Introduction
Primary intracranial aneurysm (IA) is a vascular
disease that frequently leads to fatal vascular rupture and
subarachnoid hemorrhage, which is an acute stroke and has a
mortality rate of nearly 50% (1).
Primary multiple IA (MIA) is defined as the presence of two or more
aneurysms in one patient. The reported rates of MIA range between 2
and 45% of IAs (2).
With the accumulation of studies about immunization,
non-coding RNAs and epigenetics, the molecular mechanisms of IA
have been widely identified. However, although numerous studies
have explored microRNAs (miRNAs) (3-5)
or long non-coding RNAs (lncRNAs) (6,7) in IA,
circRNAs in IA, particularly in MIA, have remained largely elusive.
The genetic pathology of MIA, particularly in terms of circular
RNAs (circRNAs) in MIA, remains to be determined.
Since their 3 and 5' ends are connected to form a
circle, circRNA molecules cannot be broken down by RNase R. This
feature leads to their high stability and abundance. Furthermore,
circRNA expression is frequently cell- or tissue-specific. In
addition, circRNAs may be potential biomarkers for diseases
(8), as they are conserved between
species. Previous studies have demonstrated that circRNAs have
numerous functions, such as sponging of miRNAs and gene regulation,
and are closely associated with cell function and diseases. Due to
rapid developments in sequencing and microarray technology,
systematic investigations of circRNA expression profiles in MIA
development are now possible.
Using high-throughput sequencing technology, the
present study aimed to define the circRNA expression profiles of
MIA and to explore novel pathological mechanisms of MIA. Compared
with microarrays, sequencing technology provides more comprehensive
and reliable data (9), which may
improve the current knowledge of the epigenetic mechanisms of MIA.
The present study revealed that certain circRNAs are involved in
the pathogenesis of MIA and mostly associated with inflammation.
These results were further verified using PCR assays in an enlarged
cohort of patients with MIA. In summary, the present results may
give novel insight into the molecular mechanisms of MIA, provide
ideas for novel treatments and promote the exploration of circRNAs
as biomarkers.
Materials and methods
Patient selection
Blood samples of three pairs of patients with
primary MIA and matched healthy individuals were collected at the
Department of Neurosurgery, Longyan First Hospital Affiliated to
Fujian Medical University (Longyan, China) between April 2018 and
June 2018. Patients in the MIA group were selected from patients
with onset within 24 h who were confirmed to have one ruptured IA
by head computed tomography angiography (CTA) or digital
subtraction angiography (DSA). The patients received microsurgical
clipping or interventional therapy within the acute phase (24 h)
after one of the aneurysms was ruptured. Those patients who had
cerebrovascular diseases other than IA, systemic malignant tumor or
severe complications were excluded. Individuals in the matched
group were selected among individuals who were attending a regular
health check-up and underwent head CTA/DSA to exclude IA. To ensure
the quality of samples, the present study excluded patients with
factors that may have affected the state of peripheral blood,
including pregnancy, chemotherapy and fever (≥37.3˚C). Individuals
in the MIA and matched groups were matched based on age, sex and
past medical history, such as hypertension and smoking. Baseline
information and clinical characteristics of the two groups are
presented in Table I. Another 20
individuals were recruited as the validation cohort following the
same criteria as those for the initial study group used for RNA
sequencing (RNA-seq). They were divided into 10 pairs of matched
patients with primary MIA as a test group and healthy individuals
as a control group. The baseline information and clinical features
of the two groups are presented in Table II. All volunteers included provided
written informed consent and the present study was approved by the
Ethics Committee of Longyan First Hospital Affiliated to Fujian
Medical University (Longyan, China).
 | Table IBaseline data of patients for
sequencing and the clinical features of MIA. |
Table I
Baseline data of patients for
sequencing and the clinical features of MIA.
