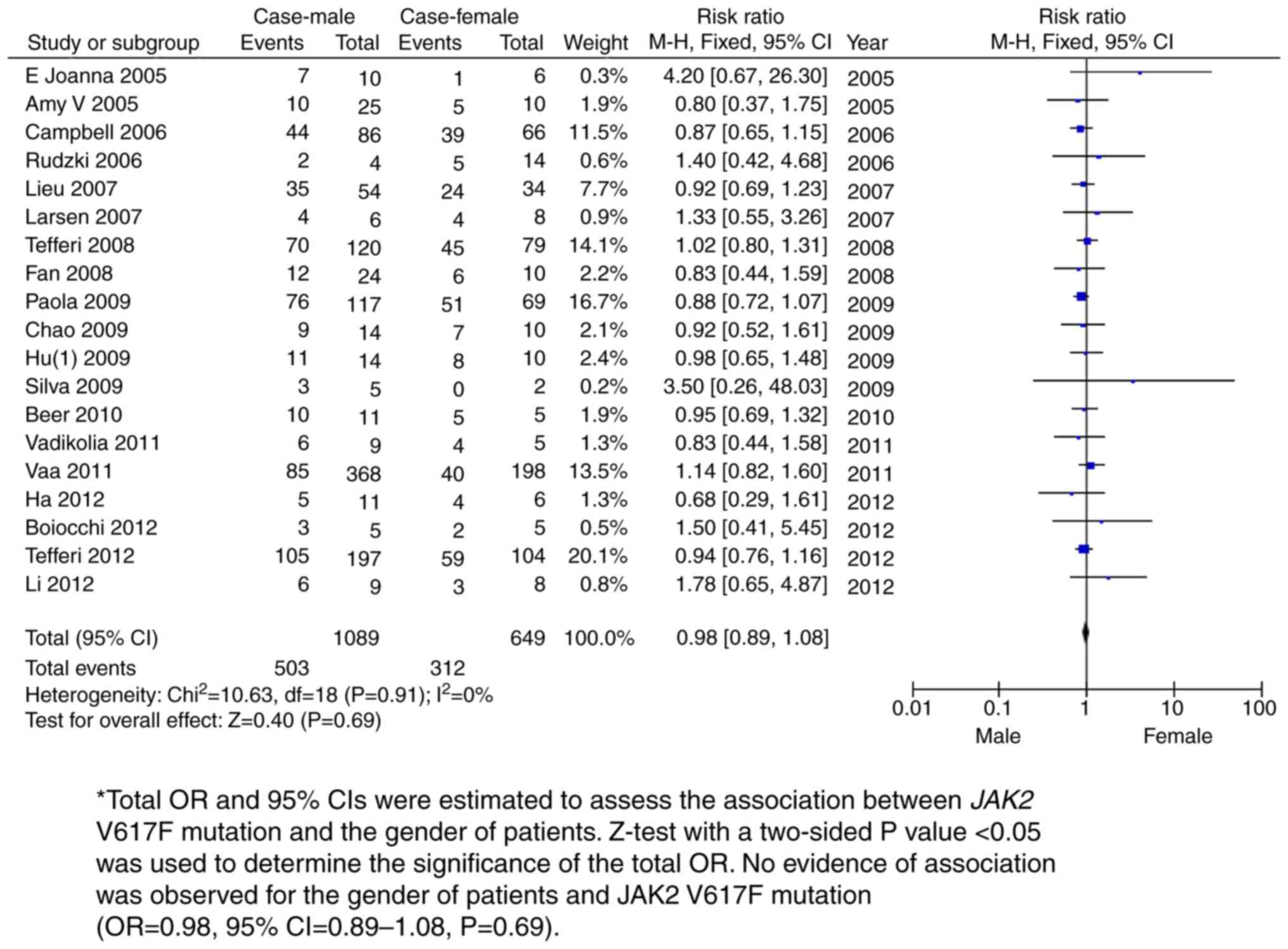
sex-based subgroup meta-analysis amongst 1,089 male and 649 female
PMF patients revealed no association between sex and the JAK2 V617F
mutation with PMF (OR=0.98, 95% CI=0.89-1.08; P=0.69; Fig. 5 and Table II). A follow-up survey demonstrated
that the JAK2 V617F mutation was not associated with PMF-related
mortality in 6 independent studies (OR=1.43, 95% CI=0.94-2.18;
P=0.09; Fig. 6 and Table II).
