Introduction
Immunoglobulin G4-related disease (IgG4-RD) is a
regional or systemic fibroinflammatory disease of unknown etiology
(1) that predominantly occurs in
middle-aged and elderly males (40-70 years old). It is
characterized by compressive lesions with inflammation or
neoplastic swelling, usually but not always elevated serum IgG4
levels and a typical histopathological appearance. The key
pathological features are a dense lymphoplasmacytic infiltrate,
particularly IgG4-positive cells, a storiform pattern of fibrosis
and obliterative phlebitis (1,2). The
disease may involve the eyes, salivary glands, lymph nodes,
pituitary glands, dura mater, thyroid, lung, pleura, breast,
pericardium, aorta, retroperitoneum, pancreas, bile duct, kidney,
prostate and skin.
It was reported that the prevalence of autoimmune
pancreatitis (AIP) was 10.1/100,000 individuals in Japan in
2016(3). When IgG4-positive cells
infiltrate the lacrimal glands, orbital soft tissues or extraocular
muscles, it is referred to as IgG4-related ophthalmopathy
(IgG4-ROD) (4). Due to the rare
involvement of the optic nerve, visual impairment is an uncommon
symptom. The present study reported on the case of a patient
diagnosed with IgG4-RD probably involving at least 10 organs with
visual impairment as the major complaint, who exhibited sensitivity
to hormone therapy with obvious therapeutic effects.
Case report
A 50-year-old male presented at the First Affiliated
Hospital of China Medical University (Shenyang, China) with
repeated diplopia for 14 years, visual impairment for 2 months and
subxiphoid pain for 1 month in May 2019. The patient had suffered
from periocular swelling and diplopia repeatedly since 2005, but he
did not complain of visual impairment until nearly two months prior
to admission. The patient was treated several times for the above
symptoms. He was diagnosed with an inflammatory pseudotumor and
then subjected to pseudotumor resection and postoperative
radiotherapy. The patient had also been diagnosed with Graves'
ophthalmopathy with normal thyroid function and given hormone shock
therapy numerous times. However, the prognosis of the patient was
unsatisfactory, and the symptoms kept on recurring. At one month
prior to admission, the patient developed subxiphoid pain lasting
~3 h after eating yogurt, accompanied by nausea and vomiting. The
symptoms occurred repeatedly thereafter, more frequently after
eating. The pain lasted ~2-3 min and improved by itself. The
patient also had problems with urinary weakness. The patient
exhibited a reduction in body weight by 5 kg over the past 2
months. He had a history of sinusitis for 30 years, secretory
otitis media for 7 years and skin eczema for 7 years. An endoscopic
operation was performed for sinusitis and secretory otitis media in
2013.
Physical examination revealed the following:
Multiple enlarged lymph nodes in the left mandible, bilateral neck
and bilateral supraclavicular fossa were palpated; these nodes were
of medium hardness, movable and painless. Bilateral eyelid edema
and a left lower eyelid mass were discovered.
Laboratory investigation revealed the following: All
enzymes representing liver and bile duct damage were elevated,
including alanine aminotransferase (ALT) at 160 U/l (normal range,
9-50 U/l), aspartate aminotransferase (AST) at 142 U/l (normal
range, 15-40 U/l), γ-glutamyl transpeptidase (GGT) at 742 U/l
(normal range, 10-60 U/l) and alkaline phosphatase (ALP) at 467 U/l
(normal range, 45-125 U/l). Direct bilirubin was slightly elevated
to 8.3 µmol/l (normal range, 0-6.8 µmol/l), while albumin was
severely reduced to 21.2 g/l (normal range, 40-55 g/l). Both
amylase (AMY) and lipase (LPS) were increased to 120 U/l (normal
range, 28-100 U/l) and 136.1 U/l (normal range, 13-60 U/l),
respectively. The erythrocyte sedimentation rate and procalcitonin
were also increased. The T cell spot test was positive. Except for
elevated thyroid stimulating hormone (TSH) and anti-thyroglobulin
antibody, there were no thyroid function abnormalities [free
thyroid (FT) hormone 3, FT4 and anti-thyroid peroxidase antibody
were all normal]. Both IgG and IgG4 were elevated to 85.44 g/l
(normal range, 7-17 g/l) and 36.9 g/l (normal range, 0.03-2.01
g/l), respectively. Antinuclear antibody was positive. The antigen
spectrum of autoimmune liver disease (e.g., anti-mitochondrial
antibody-M2), anti-hepatorenal microsomes-1, anti-hepatocyte
cytoplasmic antigen type 1 antibody, anti-soluble liver
antigen/hepatopancreas antigen antibody, Ro-52, anti-promyelocytic
leukemia protein antibody, nuclear autoantigen Sp-100, nuclear pore
protein gp210 and 2-ketoacid dehydrogenase complex was negative.