| A,
Clinicopathological parameters |
|---|
| Item | MIA (n=3) | Matched control
(n=3) |
t/χ2 | P-value |
|---|
| Age (years) | 69.33±8.39 | 68.67±9.29 | 0.092 | 0.931 |
| Female sex | 3(100) | 3(100) | 0.000 | 1.000 |
| Hypertension | 3(100) | 3(100) | 0.000 | 1.000 |
| Smoking | 0(100) | 0(100) | 0.000 | 1.000 |
| SBP (mmHg) | 156.67±10.60 | 153.00±11.79 | 0.401 | 0.709 |
| DBP (mmHg) | 83.67±5.13 | 84.33±9.07 | -0.111 | 0.917 |
| TG (mmol/l) | 0.67±0.05 | 0.75±0.07 | -1.595 | 0.186 |
| LDL (mmol/l) | 1.81±0.16 | 1.88±0.13 | -0.632 | 0.561 |
| HDL (mmol/l) | 1.33±0.10 | 1.38±0.14 | -0.511 | 0.636 |
| B, Details of
individual IAs |
| Case/IA | Location | Ruptured | Type | Size (mm) |
| Case 1 |
|
IA1 | Right PcoA | Yes | Saccular | 3x5 |
|
IA2 | Left PcoA | No | Saccular | 2x5 |
| Case 2 |
|
IA1 | BA | Yes | Saccular | 7x9 |
|
IA2 | Left MCA | No | Saccular | 3x4 |
| Case 3 |
|
IA1 | Right PcoA | Yes | Saccular | 9x3 |
|
IA2 | AcoA | No | Saccular | 3x4 |
| Table IIBaseline data of the two groups of
the validation cohort and the clinical features of multiple IA. |
Table II
Baseline data of the two groups of
the validation cohort and the clinical features of multiple IA.
| A, Baseline data of
the test group and the control group of the validation cohort |
|---|
| Item | Test group
(n=10) | Control (n=10) |
t/χ2 | P-value |
|---|
| Age (years) | 57.0±9.31 | 59.8±8.99 | -0.684 | 0.503 |
| Female sex | 6(60) | 6(60) | 0.000 | 1.000 |
| Hypertension | 8(80) | 8(80) | 0.000 | 1.000 |
| Smoking | 6(60) | 6(60) | 0.000 | 1.000 |
| SBP (mmHg) | 147.60±14.42 | 149.40±18.31 | -0.244 | 0.810 |
| DBP (mmHg) | 83.90±4.68 | 84.20±8.94 | -0.094 | 0.926 |
| TG (mmol/l) | 0.70±0.08 | 0.71±0.09 | -0.274 | 0.787 |
| LDL (mmol/l) | 1.78±0.15 | 1.82±0.18 | -0.518 | 0.610 |
| HDL (mmol/l) | 1.41±0.10 | 1.35±0.08 | 1.414 | 0.174 |
| B, Details of
individual IAs |
| Subject/IA | Location | Ruptured | Type | Size (mm) |
| Test 1 |
|
IA1 | Right PcoA | Yes | Saccular | 4x4 |
|
IA2 | Left PcoA | No | Saccular | 4x2 |
| Test 2 |
|
IA1 | BA | Yes | Saccular | 6x5 |
|
IA2 | Left MCA | No | Saccular | 4x3 |
| Test 3 |
|
IA1 | Right PcoA | Yes | Saccular | 5x3 |
|
IA2 | AcoA | No | Saccular | 5x2 |
| Test 4 |
|
IA1 | Left PcoA | Yes | Saccular | 8x7 |
|
IA2 | Right PcoA | No | Saccular | 6x4 |
| Test 5 |
|
IA1 | Left PcoA | Yes | Saccular | 12x6 |
|
IA2 | Left MCA | No | Saccular | 5x2 |
| Test 6 |
|
IA1 | Left MCA | Yes | Saccular | 9x7 |
|
IA2 | AcoA | No | Saccular | 7x6 |
| Test 7 |
|
IA1 | AcoA | Yes | Saccular | 13x10 |
|
IA2 | Right PcoA | No | Saccular | 9x6 |
| Test 8 |
|
IA1 | Right PcoA | Yes | Saccular | 13x9 |
|
IA2 | BA | No | Saccular | 4x3 |
| Test 9 |
|
IA1 | Righ MCA | Yes | Saccular | 9x5 |
|
IA2 | Right PcoA | No | Saccular | 6x4 |
| Test 10 |
|
IA1 | AcoA | Yes | Saccular | 9x3 |
|
IA2 | Left PcoA | No | Saccular | 4x3 |
RNA isolation and purification
Total RNA was extracted from peripheral blood
mononuclear cell samples using TRlzol reagent (Takara Bio, Inc.)