| Table IICharacteristics of the meta-analyses
for JAK2 V617F mutation in primary myelofibrosis studies. |
Table II
Characteristics of the meta-analyses
for JAK2 V617F mutation in primary myelofibrosis studies.
| Comparison | Studies, n | Cases, n | Controls, n | Odds ratio 95%
confidence intervala | I2 | P-value |
|---|
| Case vs.
control | 44 | 605 | 964 | 17.12 (11.32,
25.89) | 0 | <0.00001 |
| Male vs. female
case | 19 | 1,089 | 649 | 0.98 (0.89,
1.08) | 0 | 0.69 |
| Mutation and
death | 6 | 818 | 481 | 1.43 (0.94,
2.18) | 0.68 | 0.09 |
Discussion
Reports on familial PMF are rare, but first-degree
relatives may acquire one or several MPDs (32). It is reasonable to assume that
germ-line mutations or a patient's genetic background can
facilitate one or several somatic mutations that result in PMF or
other forms of MPD. The present study reported the cases of two
sisters with PMF. This case report can shed new light on PMF. A
meta-analysis was also performed to clarify the correlation between
JAK2 mutations with other clinical phenotypes, including PMF risk,
sex and PMF-related mortality.
Autosomal recessive inheritance remains a logical
explanation for PMF. The two sisters were diagnosed with PMF and
multiple hemangiomas (13); given
the fact that the parents showed no signs of PMF, recessive
inheritance may have been involved. A total of 14 patients in this
sister's family had PMF, but the previously published article did
not describe the 14 cases (33).
Perez-Encinas et al (34)
reported 5 cases of familial CMPD. Cases 1-3 were siblings. Case 5
was the daughter of case 1, and case 4 was the cousin of cases 1
and 3. This suggested that the genetic pathogenesis of CMPD may be
heritable. Sheikha (35) described
a family whose 4 consecutive children died of acute myelofibrosis.
The fact that their mother had another 2 healthy girls, ruled out
sex-linked inheritance for PMF. The present study reported the case
of two sisters with PMF who acquired a JAK2 V617F mutation that
their healthy parents did not harbor. The present case report thus
supports the notion that PMF is a recessive inheritable
disease.
The molecular basis of PMF is complex. Of the PMF
patients, ~30% had clonal chromosomal abnormalities, such as del
(13) (q12-22) or der (6) t (1;6) (q21-23;p21.3). The development
of molecular biology enabled the identification of various
important pathological events involved in PMF (7). The common mutations in the
pathogenesis of PMF included JAK2, myeloproliferative leukemia
virus oncogene (36), calreticulin
(37), tet methylcytosine
dioxygenase 2(38), additional sex
combs like 1(39), enhancer of
zeste 2 polycomb repressive complex 2 subunit (40), serine and arginine-rich splicing
factor 2(41), isocitrate
dehydrogenase (42), splicing
factor 3b subunit 1(43) and U2
small nuclear RNA auxiliary factor 1(44). Among these, the JAK2 mutation was
identified in 50-60% of PMF patients (45). In the present case report, the two
sisters were found to harbor a JAK2 mutation in the bone marrow,
which their parents did not possess. This suggested that the JAK2
mutation was somatically acquired by the two sisters.
A previous in vitro studies have found that
cells with a JAK2 V617F mutation possess proliferative and survival
advantages over wild-type JAK2 cells (46). JAK2 activation may trigger
downstream signaling pathway activation, including that of cell
survival and proliferation pathways, promoting myeloproliferation
and resistance to cell death (47).
JAK2 V617F may contribute to PMF through the deregulation of the
apoptotic pathway (48). JAK2 V617F
was also shown to work as a potentially useful biomarker for
prognosis and treatment response (4). In the present study, disease
progression in both PMF patients with the JAK2 V617F mutation were
once well controlled. This may have been due to their JAK2 V617F
mutation in the bone marrow. The worse outcome of the older sister
may have been due to her abnormal karyotype (47, XX, +8).
A comprehensive overview of mutation studies was
also performed to evaluate the overall contribution of the JAK2
V617F mutation to the risk of PMF patients in the present study.