Other markers, such as C-reactive protein, complement,
immunofixation electrophoresis, coagulation, hepatitis, viral
antibody and tumor markers, were normal. The right intraocular
pressure was 14 mmHg and the left intraocular pressure was 16 mmHg.
The visual acuity of both eyes was 0.6. Defects in the right lower
half, right upper half and left infratemporal region were detected
in the 30-degree visual field. The synoptic muscle exhibited left
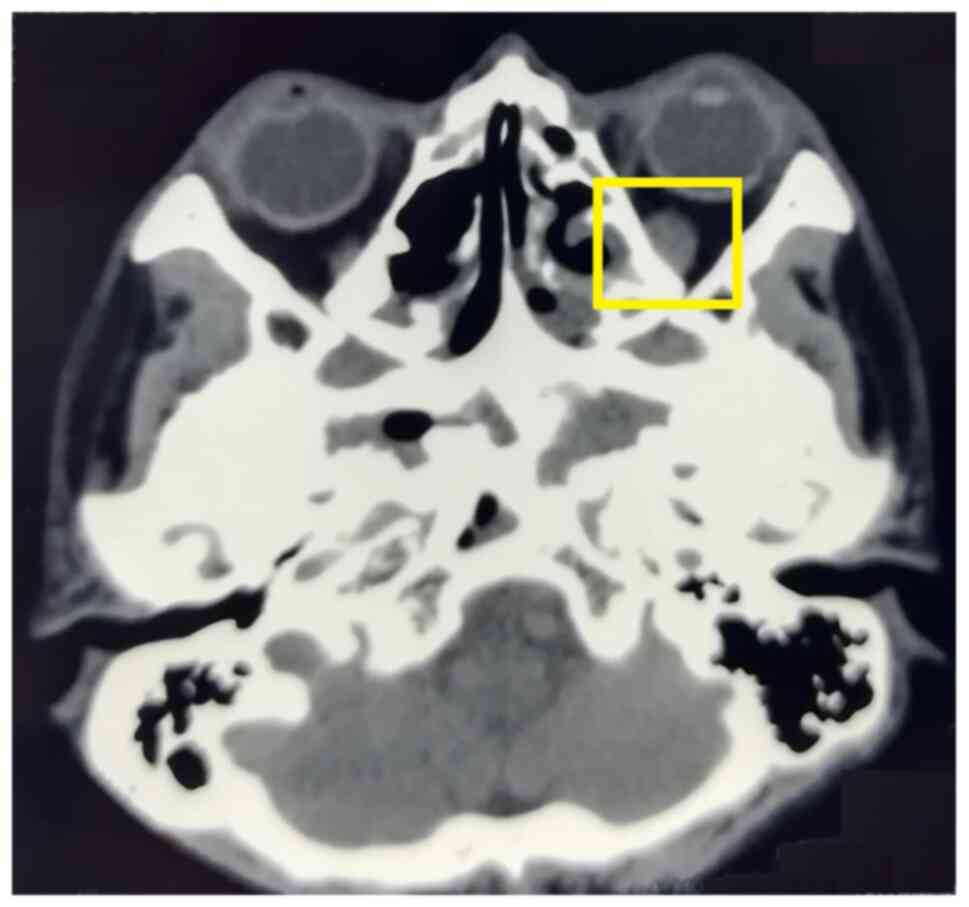
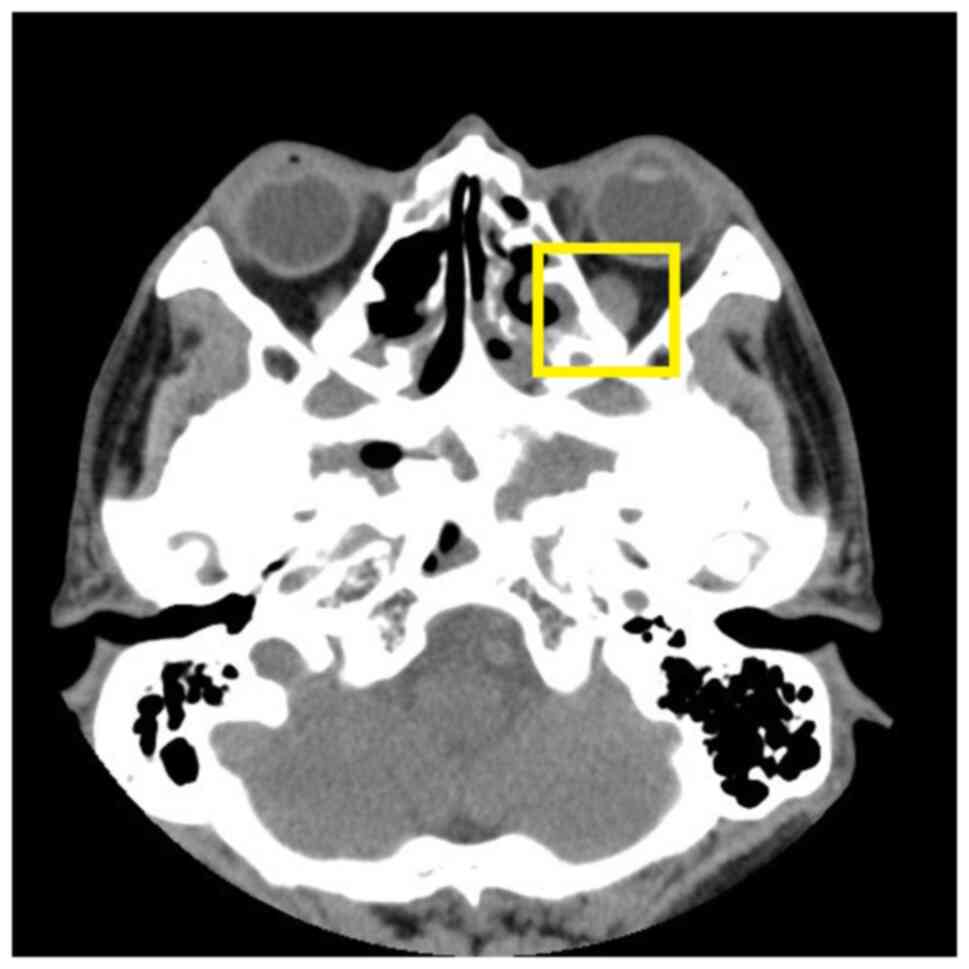
inferior oblique paralysis. Orbital CT (Fig. 1) revealed a slightly thickened left
inferior rectus muscle and paranasal sinusitis. Mild interstitial
changes, slight chronic inflammation in both lungs, small nodules