according to the kit's instructions, as described previously
(10). The NanoDrop-1000 (Thermo
Fisher Scientific, Inc.) was used to quantify total RNA, and
optical density at 260 nm (OD260)/OD280 ratios between 1.8 and 2.1
were considered acceptable. Agarose gel electrophoresis and the
Nanodrop spectrophotometer were used to check the quantity of RNA
prior to sequencing and reverse transcription-quantitative PCR
(RT-qPCR).
circRNA sequencing analysis
RNase R (Epicentre; Illumina, Inc.) was used to
degrade linear RNA and enrich circRNAs in total RNA. Subsequently,
the circRNA library was constructed according to the manufacturer's
protocols (NEBNext Ultra Directional RNA Library Prep kit; New
England BioLabs, Inc.). The Library Quantification kit (Kapa
Biosystems; Roche Diagnostics) was used to identify and quantify
sequences using the Illumina HiSeq 4000 system (Illumina, Inc.). By
using paired-end sequencing, the nucleotide length of sequencing
was 150 bp. Single-stranded DNA was generated using 0.1 M NaOH and
its loading concentration was 8 pM, which was measured by qPCR
(11,12). Sequencing quality was evaluated by
FastQC v0.11.7 software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
Adaptors and poor-quality bases were trimmed with Cutadapt v1.14
(https://cutadapt.readthedocs.io/en/stable/). In order
to ensure the quality and reliability of data analysis, it was
necessary to filter the original data. The filtering comprised the
following: Reads with adapter were removed; reads with a ratio of
indeterminable base information of >0.002 were removed; when the
number of low-quality bases in a single-ended read exceeded 50% of
read length, this paired read was removed. Furthermore, genes with
low expression levels were filtered in the analysis. The threshold
values were as follows: 1.5-fold change (FC), P≤0.05 and mean value
of fragments per kilobase of exon model per million reads mapped
≥0.5.
Base calling and image processing were performed
using Solexa pipeline v1.8 (Off-Line Base Caller software, v1.8).
Quantile normalization and subsequent data processing were
performed using the Ballgown package v2.8.4 (http://www.bioconductor.org/packages/release/bioc/html/ballgown.html)
in R software v3.4.1 (www.r-project.org). Differential expression patterns
of circRNAs in the samples were visualized using hierarchical
clustering and a scatter plot. Significantly differentially
expressed circRNAs (FC >2 and P<0.05) were identified using
volcano plot filtering.
Validation by RT-qPCR
To validate the changes in circRNA expression
identified by RNA-sequencing, 10 circRNAs, including 5 upregulated
and 5 downregulated circRNAs, were selected. They met the following
requirements: i) Length of circRNA between 200 and 3,000 bp; ii)
|log2 FC|>2; iii) P<0.05; and iv) exon-related circRNA. The
expression levels of circRNAs were evaluated by fluorescence
real-time PCR (C1000; Applied Biosystems; Thermo Fisher Scientific,
Inc.) in 10 pairs of matched patients with primary MIA as a test
group and healthy individuals as a control group using SYBR Green
as the probe, as previously described (13). The housekeeping gene β-actin was
used as an internal control. The results were calculated using the
2-ΔΔCq method (14).
Gene Ontology (GO) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) analysis
GO enrichment analysis (https://david-d.ncifcrf.gov/) was conducted to explore
the potential biological functions of the target genes of the
circRNAs identified. KEGG pathway analysis (http://www.genome.jp/kegg/) revealed the signaling
pathways the target genes of identified circRNAs were involved in
at the molecular level. Genes with a corrected P<0.05 were
considered to be enriched.
miRNA prediction and
circRNA-miRNA-mRNA network construction
miRNA binding sites of circRNAs were predicted using
TargetScan v7.2(15), circBank
v2014(16) and miRanda v3.3a.