Following a 3-step filtration process, a total of 605 PMF patients
and 964 non-PMF controls from 71 eligible studies were included in
the present meta-analysis.
The meta-analysis results showed that the JAK2 V617F
mutation was significantly associated with an increased risk of
PMF. However, subgroup meta-analysis by sex showed no evidence of
an association between the sex of the patients and the JAK2 V617F
mutation. The follow-up analysis revealed that PMF-related
mortality was not associated with the JAK2 V617F mutation. The
present study demonstrated a significant association between JAK2
V617F mutation and PMF risk. Additionally, the meta-analysis showed
no association between sex and PMF-related mortality with the JAK2
V617F mutation. These data are generally consistent with previous
studies which showed that JAK2 V617F mutation was significantly
associated with fibrotic progression and histology (49,50).
Some clinical signs of the disease, such as anemia and
splenomegaly, and the risk of transformation to AML have been shown
to be associated with JAK2 V617F mutational status or the JAK2
V617F allele burden (51). As the
most important mutation, JAK2 V617F was present in over half of the
patients with PMF, and serves as a potential molecular target for
therapeutic intervention (51),
such as through the development of ruxolitinib, an inhibitor of
JAK2, which has transformed therapy for myelofibrosis (52).
In the present study, a familial case of PMF is
described; two sisters with IMF and their healthy parents were
observed. The molecular basis of PMF was complex, and the present
report may provide additional evidence of familial PMF. The present
meta-analysis however was not without its limitations. First,
selection bias may have existed, since only English and Chinese
publications were assessed. Secondly, the primary ethnicities of
the studied population were Caucasians and Asians; other ethnic
populations should be included in the future. Well-designed studies
with larger samples may help elucidate the contribution of the JAK2
V617F mutation to disease outcomes. The majority of the studies
selected for the meta-analysis were performed using allele-specific
PCR, which may have limited the scope of the analysis. More samples
should be tested in the future in order to draw a more reliable
result. Finally, in the present study, the two sisters were
diagnosed as having PMF in 2008 and 2012 according to their
clinical manifestations, bone marrow biopsy and JAK2 mutation. The
diagnosis of familial MPN is involved in genomic aberrations of
TERT, GSKIP, ATG2B and RBBP6. However, basic genomic screenings
were not performed due to the lack of facilities at the primary
hospital.
In conclusion, the present study reported the case
of two sisters with PMF that harbored a JAK2 V617F mutation. In
addition, the meta-analysis showed that JAK2 V617F was a risk
factor for PMF, and no sex dimorphism was observed in the JAK2
V617F mutation amongst all PMF cases. There was also a lack of
association between PMF-related mortality and the JAK2 V617F
mutation.
Acknowledgements
The authors gratefully thank the undergraduate
students of the Medical Genetics Center (School of Medicine of
Ningbo University, Ningbo, China) Miss Xuer Bi, Miss Chunsheng Shu,
Miss Weicong Hua, Mr. Zhangbo Xu and Mr. Nan Wang for the
collection and collation of existing data.
Funding
This research was supported by grants from the K. C.
Wong Magna Fund of Ningbo University.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
SD designed the study; YX and QH wrote the
manuscript; YX recruited the patients and analyzed their
information; QH, ZG and SW analyzed the data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The study protocol was approved by the Ethics
Committee of Yuyao People's Hospital. Both sisters provided written
informed consent for participation in the present study.
Patient consent for publication
Both sisters provided written informed consent for
publication of the case report.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bavikar RR, Kulkarni RK, Rathod AD and
Hastak MS: Idiopathic myelofibrosis in an infant. Indian J Pediatr.
78:734–736. 2011.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bhagwat N, Levine RL and Koppikar P:
Sensitivity and resistance of JAK2 inhibitors to myeloproliferative
neoplasms. Int J Hematol. 97:695–702. 2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Cox MC, Panetta P, Venditti A, Abruzzese
E, Del Poeta G, Cantonetti M and Amadori S: New reciprocal
translocation t(6;10) (q27;q11) associated with idiopathic
myelofibrosis and eosinophilia. Leuk Res. 25:349–351.