in both lungs, and mediastinal and bilateral axillary
lymphadenopathy were detected on pulmonary CT. Prostate hypertrophy
and calcification and lymphadenopathy around the bilateral internal
iliac vessels were also observed on abdomen-enhanced CT. An
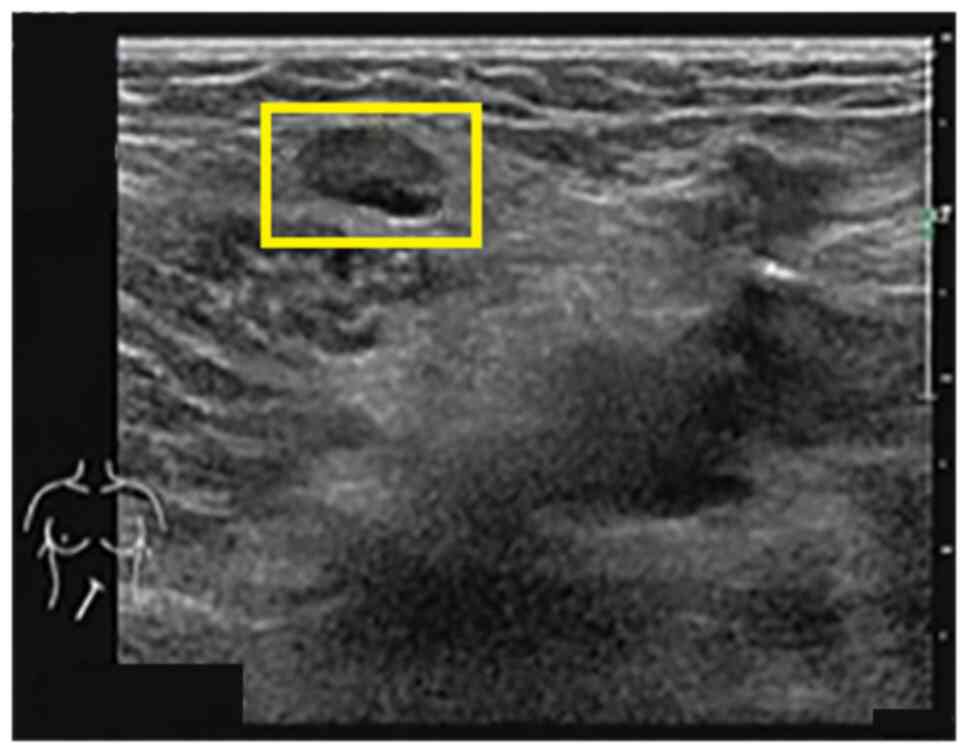
ultrasound scan of the superficial lymph nodes revealed that the
bilateral neck, bilateral supraclavicular fossa, bilateral
subclavian fossa, submental, left axilla, right groin (grades 3-4,
suspected malignancy) (Fig. 2),
right axilla and left groin (grade 3) exhibited multiple enlarged
lymph nodes. A hypoechoic mass was detected in the left
submandibular gland. Rough echo of the liver parenchyma and
increased elasticity of the liver (10.6 kpa) were observed in the
abdominal ultrasound scan.