Information on miRNA-mRNA regulation was obtained using miRTarBase
(17) and TargetScan. The potential
effects of the differentially expressed circRNAs associated with
‘leukocyte transendothelial migration’ were further predicted by
constructing a competing endogenous RNA (ceRNA) network of
circRNA-miRNA-mRNA interactions. Cytoscape v3.6.1 was used to
construct the graph of the circRNA-miRNA-mRNA network.
Statistical analysis
Normally distributed data were analyzed using a
two-tailed t-test and a Mann-Whitney U test was used for skewed
data. All statistical analyses were performed using SPSS v19.0 (IBM
Corp.). P<0.05 (two-tailed) was considered to indicate a
statistically significant difference.
Results
circRNA expression profiles in
peripheral blood mononuclear cells
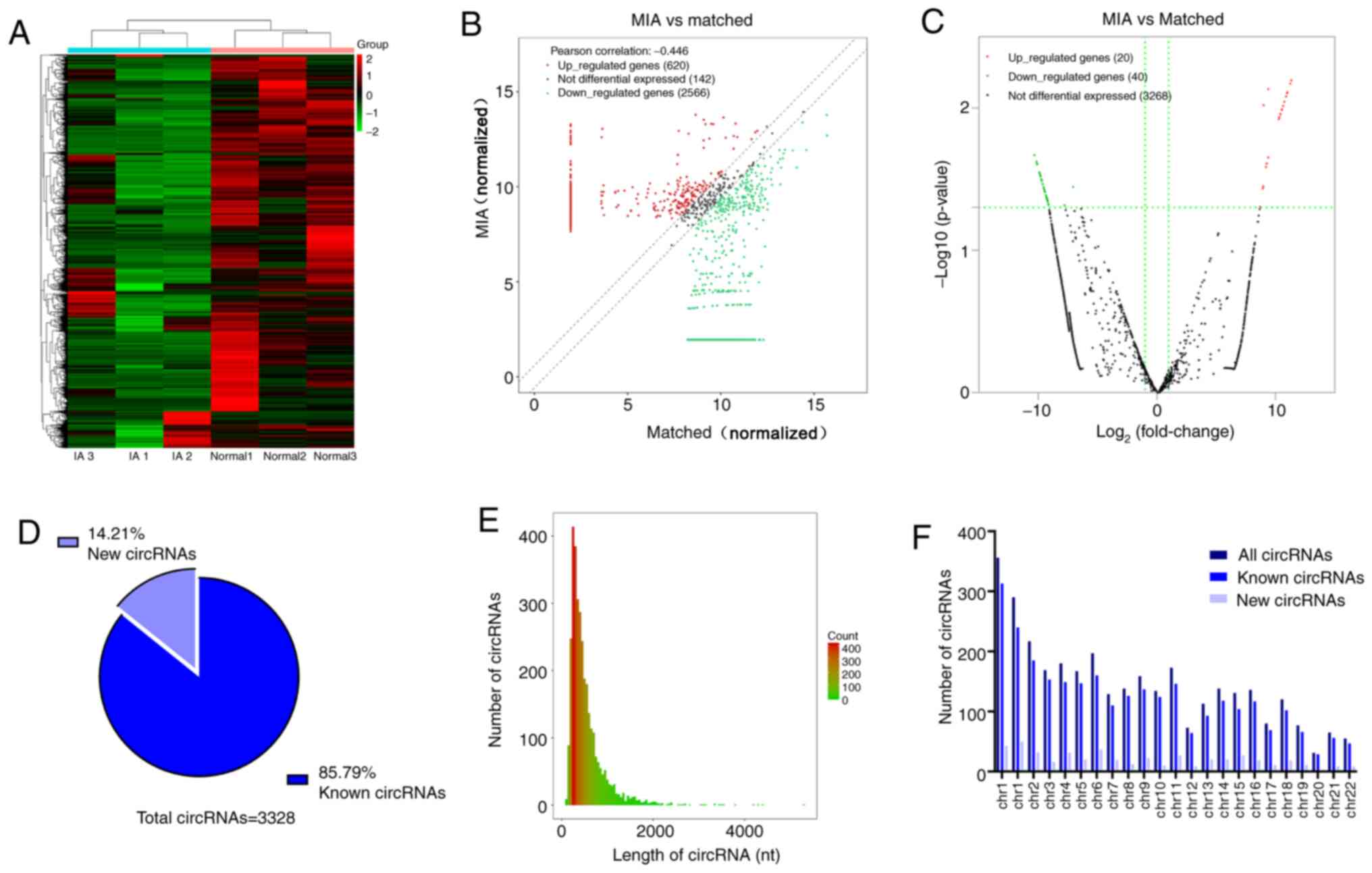
The normalized expression profiles are presented in
Fig. 1. Hierarchical clustering
revealed the difference in circRNA expression profiles between the
two groups (Fig. 1A). A total of
3,328 differentially expressed circRNAs between the MIA group and
the matched group were identified by circRNA sequencing analysis.
