Introduction
Acute myeloid leukemia (AML) is a heterogeneous
clonal hematopoietic neoplasm characterized by maturation arrest in
the myeloid lineage. AML accounts for 70% of all cases of acute
leukemia (1) and it is thought to
be propagated by a subpopulation of leukemia stem cells (LSCs) with
the ability to self-renew, an unlimited repopulating potential and
residence in a quiescent state in G0/G1 phase (2-6).
The standard of care regimen for AML has remained almost the same
over the last 40 years, with only slight amendments, and consists
of an anthracycline and cytarabine combination, followed by
consolidation therapy (7,8). However, current treatments do not lead
to long-term complete remission. Relapse remains a major hurdle for
successful AML chemotherapy (9),
and thus, the discovery of effective alternative AML treatment
strategies is a priority.
Aberrant epigenetic modifications, including DNA and
histone methylation, as well as histone acetylation, are key
contributors to leukemia initiation and maintenance (6). Epigenome-modifying drugs, including
decitabine (Dacogen®, DAC; chemical name,
5-aza-2'-deoxycytidine), have shown promise as single therapeutic
agents for AML at low doses, while higher doses are associated with
cellular differentiation and cytotoxicity (10-12).
DAC is a DNA methyltransferase inhibitor (DNMTi) leading to DNA
hypomethylation, gene activation, cell differentiation and
apoptosis (13,14), and is approved by the US Food and
Drug Administration for the treatment of myelodysplastic syndrome
(MDS) and AML.
Oxidative stress inducers comprise another class of
drugs that has been strongly implicated in the targeting of LSCs,
since increased oxidation is associated with reduced self-renewal,
which in turn leads to either differentiation or death of
haemopoietic cells. However, induction of oxidative stress alone is
not sufficient for AML treatment. Bortezomib (BZ) is a proteasome
inhibitor approved for the treatment of multiple myeloma and mantle
cell lymphoma with the ability to induce oxidative stress (15,16).
In AML, BZ lacks activity as a single agent (17) but produces promising results when
used in combination regimens (with idarubicin and cytarabine)
(18). Of note, in AML, BZ is also
considered as an indirect transcriptional inhibitor, since it
appears to disrupt the interactions among microRNA (miR)-29b, the
transcription factor signaling protein 1 (SP1) and NF-κB(p65),
affecting the expression of several genes in myeloid leukemia
cells, including DNMTs and the receptor tyrosine kinase KIT
(19,20). Activating KIT mutations, which are
frequently observed in core-binding factor (CBF) AML, have been
indicated to induce MYC-dependent miR-29b repression, which results
in increased levels of SP1 (a miR-29b target), leading to
NF-κB(p65)-mediated KIT transactivation and induction of its own
transcription via miR-29b. BZ targets the miR-29b/SP1/NF-κB(p65)
complex-dependent KIT overexpression and therefore manages to
inhibit the growth of leukemic cells via upregulation of miR-29b
(19).
While only ~50% of patients treated with DNMTis have
a haematological improvement (HI) or better, few alternative
treatments exist for patients who fail to respond. Therefore, we
investigated the possible sensitization of AML cells to DAC,
through its combination with an oxidative stress-inducing agent,
namely the proteasome inhibitor BZ.
Materials and methods
Cell culture
The human CBF AML Kasumi-1 cell line, carrying the
t(8;21) and the KIT mutation N822 was purchased from the German
Collection of Microorganisms and Cell Cultures GmbH (Leibniz
Institute). Cells were cultured in RPMI-1640 (Gibco; Thermo Fisher
Scientific, Inc.), supplemented with 20% fetal bovine serum (FBS;
Gibco; Thermo Fischer Scientific, Inc.), 100 µg/ml penicillin
(Invitrogen; Thermo Fischer Scientific, Inc.) and 100 µg/ml
streptomycin (Invitrogen; Thermo Fischer Scientific, Inc.) in 5%
CO2 at 37˚C. The cells were passaged at a ratio of 1:4
every 4 days.
Drug treatment
For drug treatment, Kasumi-1 cells were cultured as
above and treated with low doses of DAC (10, 50, 100, 200 and 400
nΜ), (21), BZ (10 nM) (22), or their combination, at 37˚C in a
humidified atmosphere with 5% CO2, for 24 h for cell
apoptosis and cell cycle profiling and for 6 days for
immunophenotype analysis.
Flow cytometric analysis of cell
apoptosis
Kasumi-1 cells were plated in 6-well-plates
(5x105 cells/well) and treated with different
concentrations of DAC and/or BZ, as described above, for 24 h.
Apoptosis was estimated through Annexin V/propidium iodide staining
(Biolegend) according to the manufacturer's protocol. Flow
cytometric analysis was performed using a CyFlow ML (Partec).
Cell cycle profiling
Kasumi-1 cells were plated in 6-well-plates
(5x105 cells/well) and treated with the indicated
concentrations of DAC and/or BZ for 24 h, harvested, washed in PBS
and fixed in ice-cold methanol for 10 min. Cells were then washed
in PBS and stained with 1 µg/ml of DAPI in PBS for 15 min at room
temperature. The cell-cycle distribution was analyzed on a CyFlow
ML (Partec) equipped with a violet laser.
The synergistic effect between low doses of DAC and
BZ was evaluated by determining the combination index (CI) based on
the Chou-Talalay method (23). The
CI value was calculated using the following equation: CI=(%
decrease of viable cell number by compound A + % decrease of viable
cell number by compound B)/(% decrease of viable cell number by
combination of compound A and B). The combination effect was
defined as ‘synergistic’, ‘additive’ or ‘antagonistic’ when CI was
<1, 1 or >1, respectively (24,25).
Immunophenotypic characterization
Kasumi-1 cells were plated in 6-well-plates
(5x105 cells/well) and treated for 6 days with the
indicated concentrations of DAC and/or BZ. DAC and/or BZ were
administrated fresh every 24 h, along with fresh medium. Untreated
cells were plated at a density of 3x105 cells/well.