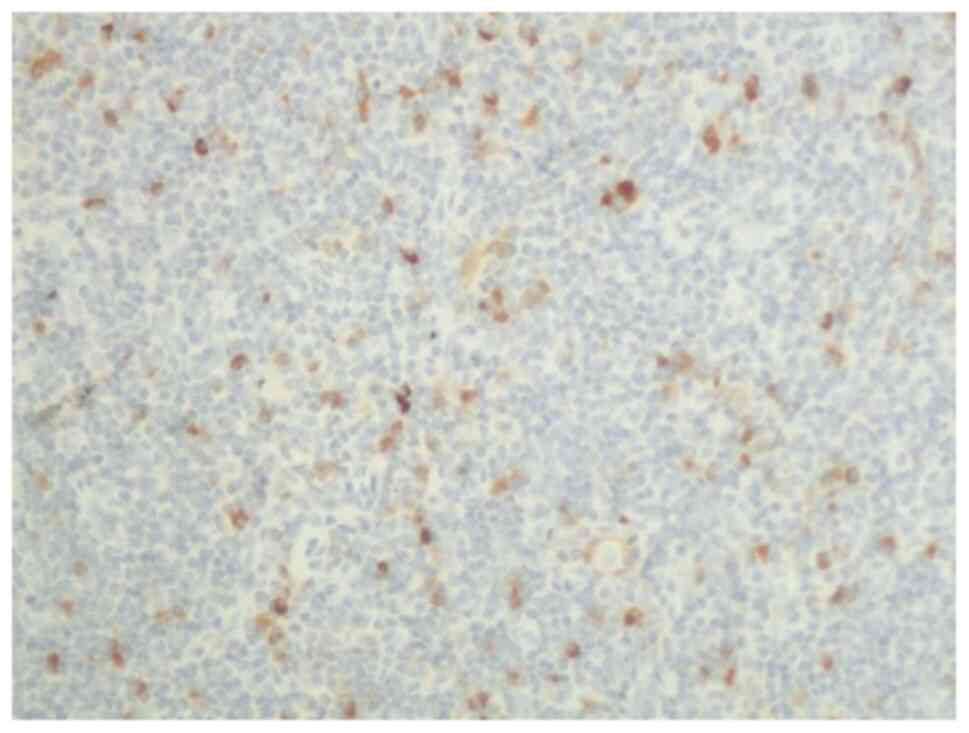
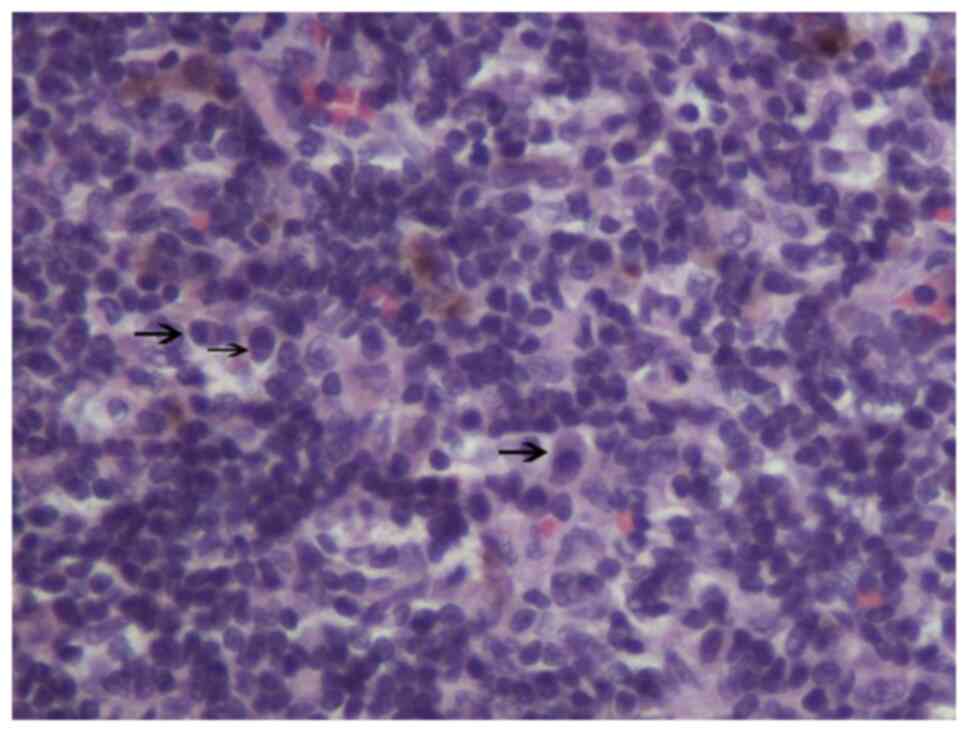
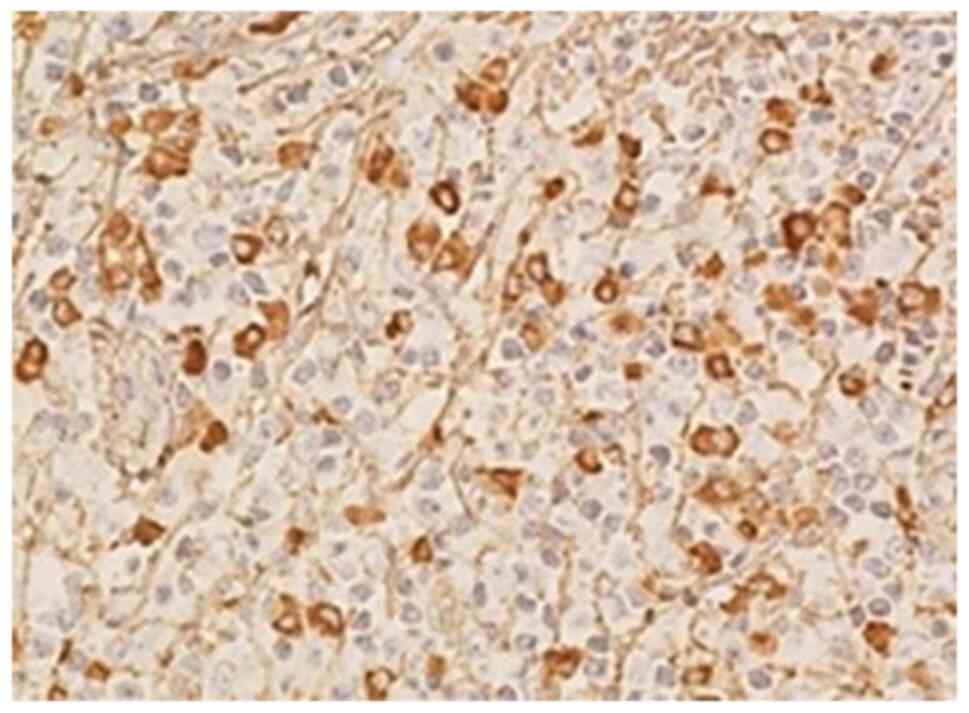
The patient underwent biopsy of the left inguinal
lymph node. Pathological analysis indicated >10 IgG4-positive
cells in the marginal sinus per high-power field (magnification,
x200; Fig. 3) and scattered
plasma-cell infiltration was observed per high-power field
(magnification, x400; Fig. 4). The
immunohistochemistry results were as follows: Creatine kinase (-),
CD3 (diffuse +), CD20 (germinal center +), CD30 (scatter +), paired
box protein Pax-5 (germinal center +), Bcl-2 (follicular -), CD21
(dendritic cells +), CD68 (scatter +), Ki-67 (~15%), CD138 (focal
+), CD38 (partial +), CD1a (scatter +), S-100 (focal +) and IgG4
(scatter + cells could be observed in the subcapsular sinus and
medullary sinus). According to the latest diagnostic criteria of
the Japanese Study Group in 2011(5), the patient was diagnosed with IgG4-RD
involving the lymph nodes.
The patient's medical records from visits to other
hospitals in 2013 were collected. A binocular MRI scan and
enhancement indicated protruding bilateral eyeballs, bilateral
intraorbital lacrimal glands and a lower left orbital mass with a
slightly longer T1 signal, a slightly shorter T2 signal, poor
display of lacrimal glands, displacement of corresponding
horizontal extraocular muscles and eyeballs, local enlargement and
irregularly shaped extraocular muscles. Following injection of
GD-DPTA (a contrast agent), the lesions displayed with moderate
contrast and homogeneous enhancement. In the pathological sections
of the periocular mass, >10 IgG4-positive cells per high-power
field were determined (magnification, x200; Fig. 5), which was consistent with the
pathological features of IgG4-RD. Finally, the patient was
diagnosed with IgG4-ROD. The previous misdiagnosis may have been
due to an insufficient understanding of IgG4-RD.