2001.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Tognon R, Gasparotto EP, Leroy JM,
Oliveira GL, Neves RP, Carrara Rde C, Kashima S, Covas DT, Santana
M, Souto EX, et al: Differential expression of apoptosis-related
genes from death receptor pathway in chronic myeloproliferative
diseases. J Clin Pathol. 64:75–82. 2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Bouabdallah R, Coso D, Gonzague-Casabianca
L, Alzieu C, Resbeut M and Gastaut JA: Safety and efficacy of
splenic irradiation in the treatment of patients with idiopathic
myelofibrosis: A report on 15 patients. Leuk Res. 24:491–495.
2000.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kerbauy DM, Gooley TA, Sale GE, Flowers
ME, Doney KC, Georges GE, Greene JE, Linenberger M, Petersdorf E,
Sandmaier BM, et al: Hematopoietic cell transplantation as curative
therapy for idiopathic myelofibrosis, advanced polycythemia vera,
and essential thrombocythemia. Biol Blood Marrow Transplant.
13:355–365. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wolf D, Rudzki J and Gastl G: Current
treatment concepts of Philadelphia-negative MPN. Curr Cancer Drug
Targets. 11:44–55. 2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Cervantes F: Modern management of
myelofibrosis. Br J Haematol. 128:583–592. 2005.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Cervantes F, Barosi G, Demory JL, Reilly
J, Guarnone R, Dupriez B, Pereira A and Montserrat E: Myelofibrosis
with myeloid metaplasia in young individuals: Disease
characteristics, prognostic factors and identification of risk
groups. Br J Haematol. 102:684–690. 1998.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Rumi E: Familial chronic
myeloproliferative disorders: The state of the art. Hematol Oncol.
26:131–138. 2008.PubMed/NCBI View
Article : Google Scholar
|
|
11
|
Rumi E, Passamonti F, Della Porta MG,
Elena C, Arcaini L, Vanelli L, Del Curto C, Pietra D, Boveri E,
Pascutto C, et al: Familial chronic myeloproliferative disorders:
Clinical phenotype and evidence of disease anticipation. J Clin
Oncol. 25:5630–5635. 2007.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Quintas-Cardama A and Verstovsek S:
Molecular pathways: Jak/STAT pathway: Mutations, inhibitors, and
resistance. Clin Cancer Res. 19:1933–1940. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Bennett M and Stroncek DF: Recent advances
in the bcr-abl negative chronic myeloproliferative diseases. J
Transl Med. 4(41)2006.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ishida S, Akiyama H, Umezawa Y, Okada K,
Nogami A, Oshikawa G, Nagao T and Miura O: Mechanisms for mTORC1
activation and synergistic induction of apoptosis by ruxolitinib
and BH3 mimetics or autophagy inhibitors in JAK2-V617F-expressing
leukemic cells including newly established PVTL-2. Oncotarget.
9:26834–26851. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
He ZP, Tian HY, Tan M and Wu Y: Clinical
Analysis of Driver Mutations in Patients with Ph Negative
Myeloproliferative Neoplasms. Zhongguo Shi Yan Xue Ye Xue Za Zhi.
26:842–848. 2018.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
16
|
Tefferi A: Primary myelofibrosis: 2017
update on diagnosis, risk-stratification, and management. Am J
Hematol. 91:1262–1271. 2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kvasnicka HM: The differential diagnosis
of classical myeloproliferative neoplasms (MPN): The updated WHO
criteria. Rinsho Ketsueki. 60:1166–1175. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Baxter EJ, Scott LM, Campbell PJ, East C,
Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N,
et al: Acquired mutation of the tyrosine kinase JAK2 in human
myeloproliferative disorders. Lancet. 365:1054–1561.
2005.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Ayatollahi H, Keramati MR, Shirdel A,
Kooshyar MM, Raiszadeh M, Shakeri S and Sadeghian MH: BCR-ABL
fusion genes and laboratory findings in patients with chronic
myeloid leukemia in northeast Iran. Caspian J Intern Med. 9:65–70.
2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Fatema K, Tabassum S, Nessa A and Jahan M:
Development and evaluation of an in-house ELISA to detect hepatitis
B virus surface antigen in resource-limited settings. Bangladesh
Med Res Counc Bull. 39:65–8. 2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Friedrich-Rust M, Rosenberg W, Parkes J,
Herrmann E, Zeuzem S and Sarrazin C: Comparison of ELF, FibroTest
and FibroScan for the non-invasive assessment of liver fibrosis.