After washing and blocking with FcBlocker (cat. no. 422302;
BioLegend, Inc.) for 10 min at room temperature, cells were stained
in three 4-color tubes with FITC-conjugated anti-CD11b (cat. no.
301329; BioLegend, Inc.), anti-CD14 (cat. no. CYT-14F6; Cytognos),
anti-CD15 (cat. no. CYT-15F4; Cytognos), phycoerythrin (PE)-Cyanine
5-conjugated anti-CD16 (cat. no. 302010; BioLegend, Inc.) and
anti-HLA-DR (cat. no. 307608; BioLegend, Inc.),
allophycocyanin-conjugated anti-CD45 (cat. no. CYT-45AP5; Cytognos)
and PE-conjugated anti-CD13 (cat. no. 301704; BioLegend, Inc.),
anti-CD117 (cat. no. 313204; BioLegend, Inc.) and anti-CD193 (cat.
no. 310706; BioLegend, Inc.). The analysis was performed on a
CyFlow ML (Partec).
Statistical analysis
Statistical analyses were performed with SPSS
software (version 25.0; IBM Corp.). All experiments were performed
three times and values are expressed as the mean mean ± standard
deviation. One-way analysis of variance followed by the
least-significant differences/Bonferroni's multiple-comparison
tests were used to compare the means among more than two different
groups. A two-sided P<0.05 was considered to indicate
statistical significance.
Results
BZ synergizes the apoptotic effect of
low-dose DAC on Kasumi-1 cells
Initially, the possible synergistic effect of BZ on
DAC in the induction of apoptosis in AML (M2) Kasumi-1 cells was
assessed.
Single-agent treatment of Kasumi-1 cells with
various concentrations of low-dose DAC (10, 50, 100, 200 or 400 nΜ)
or BZ (10 nM) for 24 h resulted in a statistically significant
increase in the proportion of apoptotic cells from 15.15±0.21% in
the untreated cells to 27.2±7.91 and 28.25±0.77% after treatment
with 100 and 400 nM of DAC (P=0.037 and P=0.026, respectively) and
30.5±3.39% following treatment with 10 nM BZ (P=0.011; Fig. 1).
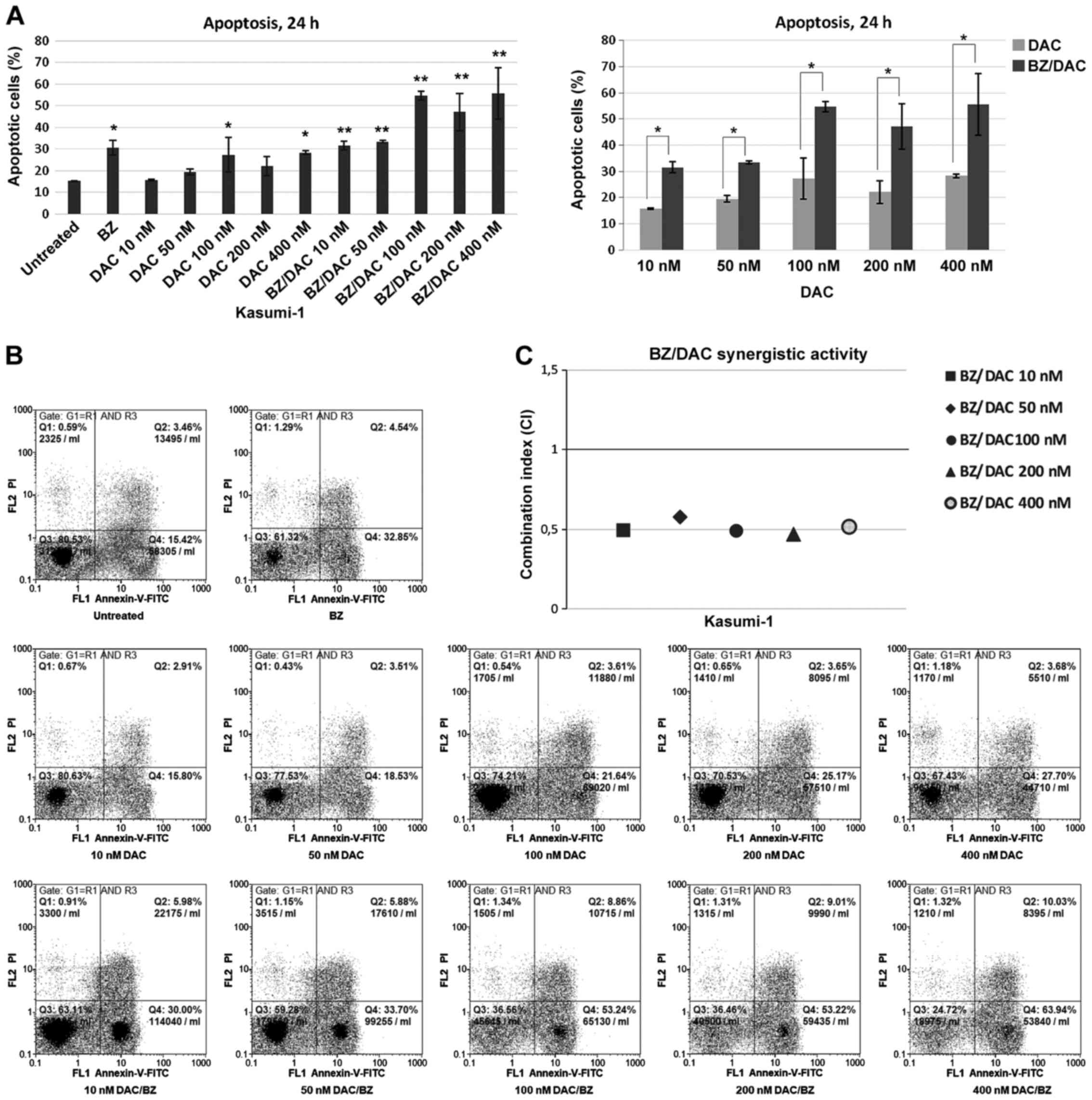 | Figure 1Apoptosis of the acute myeloid
leukemia cell line Kasumi-1 following exposure to low doses of DAC
and/or BZ. (A) Percentage of Annexin V-positive untreated cells and
cells treated with 10, 50, 100, 200 and 400 nM DAC and/or 10 nM BZ
for 24 h. Comparisons between single DAC treatment and combination
treatment with BZ, for all decitabine concentrations examined, are
also presented. Values are expressed as the mean ± standard
deviation of three independent experiments. *P<0.05,
**P<0.01 as indicated or vs. untreated. (B)
Representative dot plots of cells with Annexin V/PI staining for
each drug treatment. Cells were gated on FSC/side scatter to