Based on the patient's medical history and previous
studies (1,6-8),
it was indicated that the submandibular glands, sinuses, middle
ears, bile duct, liver, pancreas, prostate and skin may also be
involved in IgG4-RD. IgG4-RD is most prevalent in older males
(1), and in this patient population
prostate problems are very common, therefore there may be a
connection.
The patient was treated with 120 mg of intravenous
methylprednisolone daily, which was gradually reduced to 40 mg/day
after three weeks, and then decreased by 5 mg every two weeks.
Considering the latent infection with tuberculosis bacterium, 0.3 g
oral isoniazid was administered once daily to prevent tuberculosis.
After oral administration of methylprednisolone for two months, the
eyelid edema was relieved, the left lower eyelid mass became
smaller and the bilateral neck and left submandibular lymph nodes
were significantly reduced. The symptoms of urination and eczema,
smelling caused by sinusitis and hearing caused by secretory otitis
media were all improved and no nausea or vomiting symptoms
occurred. ALT, AST, GGT and ALP were all decreased to 25, 17, 80
and 70 U/l, respectively. AMY and LPS became normal. IgG4 decreased
to 1.32 g/l (normal level) and albumin increased to 37 g/l.
The lymph nodes of the bilateral neck, bilateral
supraclavicular fossa and submandibular gland became smaller on the
ultrasound scan. The lymph node grade at the first two sites
changed to grade 2, excluding malignancy. Orbital CT revealed that
the enlarged left inferior rectus muscle became slightly narrowed
(Fig. 6). The visual acuity of both
eyes exhibited marked improvements and the vision of both eyes
increased to 1.0.
Discussion
The patient described in the present study had a
long history of hospital visits with large expenses. He was
misdiagnosed with an ‘inflammatory pseudotumor’ and ‘Graves'
ophthalmopathy’ and the treatment effects were poor. An accurate
diagnosis of IgG4-RD was difficult. The proposed IgG4-RD was based
on cumulative evidence of AIP and related extrapancreatic lesions.
In 2001 and 2002, Hamano et al (9,10)
reported an elevation in the serum IgG4 concentration in patients
with AIP and the infiltration of IgG4-positive plasma cells in the
pancreas and retroperitoneal tissues, respectively. In 2003,
Kamisawa et al (11)
reported that IgG4-positive plasma cells invaded the pancreas and
extrapancreatic tissue in type 1 AIP and suggested that this
phenomenon may be referred to as IgG4-RD. Ultimately, the
diagnostic criteria for IgG4-RD were established in 2011(5). However, articles on the incidence of
IgG4-RD remain rare. In 2016, a national epidemiological survey was
performed in Japan. The prevalence of AIP was 10.1/100,000
individuals, which more than doubled in 2011(3).
A retrospective analysis of 103 patients previously
diagnosed with an inflammatory pseudotumor was recently performed
(12). Immunohistochemical analysis
of IgG and IgG4 was performed on 16 biopsy samples and six cases of
IgG4-ROD and 10 cases of orbital inflammatory pseudotumor were
diagnosed. The aforementioned study (12) has also indicated that lesions in the
lacrimal gland, IgG4-positive plasma-cell counts and ratios, as
well as collagen fibrosis, are helpful for the diagnosis of
IgG4-ROD in patients with orbital mass-like lesions. In addition,
as an idiopathic inflammatory response, inflammatory pseudotumors
are frequently accompanied by obvious pain. Tiegs-Heiden et
al (13) reviewed the imaging
features of 27 IgG4-ROD cases confirmed by biopsy. The vast
majority of patients (89%) had enlarged extraocular muscles. The
external rectus was the most frequently involved muscle, while the
inferior rectus and medial rectus were frequently involved in
Graves' ophthalmopathy. A small number of patients may exhibit
characteristics that differ from those described above, such as the
case of the present study. If TSH receptor antibody is negative in
a patient suspected of having Graves' orbital lesions with normal
thyroid function, the presence of IgG4-ROD must be considered. In
conclusion, when orbital masses occur, in addition to basic
examinations, the state of the whole body should be assessed and
biopsy tissues should be stained with IgG and IgG4 to exclude
IgG4-ROD.
Painless eyelid swelling, mild exophthalmos and
diplopia are common symptoms of IgG4-ROD. Due to the rare
involvement of the optic nerve, visual impairment is an uncommon
symptom (14,15). In the reported cases, the symptoms
of visual impairment were mostly due to the oppression of the optic
nerve by the surrounding mass (16-19),
which was not observed in the present case. Zhang et al
(20) reported on the case of a
79-year-old woman with skin changes, pancreatic tumors,
lymphadenopathy an eyelid mass and interstitial pneumonia for
>30 years. Rapid vision loss occurred in the left eye 2 months
prior to admission. On T2-weighted MRI, left optic atrophy with
focal hyperintense lesions was observed and the latency time of
visually evoked potential was prolonged. In addition, the patient
did not have any compressive mass around the optic nerve, similar
to the patient of the present study, suggesting that there may be
novel mechanisms of visual impairment in IgG4-ROD. Li et al
(21) reviewed 225 patients
diagnosed with IgG4-RD between January 2014 and December 2017.