The distribution of circRNAs in three pairs of matched samples is
presented in a scatter plot (Fig.
1B). Compared to the matched group, 60 circRNAs were revealed
to be significantly differentially expressed in the MIA group
(|log2 FC|≥2; P<0.05). Among them, 20 were upregulated and 40
were downregulated (Fig. 1C).
Furthermore, 85.79% of the circRNAs were previously identified in
circbase (18) and 14.21% were
novel (Fig. 1D). Most circRNAs had
a predicted spliced length <1,000 nt (Fig. 1E). These circRNAs were widely
distributed among all chromosomes except the Y chromosome, since
only samples from female patients were collected (Fig. 1F). The top 10 most upregulated and
downregulated circRNAs are listed in Table III.
| Table IIITop 10 upregulated and downregulated
circRNAs. |
Table III
Top 10 upregulated and downregulated
circRNAs.
| CircBase_ID | Direction of
change | Host gene | Chromosomal
location | Length (nt) | log2FC | P-value |
|---|
|
hsa_circ_0135895 | Upregulation | PTK2 | Chr8 | 853 | 11.34 | 0.006 |
|
hsa_circ_0008911 | Upregulation | ZNF512 | Chr2 | 195 | 11.01 | 0.008 |
|
hsa_circ_0008122 | Upregulation |
TCONS_l2_00012420 | Chr19 | 223 | 10.90 | 0.008 |
|
hsa_circ_0074837 | Upregulation | PWWP2A | Chr5 | 965 | 10.75 | 0.009 |
|
hsa_circ_0078380 | Upregulation | SCAF8 | Chr6 | 1,159 | 10.64 | 0.010 |
|
hsa_circ_0093067 | Upregulation | CAMK1D | Chr10 | 192 | 10.53 | 0.010 |
|
hsa_circ_0037572 | Upregulation | SRRM2 | Chr16 | 93 | 10.33 | 0.012 |
|
hsa_circ_0089775 | Upregulation | PPP2R3B | Chrx | 1,253 | 10.28 | 0.012 |
|
hsa_circ_0000192 | Upregulation | GALNT2 | Chr1 | 248 | 9.40 | 0.022 |
|