include cells and on FSC area/FSC width to remove doublets. (C) The
CI was calculated to determine drug interactions. Effects were
defined as synergistic, additive and antagonistic when CI<1,
CI=1 and CI>1, respectively. BZ, bortezomib; DAC,
Dacogen®/decitabine; PI, propidium iodide; q, quadrant;
FSC, forward scatter; CI, combination index. |
When Kasumi-1 cells were subjected to DAC/BZ
combination treatment, increased apoptotic rates compared to
untreated cells were observed for all DAC/BZ combinations tested,
with a higher apoptotic ratio observed after treatments with 100 or
400 nM of DAC with BZ (54.65±1.9 and 55.55±11.8% of apoptotic
cells, respectively; P<0.001). Furthermore, compared to single
DAC treatments, the DAC/BZ combinations significantly increased
apoptosis by 101.92, 72, 100.9, 113.12 and 96.63% (P=0.009 for 10
nM DAC; P=0.019 for 50 nM DAC; P<0.001 for 100, 200 and 400 nM
DAC; Fig. 1) and reduced the live
cell population by 23.51, 22.21, 54.9, 41 and 49.81% for 10, 50,
100, 200 and 400 nΜ of DAC in the DAC/BZ treatments, respectively
(P=0.006 and P=0.011 for 10 and 50 nM of DAC, respectively, and
P<0.001 for the remaining ones; data not shown). Most
importantly, calculation of the CI revealed a synergistic effect
for all low-dose DAC and BZ combinations (CI<1), with the 100
and 200 nM DAC with BZ combinations exhibiting maximum synergy
(CI=0.636 and 0.631, respectively; Fig.
1C). Therefore, these results indicated that DAC/BZ combination
treatment led to significant increases in the apoptotic rate of
Kasumi-1 cells compared with untreated cells and single
treatments.
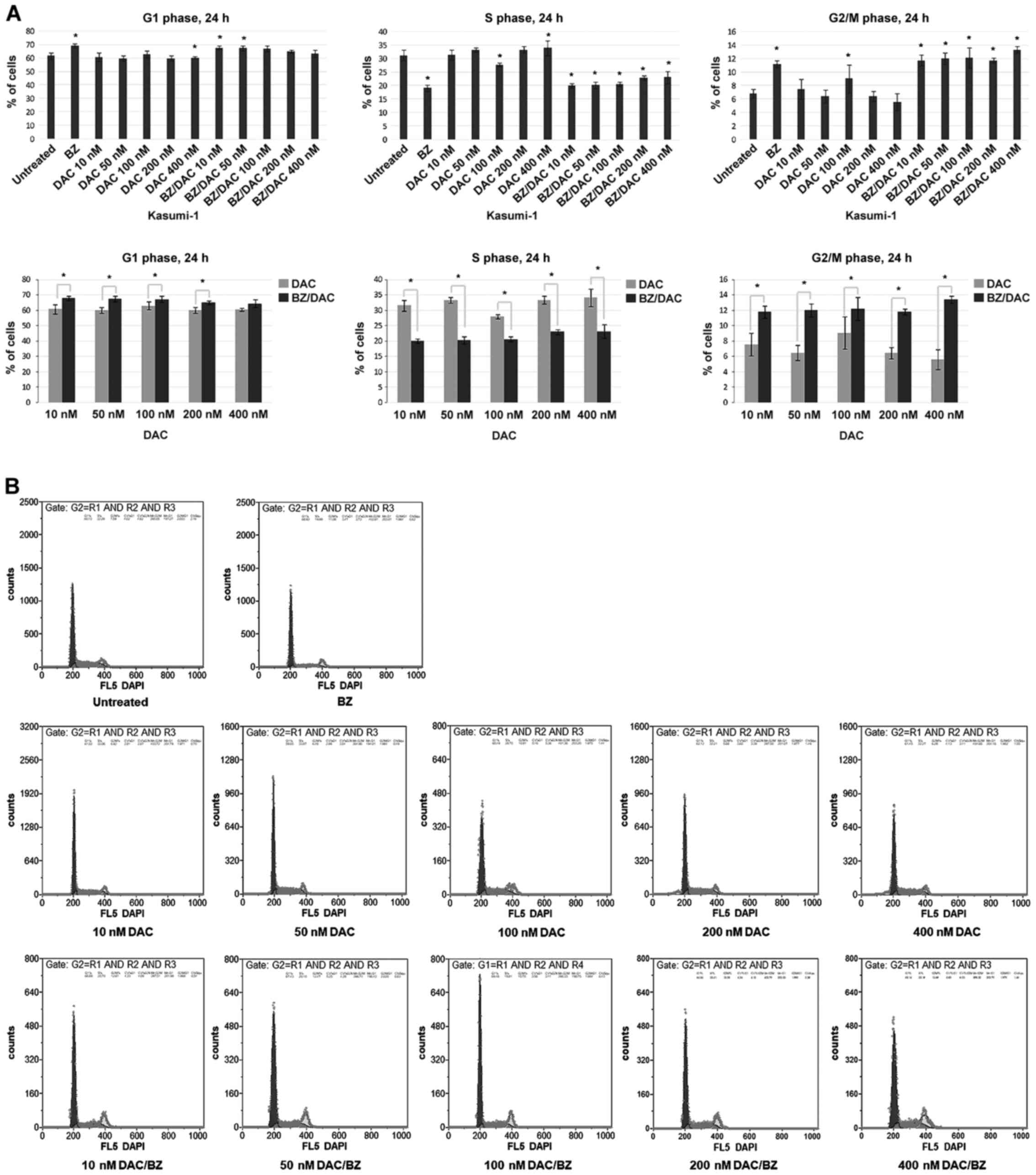
DAC has a higher impact on the cell
cycle in combination with BZ and induces G0/G1- and G2/M-phase
arrest in Kasumi-1 cells
After having demonstrated that the DAC/BZ
combination leads to enhanced apoptosis in Kasumi-1 cells, its
impact on the cell cycle was next investigated. The results
indicated a significantly increased sensitivity of Kasumi-1 cells
exposed to DAC/BZ combinations compared to those subjected to
single treatments. More specifically, cells treated with 10, 50,
100 and 200 nM DAC/BZ appeared with a statistically significant
increase in the G0/G1 population, compared to single treatment, by
11.84% (P<0.001), 12.55% (P<0.001), 6.62% (P=0.021) and 8.38%
(P=0.006), respectively. It should be noted that the 400 nM DAC/BZ
combination also produced an increase by 4.68% in the G0/G1
population compared to single treatment, although this was not
statistically significant (Fig. 2).