Among these patients, 26 with decreased vision underwent orbital
MRI and optic nerve injury was detected in all of them prior to
treatment. The most common causes of optic nerve injury in these
patients with IgG-4-ROD were inflammation of the optic nerve sheath
(12 patients), compression of extraocular muscles and a pseudotumor
mass (14 patients), hypertrophic meningitis (2 patients) and
pituitary involvement in the optic chiasm (1 patient), where a
patient may simultaneously exhibit ≥2 causes of optic nerve injury,
which suggests that pathological changes in the optic nerve,
meninges and pituitary glands should not be ignored when
investigating the mechanisms of IgG4-ROD.
In a large multicenter study by Yamada et al
(22), the clinical and laboratory
parameter characteristics of 334 patients with IgG4-RD were
analyzed. A total of 205 patients were male and the average age at
diagnosis was 65 years. The average number of organs affected at
diagnosis was 3.2. The most frequently involved organs were the
salivary glands, followed by the lacrimal glands, lymph nodes,
pancreas, retroperitoneal/peri-aorta area, kidneys and lungs. It is
rare for a patient to have 10 organs involved (in the patient
described here: The eyes, submandibular glands, sinuses, middle
ears, bile ducts, liver, pancreas, prostate, skin and lymph nodes
were involved). Due to the early stages of disease progression in
certain organs, only enzymatic changes were present and no changes
were observed on imaging. Among these changes, liver injury was
severe. The albumin concentration was as low as 21.2 g/l at
diagnosis. Hypoalbuminemia mimicked cirrhosis but returned to
normal after hormone treatment.
The reported treatment options for IgG4-RD include
systemic glucocorticoids, immunosuppressive drugs, biological
agents and surgical resection of affected tissues. Except for
contraindications, glucocorticoids are recommended as first-line
drugs for remission induction in all active or untreated patients
with IgG4-RD (23). According to
the international consensus on the treatment of autoimmune
pancreatitis, the initial dose of prednisone should be 0.6-1.0
mg/kg/day and the minimum dose 20 mg/day. The therapeutic effect
may be evaluated for the first time after 2 weeks. Hormone
reduction may be considered after 2-4 weeks as follows: A reduction
by 5-10 mg/day every 1-2 weeks and a reduction by 5 mg/day every 2
weeks after 20 mg/day until drug withdrawal (24). Glucocorticoid retreatment is
suitable for patients who relapse after successful remission.
Following relapse, the continuous use of steroid-sparing agents
should be considered during remission maintenance periods (23).
In conclusion, IgG4-RD should be considered a
differential diagnosis for patients who suffer longstanding
symptoms involving multiple organs. Steroid therapy should be
considered the first choice of treatment.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XW and XS analyzed the data, performed a literature
review and were major contributors in writing the manuscript. DL
and YL encountered and treated the patient and directed the writing
of the article. RA and ZZ collected and confirmed all data for this
patient. All authors confirm the authenticity of the raw data and
have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Informed written consent was obtained from the
patient for publication of this case report and accompanying
images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kubo K and Yamamoto K: IgG4-related
disease. Int J Rheum Dis. 19:747–762. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Deshpande V, Zen Y, Chan JK, Yi EE, Sato
Y, Yoshino T, Klöppel G, Heathcote JG, Khosroshahi A, Ferry JA, et
al: Consensus statement on the pathology of IgG4-related disease.
Mod Pathol. 25:1181–1192. 2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Masamune A, Kikuta K, Hamada S, Tsuji I,
Takeyama Y, Shimosegawa T and Okazaki K: Collaborators. Nationwide
epidemiological survey of autoimmune pancreatitis in Japan in 2016.
J Gastroenterol. 55:462–470. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Mejico LJ: IgG4-related ophthalmic
disease. Saudi J Ophthalmol. 29:53–56. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Umehara H, Okazaki K, Masaki Y, Kawano M,
Yamamoto M, Saeki T, Matsui S, Yoshino T, Nakamura S, Kawa S, et
al: Comprehensive diagnostic criteria for IgG4-related disease
(IgG4-RD), 2011. Mod Rheumatol. 22:21–30. 2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Tokura Y, Yagi H, Yanaguchi H, Majima Y,
Kasuya A, Ito T, Maekawa M and Hashizume H: IgG4-related skin
disease. Br J Dermatol. 171:959–967. 2016.
|
|
7
|
Agaimy A and Ihrler S: Immunoglobulin G4
(IgG4)-related disease. A review of head and neck manifestations.