BMC Gastroenterol. 10(103)2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Bain BJ: Bone marrow aspiration. J Clin
Pathol. 54:657–63. 2001.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wang YL, Wang T, Xu F, Gang Y and Wang J:
Analysis of DEK-CAN fusion gene expression in acute myeloid
leukemia patients with 6; 9 chromosome translocation. Zhongguo Shi
Yan Xue Ye Xue Za Zhi. 14:232–236. 2006.PubMed/NCBI(In Chinese).
|
|
24
|
Khan SA, Yasmeen S, Adel H, Adil SO, Huda
F and Khan S: Sonographic evaluation of normal liver, spleen, and
renal parameters in adult population: A multicenter study. J Coll
Physicians Surg Pak. 28:834–839. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Humphries JE: Dry tap bone marrow
aspiration: Clinical significance. Am J Hematol. 35:247–250.
1990.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Hirri HM and Green RJ: Myelodysplasia and
bone marrow fibrosis treated with calcitriol and venesection. Leuk
Lymphoma. 43:1489–1491. 2002.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Guo Y, Xu WW, Dong L, Huang N and Bi KH:
Progression of primary myelofibrosis to polycythemia vera: A case
report. Medicine (Baltimore). 96(e7464)2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Tefferi A: The forgotten
myeloproliferative disorder: Myeloid metaplasia. Oncologist.
8:225–231. 2003.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Narendran A, Hawkins LM, Ganjavi H, Vanek
W, Gee MF, Barlow JW, Johnson G, Malkin D and Freedman MH:
Characterization of bone marrow stromal abnormalities in a patient
with constitutional trisomy 8 mosaicism and myelodysplastic
syndrome. Pediatr Hematol Oncol. 21:209–221. 2004.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Tavares RS, Nonino A, Pagnano KBB,
Nascimento ACKVD, Conchon M, Fogliatto LM, Funke VA, Bendit I,
Clementino NC, Chauffaille MLLF, et al: Guideline on
myeloproliferative neoplasms: Associacão brasileira de hematologia,
hemoterapia E terapia cellular: Project guidelines: Associação
médica brasileira-2019. Hematol Transfus Cell Ther. 41
(Suppl):S1–S73. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Alshemmari SH, Rajan R and Emadi A:
Molecular pathogenesis and clinical significance of driver
mutations in primary myelofibrosis: A review. Med Princ Pract.
25:501–509. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Skoda R and Prchal JT: Lessons from
familial myeloproliferative disorders. Semin Hematol. 42:266–273.
2005.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Malak S, Labopin M, Saint-Martin C,
Bellanne-Chantelot C and Najman A: French Group of Familial
Myeloproliferative Disorders. Long term follow up of 93 families
with myeloproliferative neoplasms: Life expectancy and implications
of JAK2V617F in the occurrence of complications. Blood Cells Mol
Dis. 49:170–176. 2012.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Perez-Encinas M, Bello JL, Perez-Crespo S,
De Miguel R and Tome S: Familial myeloproliferative syndrome. Am J
Hematol. 46:225–229. 1994.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Sheikha A: Fatal familial infantile
myelofibrosis. J Pediatr Hematol Oncol. 26:164–168. 2004.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Bruchova H, Merkerova M and Prchal JT:
Aberrant expression of microRNA in polycythemia vera.
Haematologica. 93:1009–1016. 2008.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Panagiota V, Thol F, Markus B, Fehse B,
Alchalby H, Badbaran A, Lehmann U, Koenecke C, Shahswar R,
Chaturvedi A, et al: Prognostic effect of calreticulin mutations in
patients with myelofibrosis after allogeneic hematopoietic stem
cell transplantation. Leukemia. 28:1552–1555. 2014.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Saint-Martin C, Leroy G, Delhommeau F,
Panelatti G, Dupont S, James C, Plo I, Bordessoule D, Chomienne C,
Delannoy A, et al: Analysis of the ten-eleven translocation 2
(TET2) gene in familial myeloproliferative neoplasms. Blood.