A significant decrease in the S-phase population was observed after
all DAC/BZ combination treatments tested compared to single ones,
by 36.26, 39.25, 26, 30.99 and 30.8% from the lower to the higher
DAC concentrations (10, 50, 100, 200 and 400 nM), respectively
(P<0.001; Fig. 2). Finally, the
G2/M population was increased by 56.35% (P<0.001), 85.34%
(P<0.001), 34.29% (P=0.02), 81.92% (P<0.001) and 138.45%
(P<0.001), after treatment with 10, 50, 100, 200 and 400 nM
DAC/BZ respectively, compared with single treatment (Fig. 2). Thus, the DAC/BZ treatments led to
an increase in the proportion of cells in the G0/G1 and G2/M
phases, while decreasing the S-phase population, suggesting the
induction of cell cycle arrest in G0/G1 and G2/M phases.
DAC-induced differentiation of
Kasumi-1 cells is not further enhanced by BZ
The impact of low-dose DAC treatment on Kasumi-1
cell differentiation was then examined by measuring the cell
surface levels of several myeloid, monocyte and granulocytic
antigens (Fig. 3).
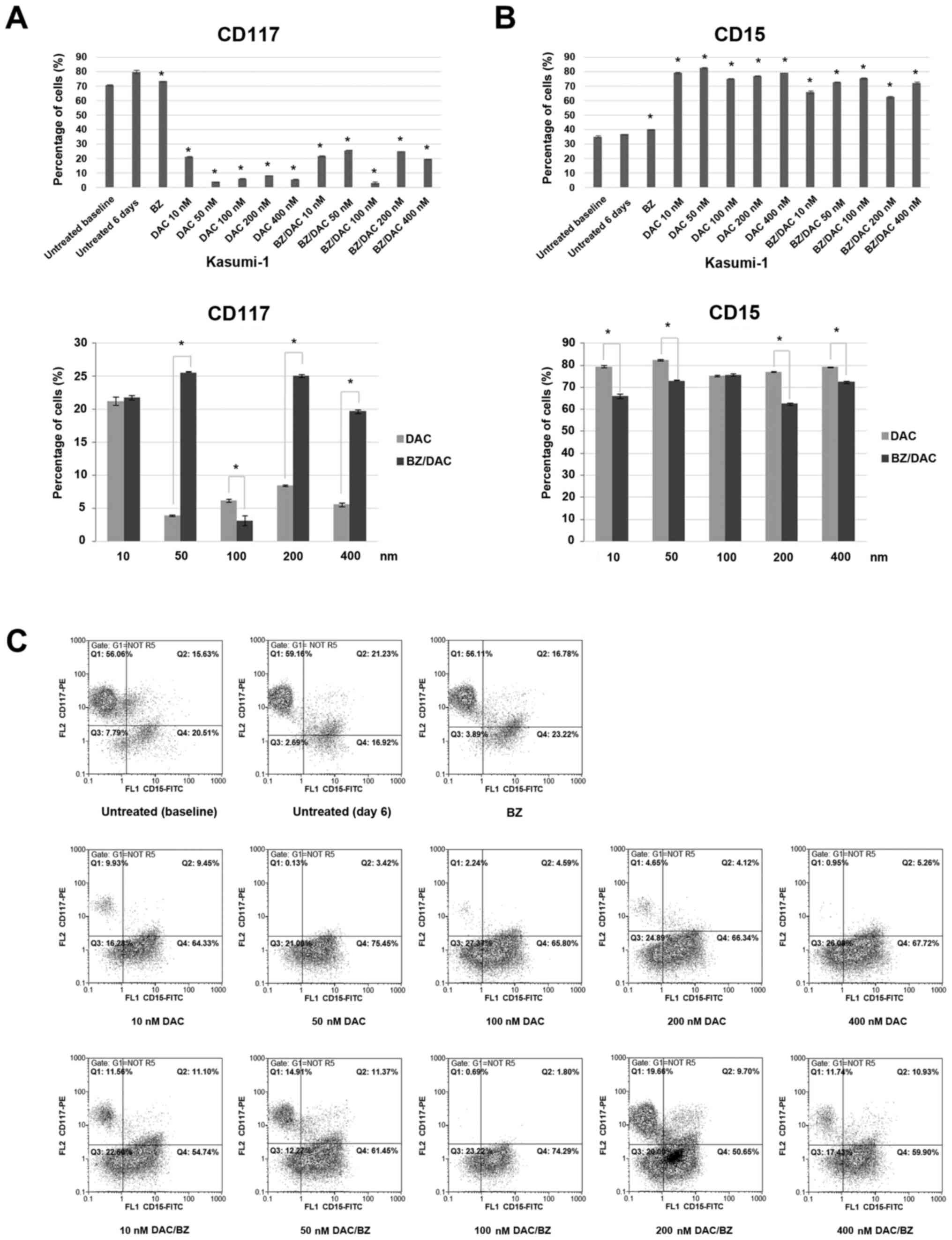 | Figure 3Effect of low-dose decitabine,
bortezomib and their combination on differentiation surface markers
on acute myeloid leukemia Kasumi-1 cells. Cells were exposed for 6
days to 10, 50, 100, 200 or 400 nM DAC and/or 10 nM of BZ, stained
with antibodies against surface markers CD117 and CD15 and analyzed
through flow cytometry. Bars in graphs represent the percentages of
cells expressing (A) the myeloid progenitor surface antigen CD117
and (B) the monocytic surface antigen CD15. Values are expressed as
the mean ± standard deviation of three independent experiments.