Pathologe. 35:152–159. 2014.PubMed/NCBI View Article : Google Scholar : (In German).
|
|
8
|
Minaga K, Watanabe T, Chung H and Kudo M:
Autoimmune hepatitis and IgG4-related disease. World J
Gastroenterol. 25:2308–2314. 2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hamano H, Kawa S, Horiuchi A, Unno H,
Furuya N, Akamatsu T, Fukushima M, Nikaido T, Nakayama Usuda N and
Kiyosawa K: High serum IgG4 concentrations in patients with
sclerosing pancreatitis. N Engl J Med. 344:732–738. 2001.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Hamano H, Kawa S, Ochi Y, Unno H, Shiba N,
Wajiki M, Nakazawa K, Shimojo H and Kiyosawa K: Hydronephrosis
associated with retroperitoneal fibrosis and sclerosing
pancreatitis. Lancet. 359:1403–1404. 2002.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kamisawa T, Funata N, Hayashi Y, Eishi Y,
Koike M, Tsuruta K, Okamoto A, Egawa N and Nakajima H: A new
clinicopathological entity of IgG4-related autoimmune disease. J
Gastroenterol. 38:982–984. 2003.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Min HK, Lee YS, Yang SW, Lee J, Kwok SK,
Ju JH, Kim WU and Park SH: Clinical outcomes and pathological
characteristics of immunoglobulin G4-related ophthalmic disease
versus orbital inflammatory pseudotumor. Korean J Intern Med.
34:220–226. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Tiegs-Heiden CA, Eckel LJ, Hunt CH, Diehn
FE, Schwartz KM, Kallmes DF, Salomão DR, Witzig TE and Garrity JA:
Immunoglobulin G4-related disease of the orbit: Imaging features in
27 patients. Am J Neuroradiol. 35:1393–1397. 2014.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Andrew N, Kearney D and Selva D:
IgG4-related orbital disease: A meta-analysis and review. Acta
Ophthalmol. 91:694–700. 2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Kubota T and Moritani S: Orbital
IgG4-related disease: Clinical features and diagnosis. ISRN
Rheumatol. 2012(412896)2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Takahashi Y, Kitamura A and Kakizaki H:
Bilateral optic nerve involvement in immunoglobulin G4-related
ophthalmic disease. J Neuroophthalmol. 34:16–19. 2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Takahira M, Ozawa Y, Kawano M, Zen Y,
Hamaoka S, Yamada K and Sugiyama K: Clinical aspects of
IgG4-related orbital inflammation in a case series of ocular
adnexal lymphoproliferative disorders. Int J Rheumatol.
2012(635473)2012.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Ramirez L, D'Auria A, Popalzai A and
Sanossian N: Bilateral vision loss secondary to pachymeningitis in
a patient with IgG4-related disease. Front Neurol.
5(192)2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Sogabe Y, Ohshima K, Azumi A, Takahira M,
Kase S, Tsuji H, Yoshikawa H and Nakamura T: Location and frequency
of lesions in patients with IgG4-related ophthalmic diseases.
Graefes Arch Clin Exp Ophthalmol. 252:531–538. 2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhang W, Luo J and Jiao J: Optic nerve
involvement in immunoglobulin G4-related disease: A case report.
Exp Ther Med. 12:111–114. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Li J, Zhang Y, Zhou H, Wang L, Wang Z and
Li H: Magnetic resonance imaging indicator of the causes of optic
neuropathy in IgG4-related ophthalmic disease. BMC Med Imaging.
19(49)2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Yamada K, Yamamoto M, Saeki T, Mizushima
I, Matsui S, Fujisawa Y, Hara S, Takahashi H, Nomura H, Kawa S and
Kawano M: New clues to the nature of immunoglobulin G4-related
disease: A retrospective Japanese multicenter study of baseline
clinical features of 334 cases. Arthritis Res Ther.
19(262)2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Khosroshahi A, Wallace ZS, Crowe JL,
Akamizu T, Azumi A, Carruthers MN, Chari ST, Della-Torre E,
Frulloni L, Goto H, et al: International consensus guidance
statement on the management and treatment of IgG4-related disease.
Arthritis Rheumatol. 67:1688–1699. 2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Okazaki K, Chari ST, Frulloni L, Lerch MM,
Kamisawa T, Kawa S, Kim MH, Lévy P, Masamune A, Webster G and
Shimosegawa T: International consensus for the treatment of
autoimmune pancreatitis. Pancreatology. 17:1–6. 2017.PubMed/NCBI View Article : Google Scholar
|