114:1628–1632. 2009.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Stein BL, Williams DM, O'Keefe C, Rogers
O, Ingersoll RG, Spivak JL, Verma A, Maciejewski JP, McDevitt MA
and Moliterno AR: Disruption of the ASXL1 gene is frequent in
primary, post-essential thrombocytosis and post-polycythemia vera
myelofibrosis, but not essential thrombocytosis or polycythemia
vera: Analysis of molecular genetics and clinical phenotypes.
Haematologica. 96:1462–1469. 2011.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Guglielmelli P, Biamonte F, Score J,
Hidalgo-Curtis C, Cervantes F, Maffioli M, Fanelli T, Ernst T,
Winkelman N, Jones AV, et al: EZH2 mutational status predicts poor
survival in myelofibrosis. Blood. 118:5227–5234. 2011.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Guglielmelli P, Lasho TL, Rotunno G, Score
J, Mannarelli C, Pancrazzi A, Biamonte F, Pardanani A, Zoi K,
Reiter A, et al: The number of prognostically detrimental mutations
and prognosis in primary myelofibrosis: An international study of
797 patients. Leukemia. 28:1804–1810. 2014.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Tefferi A: Primary myelofibrosis: 2014
update on diagnosis, risk-stratification, and management. Am J
Hematol. 89:915–925. 2014.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Lasho TL, Finke CM, Hanson CA, Jimma T,
Knudson RA, Ketterling RP, Pardanani A and Tefferi A: SF3B1
mutations in primary myelofibrosis: Clinical, histopathology and
genetic correlates among 155 patients. Leukemia. 26:1135–1137.
2012.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Tefferi A, Finke CM, Lasho TL, Wassie EA,
Knudson R, Ketterling RP, Hanson CA and Pardanani A: U2AF1
mutations in primary myelofibrosis are strongly associated with
anemia and thrombocytopenia despite clustering with JAK2V617F and
normal karyotype. Leukemia. 28:431–433. 2014.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Tanaka H, Takeuchi M, Takeda Y, Sakai S,
Abe D, Ohwada C, Sakaida E, Shimizu N, Saito Y, Miyagi S, et al:
Identification of a novel TEL-Lyn fusion gene in primary
myelofibrosis. Leukemia. 24:197–200. 2010.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Kralovics R and Skoda RC: Molecular
pathogenesis of Philadelphia chromosome negative myeloproliferative
disorders. Blood Rev. 19:1–13. 2005.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Tognon R, Gasparotto EP, Neves RP, Nunes
NS, Ferreira AF, Palma PV, Kashima S, Covas DT, Santana M, Souto
EX, et al: Deregulation of apoptosis-related genes is associated
with PRV1 overexpression and JAK2 V617F allele burden in Essential
Thrombocythemia and Myelofibrosis. J Hematol Oncol.
5(2)2012.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Kaushansky K: The chronic
myeloproliferative disorders and mutation of JAK2: Dameshek's 54
year old speculation comes of age. Best Pract Res Clin Haematol.
20:5–12. 2007.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Latagliata R, Polverelli N, Tieghi A,
Palumbo GA, Breccia M, Sabattini E, Villari L, Riminucci M, Valli
R, Catani L, et al: Comparison of JAK2V617F-positive
essential thrombocythaemia and early primary myelofibrosis: The
impact of mutation burden and histology. Hematol Oncol. 36:269–275.
2018.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Schwartz DM, Kanno Y, Villarino A, Ward M,
Gadina M and O'Shea JJ: JAK inhibition as a therapeutic strategy
for immune and inflammatory diseases. Nat Rev Drug Discov.
17(78)2017.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Verstovsek S, Kantarjian H, Mesa RA,
Pardanani AD, Cortes-Franco J, Thomas DA, Estrov Z, Fridman JS,
Bradley EC, Erickson-Viitanen S, et al: Safety and efficacy of
INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J
Med. 363:1117–1127. 2010.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Hu M, Xu C, Yang C, Zuo H, Chen C, Zhang
D, Shi G, Wang W, Shi J and Zhang T: Discovery and evaluation of
ZT55, a novel highly-selective tyrosine kinase inhibitor of
JAK2V617F against myeloproliferative neoplasms. J Exp
Clin Cancer Res. 38(49)2019.PubMed/NCBI View Article : Google Scholar
|