*P<0.05 as indicated or vs. untreated (6 days). (C)
Representative fluorescence-assisted cell sorting dot plots for
each staining condition are presented. The expression of myeloid
markers was evaluated in two parameter density plots by gating on
CD45+dim/side scatter low-medium blast cells. Q,
quadrant; unt., untreated; BZ, bortezomib; DAC,
Dacogen®/decitabine; PE, phycoerythrin. |
FACS analysis revealed decreased expression of the
myeloid progenitor surface antigen CD117 after all treatments,
single or combination, compared to 6 day-cultured untreated cells,
which exhibited a CD117 expression ratio of 80.1±1.13%. The
percentages of CD117-expressing cells after the single DAC
treatments were 21.25±0.63, 3.9±0.14, 6.15±0.21, 8.45±0.07 and
5.6±0.28%, respectively, and 21.75±0.35, 25.55±0.49, 3.15±0.07,
25.05±0.21 and 19.7±0.28% for the DAC/BZ combination treatments,
respectively, from the lower to the higher DAC concentration
(P<0.001 for all; Fig. 3A and
C). Of note, apart from the 100 nM
DAC/BZ combination, all other DAC/BZ combination treatments were
significantly less effective in reducing CD117 expression on
Kasumi-1 cells compared to the respective single DAC
treatments.
Furthermore, FACS analysis revealed an increased
expression of the granulocytic antigen CD15 after the treatments,
compared to 6 day-cultured untreated cells, which exhibited a CD15
antigen expression of 36.7±0.28%. The percentages of cells
expressing CD15 from the lower to the higher DAC concentration (10,
50, 100, 200 and 400 nM) were 79.35±0.49, 82.55±0.35, 75.15±0.35,
77.05±0.21 and 79.25±0.21% (P<0.001) for the single DAC
treatments and 65.95±1.06, 72.75±0.35, 75.65±0.49, 62.45±0.63 and
72.35±0.63% (P<0.001) for the DAC/BZ combination treatment,
respectively (Fig. 3B and C). With the exception of 100 nM DAC, all
other single DAC treatments induced a higher expression of CD15 in
Kasumi-1 cells compared to the respective DAC/BZ combinations.
Discussion
Despite the increase in the survival rates of
younger patients with AML over the past years, the 5-year survival
rate of older patients (>60 years old) still remains <8%
(8). Epigenetic alterations in
leukemia constitute an attractive pharmacological target, mostly
due to their reversible nature. DAC, as aforementioned, is an
epigenetic modifying agent, used as first-line therapy for the
treatment of high-risk MDS and AML, particularly for elderly
patients or patients unfit for intensive chemotherapy (26-29).
However, in DAC-treated patients, the estimated complete response
rate is 30-40%, while the majority of responders develop resistance
within the following year (30-33).
Therefore, the present study aimed to examine the
in vitro cellular effects of the epigenetic modifying agent
DAC in combination with BZ - a proteasome inhibitor which also acts
as an oxidative stress inducer - on apoptosis, cell cycle and cell
differentiation in the human AML cell line Kasumi-1, carrying the
t(8;21) and the KIT mutation N822, aiming to enhance the efficacy
of DAC on AML cells.
The results suggested that, although both drugs were
able to inhibit the growth of Kasumi-1 cells, their combination
appeared more potent in suppressing cell proliferation. Indeed, the
present results indicated that the addition of a low concentration
of BZ (10 nM) to low doses of DAC significantly enhanced apoptosis
and decreased the live cell population of Kasumi-1 cells, with the
100 and 200 nM DAC/BZ combinations, which produced the maximum drug
synergy according to the CI values, appearing to be the most
successful ones. Furthermore, cell cycle profiling revealed that
DAC/BZ treatment synergistically led to G0/G1- and G2/M-phase
arrest, hence prohibiting cells to either synthesize DNA (S phase)
or to complete mitosis.
Several studies have previously demonstrated that
DNMTi cause tumor cell death by inducing apoptosis. DAC has also
been reported to induce apoptosis in various leukemia cell lines
(34-36).
Recently, Zeng et al (37)
investigated the effects of DAC treatment on the myeloid MDS cell
line SKM-1 and investigated the role of FOXO3A, a potentially
tumor-suppressive transcription factor, by silencing its expression
prior to DAC treatment. They showed that the activation of FOXO3A
gene is responsible for SKM-1 cell cycle arrest, apoptosis, and
autophagy as well as for the DAC-induced differentiation of SKM-1
cells into monocytes.
Another study (38)
demonstrated that apoptosis of human leukemic cells in response to
treatment with DAC occurs through a mitochondria-mediated pathway
that requires ROS generation upstream for disruption of the
mitochondrial membrane potential, which leads to subsequent
activation of caspases. Zhang et al (39) indicated that treatment of K562
leukemic cells with DAC led to a significant increase of G0/G1-and
G2/M-phase populations, as well as a significant decrease in the
S-phase population, which is in accordance with the present data on
Kasumi-1 cells. Furthermore, they suggested that the methylation
level of death receptor 4 (DR4) gene promoters gradually decreased,
while the mRNA expression levels of DR4 genes gradually increased,
thus suggesting that DAC is able to inhibit the proliferation of
leukemia cells partly by terminating the methylation effect of DR4
gene promoters and restoring the mRNA expression of DR4 genes.
The present study indicated that BZ increased
apoptosis and caused cell cycle alterations in the CBF AML cell
line Kasumi-1 as compared to untreated cells, while it also
displayed synergistic effects with DAC. These results are in line
with the results of a previously published study, reporting that BZ
manages to inhibit the growth of CBF AML by targeting the
miR-29b/SP1/NF-κB(p65) complex-dependent overexpression of KIT
(19). These observations are
further supported by previous studies suggesting that BZ (either as
a single agent or combined with other drugs, e.g., histone
deacetylase inhibitors) inhibits the growth and induces apoptosis
in leukemia cells, even with sensitivity or resistance to
conventional chemotherapeutics (40). Relevant studies strongly suggest
that proteasome inhibition should be considered as a therapy for
leukemia (41,42).
The present study reported a synergistic effect of
DAC and BZ in the CBF AML Kasumi-1 leukemic cell line in terms of
induction of apoptosis and inhibition of cell growth. Indeed, the
combination of DAC and BZ has been previously studied in
vitro in RPMI-8226 multiple myeloma cells and was indicated to
enhance the apoptotic rate and induce G0/G1 arrest compared to
single-agent therapy. Furthermore, it increased caspase-3 and -9
activation and downregulated the expression levels of
DNMT1(43). Recently, another in
vitro study by Jin et al (44) reported the synergistic antitumor
efficacy of the combination of DAC and BZ in human multiple myeloma
cell lines and revealed that DAC was able to synergistically
enhance myeloma cell sensitivity to BZ by regulating Wnt/β-catenin
signaling. Specifically, the study proved that DAC demethylated and
induced the re-expression of the Wnt antagonists secreted
frizzled-related protein 3 and dickkopf WNT signaling inhibitor 1,
while it also reduced glycogen synthase kinase 3β (Ser9)
phosphorylation and decreased the BZ-mediated β-catenin
accumulation in the nucleus. Thus, the transcription of cyclin D1,
c-Myc and LEF/TCF was reduced, which synergistically inhibited cell
proliferation, enhanced BZ-induced apoptosis and promoted
BZ-induced cell cycle arrest in myeloma cells. Since the
dysregulation of Wnt/β-catenin signaling has been implicated in
various hematological malignancies, including AML (45), such a mechanism may also be involved
in the effects of DAC/BZ co-treatment of AML Kasumi-1 cells,
further supporting the present observations regarding the
synergistic activity of the two agents. Of note, a phase I clinical
study by Blum et al (46)
including 19 poor-risk patients AML reported promising clinical
effects of the DAC/BZ combination and suggested that in AML, BZ
acts through downregulation of fms-related receptor tyrosine kinase
3 due to upregulation of miR-29b, further validating the present
observations in the Kasumi-1 cell line. Recently, a randomized
phase 2 trial tested the efficacy of a 10-day DAC treatment with a
10-day DAC/BZ treatment, in previously untreated, newly diagnosed,
elderly patients with AML (47).
Although no clinical improvement was observed, responses were
better than those in previous trials using 5-day DAC cycles.
Currently, an ongoing phase I trial (registered at www.clinicaltrials.gov as #NCT01861314) studied the
side effects and the optimum dose of bortezomib and sorafenib to
sylate when administered together with decitabine in patients with
AML, although its results are yet to be announced.
Finally, the present results suggested that low
doses of DAC induced monocytic and granulocytic differentiation of
Kasumi-1 cells, in line with similar previous studies (13,14,37).
However, BZ did not appear to enhance the ability of DAC to induce
the differentiation of Kasumi-1 cells and therefore, the
synergistic activity of the two agents appears to rely on other
mechanisms of action, including the induction of apoptosis and cell
cycle alterations, possibly due to upstream events, such as DNA
damage and oxidative stress production, that lead to cell growth
inhibition.
In conclusion, the present study demonstrated that
in human CBF AML Kasumi-1 cells, low doses of DAC in combination
with BZ synergistically induce apoptosis and lead to G0/G1- and
G2/M-phase arrest, hence prohibiting cells to either synthesize DNA
(S phase) or complete mitosis. Although further in vitro
investigation in other DAC-resistant AML cell lines and in
vivo verifications are necessary, these results provide a
strong rationale for the integration of combination treatment with
DAC and BZ in AML therapy, followed by DAC alone, which will
hopefully lead to better clinical responses and possibly partially
overcome the frequently observed DAC resistance in patients with
AML.
Acknowledgements
Not applicable.
Funding
The present study was supported by Janssen Cilag
Pharmaceutical S.A.C.I, a Johnson & Johnson, Company, under an
Oncology & Haematology Non-Clinical IIS Research Grant.
Availability of data and materials
All data generated and analyzed in the present study
are available from the corresponding author on request.
Authors' contributions
VM conceived and designed the study, performed
experiments, analyzed data and drafted the manuscript. AS performed
part of the experiments, performed the statistical analysis of the
data and contributed to drafting the manuscript. AB, EM, SP, KG, EG
and TT designed the study and drafted the manuscript. PF supervised
and coordinated the study. VP conceived and supervised the study,
participated in its design and coordination and reviewed the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The present study was supported by Janssen Cilag
Pharmaceutical S.A.C.I, a Johnson & Johnson, Company, under an
Oncology & Haematology Non-Clinical IIS Research Grant, and
included drug supply and financial support.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Lapidot T, Sirard C, Vormoor J, Murdoch B,
Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA and
Dick JE: A cell initiating human acute myeloid leukaemia after
transplantation into SCID mice. Nature. 367:645–648.
1994.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Bonnet D and Dick JE: Human acute myeloid
leukemia is organized as a hierarchy that originates from a
primitive hematopoietic cell. Nat Med. 3:730–737. 1997.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Konopleva MY and Jordan CT: Leukemia stem
cells and microenvironment: Biology and therapeutic targeting. J
Clin Oncol. 29:591–599. 2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Lane SW, Wang YJ, Lo Celso C, Ragu C,
Bullinger L, Sykes SM, Ferraro F, Shterental S, Lin CP, Gilliland
DG, et al: Differential niche and Wnt requirements during acute
myeloid leukemia progression. Blood. 118:2849–2856. 2011.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Valent P, Sadovnik I, Eisenwort G, Bauer
K, Herrmann H, Gleixner KV, Schulenburg A, Rabitsch W, Sperr WR and
Wolf D: Immunotherapy based targeting and elimination of leukemic
stem cells in AML and CML. Int J Mol Sci. 20(4233)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yates JW, Wallace HJ Jr, Ellison RR and
Holland JF: Cytosine arabinoside (NSC-63878) and daunorubicin
(NSC-83142) therapy in acute nonlymphocytic leukemia. Cancer
Chemother Rep. 57:485–488. 1973.PubMed/NCBI
|
|
8
|
Burnett A, Wetzler M and Löwenberg B:
Therapeutic advances in acute myeloid leukemia. J Clin Oncol.
29:487–494. 2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Bose P, Vachhani P and Cortes JE:
Treatment of relapsed/refractory acute myeloid leukemia. Curr Treat
Options Oncol. 18(17)2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kantarjian H, Oki Y, Garcia-Manero G,
Huang X, O'Brien S, Cortes J, Faderl S, Bueso-Ramos C, Ravandi F,
Estrov Z, et al: Results of a randomized study of 3 schedules of
low-dose decitabine in higher-risk myelodysplastic syndrome and
chronic myelomonocytic leukemia. Blood. 109:52–57. 2007.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Oki Y, Aoki E and Issa JP:
Decitabine--bedside to bench. Crit Rev Oncol Hematol. 61:140–152.
2007.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Oki Y and Issa JP: Treatment options in
advanced myelodysplastic syndrome, with emphasis on epigenetic
therapy. Int J Hematol. 86:306–314. 2007.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ding L, Qiu L, Zhang J and Guo B:
Camptothecin-induced cell proliferation inhibition and apoptosis
enhanced by DNA methyltransferase inhibitor,
5-aza-2'-deoxycytidine. Biol Pharm Bull. 32:1105–1108.
2009.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Turcan S, Fabius AW, Borodovsky A, Pedraza
A, Brennan C, Huse J, Viale A, Riggins GJ and Chan TA: Efficient
induction of differentiation and growth inhibition in IDH1 mutant
glioma cells by the DNMT Inhibitor Decitabine. Oncotarget.
4:1729–1736. 2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Fribley A, Zeng Q and Wang CY: Proteasome
inhibitor PS-341 induces apoptosis through induction of endoplasmic
reticulum stress-reactive oxygen species in head and neck squamous
cell carcinoma cells. Mol Cell Biol. 24:9695–9704. 2004.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Fribley A and Wang CY: Proteasome
inhibitor induces apoptosis through induction of endoplasmic
reticulum stress. Cancer Biol Ther. 5:745–748. 2006.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Cortes J, Thomas D, Koller C, Giles F,
Estey E, Faderl S, Garcia-Manero G, McConkey D, Ruiz SL,
Guerciolini R, et al: Phase I study of bortezomib in refractory or
relapsed acute leukemias. Clin Cancer Res. 10:3371–3376.
2004.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Attar EC, De Angelo DJ, Supko JG, D'Amato
F, Zahrieh D, Sirulnik A, Wadleigh M, Ballen KK, McAfee S, Miller
KB, et al: Phase I and pharmacokinetic study of bortezomib in
combination with idarubicin and cytarabine in patients with acute
myelogenous leukemia. Clin Cancer Res. 14:1446–1454.
2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Liu S, Liu Z, Xie Z, Pang J, Yu J, Lehmann
E, Huynh L, Vukosavljevic T, Takeki M, Klisovic RB, et al:
Bortezomib induces DNA hypomethylation and silenced gene
transcription by interfering with Sp1/NF-kappaB-dependent DNA
methyltransferase activity in acute myeloid leukemia. Blood.
111:2364–2373. 2008.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Liu S, Wu LC, Pang J, Santhanam R, Schwind
S, Wu YZ, Hickey CJ, Yu J, Becker H, Maharry K, et al:
Sp1/NFkappaB/HDAC/miR-29b regulatory network in KIT-driven myeloid
leukemia. Cancer Cell. 17:333–347. 2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Klco JM, Spencer DH, Lamprecht TL,
Sarkaria SM, Wylie T, Magrini V, Hundal J, Walker J, Varghese N,
Erdmann-Gilmore P, et al: Genomic impact of transient low-dose
decitabine treatment on primary AML cells. Blood. 121:1633–1643.
2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Roccaro AM, Hideshima T, Raje N, Kumar S,
Ishitsuka K, Yasui H, Shiraishi N, Ribatti D, Nico B, Vacca A, et
al: Bortezomib mediates antiangiogenesis in multiple myeloma via
direct and indirect effects on endothelial cells. Cancer Res.
66:184–191. 2006.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Tomikoshi Y, Nomura M, Okudaira N,
Sakagami H and Wakabayashi H: Enhancement of cytotoxicity of three
apoptosis-inducing agents against human oral squamous cell
carcinoma cell line by benzoxazinotropone. In Vivo. 30:645–650.
2016.PubMed/NCBI
|
|
25
|
Iijima Y, Bandow K, Sano M, Hino S, Kaneko
T, Horie N and Sakagami H: In vitro assessment of antitumor
potential and combination effect of classical and
molecular-targeted anticancer drugs. Anticancer Res. 39:6673–6684.
2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Jabbour E, Issa JP, Garcia-Manero G and
Kantarjian H: Evolution of decitabine development: Accomplishments,
ongoing investigations, and future strategies. Cancer.
112:2341–2351. 2008.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Erba HP: Finding the optimal combination
therapy for the treatment of newly diagnosed AML in older patients
unfit for intensive therapy. Leuk Res. 39:183–191. 2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Welch JS, Petti AA, Miller CA, Fronick CC,
O'Laughlin M, Fulton RS, Wilson RK, Baty JD, Duncavage EJ, Tandon
B, et al: TP53 and Decitabine in Acute Myeloid Leukemia and
Myelodysplastic Syndromes. N Engl J Med. 375:2023–2036.
2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Tamamyan G, Kadia T, Ravandi F, Borthakur
G, Cortes J, Jabbour E, Daver N, Ohanian M, Kantarjian H and
Konopleva M: Frontline treatment of acute myeloid leukemia in
adults. Crit Rev Oncol Hematol. 110:20–34. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Blum W, Garzon R, Klisovic RB, Schwind S,
Walker A, Geyer S, Liu S, Havelange V, Becker H, Schaaf L, et al:
Clinical response and miR-29b predictive significance in older AML
patients treated with a 10-day schedule of decitabine. Proc Natl
Acad Sci USA. 107:7473–7478. 2010.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ritchie EK, Feldman EJ, Christos PJ, Rohan
SD, Lagassa CB, Ippoliti C, Scandura JM, Carlson K and Roboz GJ:
Decitabine in patients with newly diagnosed and relapsed acute
myeloid leukemia. Leuk Lymphoma. 54:2003–2007. 2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Bhatnagar B, Duong VH, Gourdin TS, Tidwell
ML, Chen C, Ning Y, Emadi A, Sausville EA and Baer MR: Ten-day
decitabine as initial therapy for newly diagnosed patients with
acute myeloid leukemia unfit for intensive chemotherapy. Leuk
Lymphoma. 55:1533–1537. 2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Khan N, Hantel A, Knoebel RW, Artz A,
Larson RA, Godley LA, Thirman MJ, Liu H, Churpek JE, King D, et al:
Efficacy of single-agent decitabine in relapsed and refractory
acute myeloid leukemia. Leuk Lymphoma. 58:1–7. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Stresemann C, Bokelmann I, Mahlknecht U
and Lyko F: Azacytidine causes complex DNA methylation responses in
myeloid leukemia. Mol Cancer Ther. 7:2998–3005. 2008.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Valdez BC, Li Y, Murray D, Corn P,
Champlin RE and Andersson BS: 5-Aza-2'-deoxycytidine sensitizes
busulfan-resistant myeloid leukemia cells by regulating expression
of genes involved in cell cycle checkpoint and apoptosis. Leuk Res.
34:364–372. 2010.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Tsujioka T, Yokoi A, Uesugi M, Kishimoto
M, Tochigi A, Suemori S, Tohyama Y and Tohyama K: Effects of DNA
methyltransferase inhibitors (DNMTIs) on MDS-derived cell lines.
Exp Hematol. 41:189–197. 2013.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Zeng W, Dai H, Yan M, Cai X, Luo H, Ke M
and Liu Z: Decitabine-Induced Changes in Human Myelodysplastic
Syndrome Cell Line SKM-1 Are Mediated by FOXO3A Activation. J
Immunol Res. 2017(4302320)2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Shin DY, Park YS, Yang K, Kim GY, Kim WJ,
Han MH, Kang HS and Choi YH: Decitabine, a DNA methyltransferase
inhibitor, induces apoptosis in human leukemia cells through
intracellular reactive oxygen species generation. Int J Oncol.
41:910–918. 2012.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Zhang W, Chen Y, Pei X, Zang Y and Han S:
Effects of Decitabine on the proliferation of K562 cells and the
expression of DR4 gene. Saudi J Biol Sci. 25:242–247.
2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Csizmar CM, Kim DH and Sachs Z: The role
of the proteasome in AML. Blood Cancer J. 6(e503)2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Vink J, Cloos J and Kaspers GJ: Proteasome
inhibition as novel treatment strategy in leukaemia. Br J Haematol.
134:253–262. 2006.PubMed/NCBI View Article : Google Scholar
|
|
42
|
McCloskey SM, McMullin MF, Walker B and
Irvine AE: The therapeutic potential of the proteasome in
leukaemia. Hematol Oncol. 26:73–81. 2008.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Cao Y, Qiu GQ, Wu HQ, Wang ZL, Lin Y, Wu
W, Xie XB and Gu WY: Decitabine enhances bortezomib treatment in
RPMI 8226 multiple myeloma cells. Mol Med Rep. 14:3469–3475.
2016.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Jin Y, Xu L, Wu X, Feng J, Shu M, Gu H,
Gao G, Zhang J, Dong B and Chen X: Synergistic efficacy of the
demethylation agent decitabine in combination with the protease
inhibitor bortezomib for treating multiple myeloma through the
Wnt/β-catenin pathway. Oncol Res. 27:729–737. 2019.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Grainger S, Traver D and Willert K: Wnt
signaling in hematological malignancies. Prog Mol Biol Transl Sci.
153:321–341. 2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Blum W, Schwind S, Tarighat SS, Geyer S,
Eisfeld AK, Whitman S, Walker A, Klisovic R, Byrd JC, Santhanam R,
et al: Clinical and pharmacodynamic activity of bortezomib and
decitabine in acute myeloid leukemia. Blood. 119:6025–6031.
2012.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Roboz GJ, Mandrekar SJ, Desai P, Laumann
K, Walker AR, Wang ES, Kolitz JE, Powell BL, Attar EC, Stock W, et
al: Randomized trial of 10 days of decitabine ± bortezomib in
untreated older patients with AML: CALGB 11002 (Alliance). Blood
Adv. 2:3608–3617. 2018.PubMed/NCBI View Article : Google Scholar
|