hsa_circ_0131628 | Upregulation | SLC22A23 | Chr6 | 414 | 9.40 | 0.007 |
|
hsa_circ_0009076 | Downregulation | NRD1 | Chr1 | 228 | -10.33 | 0.021 |
|
hsa_circ_0000982 | Downregulation | LOC375190 | Chr2 | 395 | -10.16 | 0.024 |
|
hsa_circ_0001492 | Downregulation | ERBB2IP | Chr5 | 364 | -10.09 | 0.025 |
|
hsa_circ_0000698 | Downregulation | PHKB | Chr16 | 518 | -9.90 | 0.028 |
|
hsa_circ_0141172 | Downregulation | DAAM1 | Chr14 | 310 | -9.89 | 0.029 |
|
hsa_circ_0002665 | Downregulation | GDI2 | Chr10 | 343 | -9.86 | 0.029 |
|
hsa_circ_0001413 | Downregulation | FIP1L1 | Chr4 | 362 | -9.86 | 0.029 |
|
hsa_circ_0006208 | Downregulation | NPAT | Chr11 | 226 | -9.83 | 0.030 |
|
hsa_circ_0001936 | Downregulation | BRWD3 | Chrx | 676 | -9.82 | 0.030 |
|
hsa_circ_0126525 | Downregulation | SLAIN2 | Chr4 | 971 | -9.81 | 0.030 |
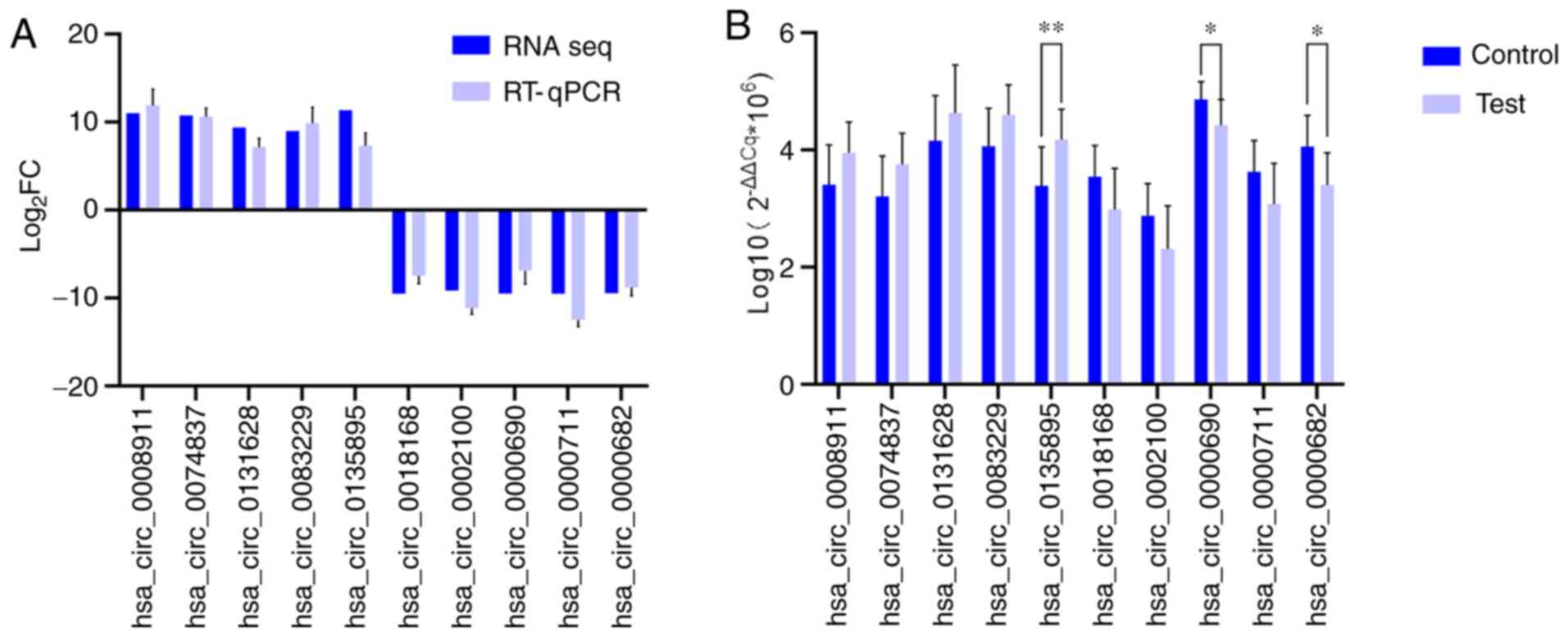
RT-qPCR validation of differential
expression of circRNAs
A total of 10 differentially expressed circRNAs,
including 5 upregulated [Homo sapiens circRNA
(hsa_circ)_0008911, hsa_circ_0074837, hsa_circ_0131628,
hsa_circ_0083229 and hsa_circ_0135895] and 5 downregulated
(hsa_circ_0018168, hsa_circ_0002100, hsa_circ_0000690,
hsa_circ_0000711 and hsa_circ_0000682) circRNAs, were selected to
confirm the sequencing data using RT-qPCR. As presented in Fig. 2A, the changes observed in all
selected circRNAs were similar between circRNA sequencing and the
RT-qPCR assays, which confirmed the accuracy of the sequencing. As
indicated in Fig. 2B,
hsa_circ_0135895, hsa_circ_0000690 and hsa_circ_0000682 were
significantly differentially expressed between samples from 10
patients with MIA and 10 healthy individuals (P<0.05).
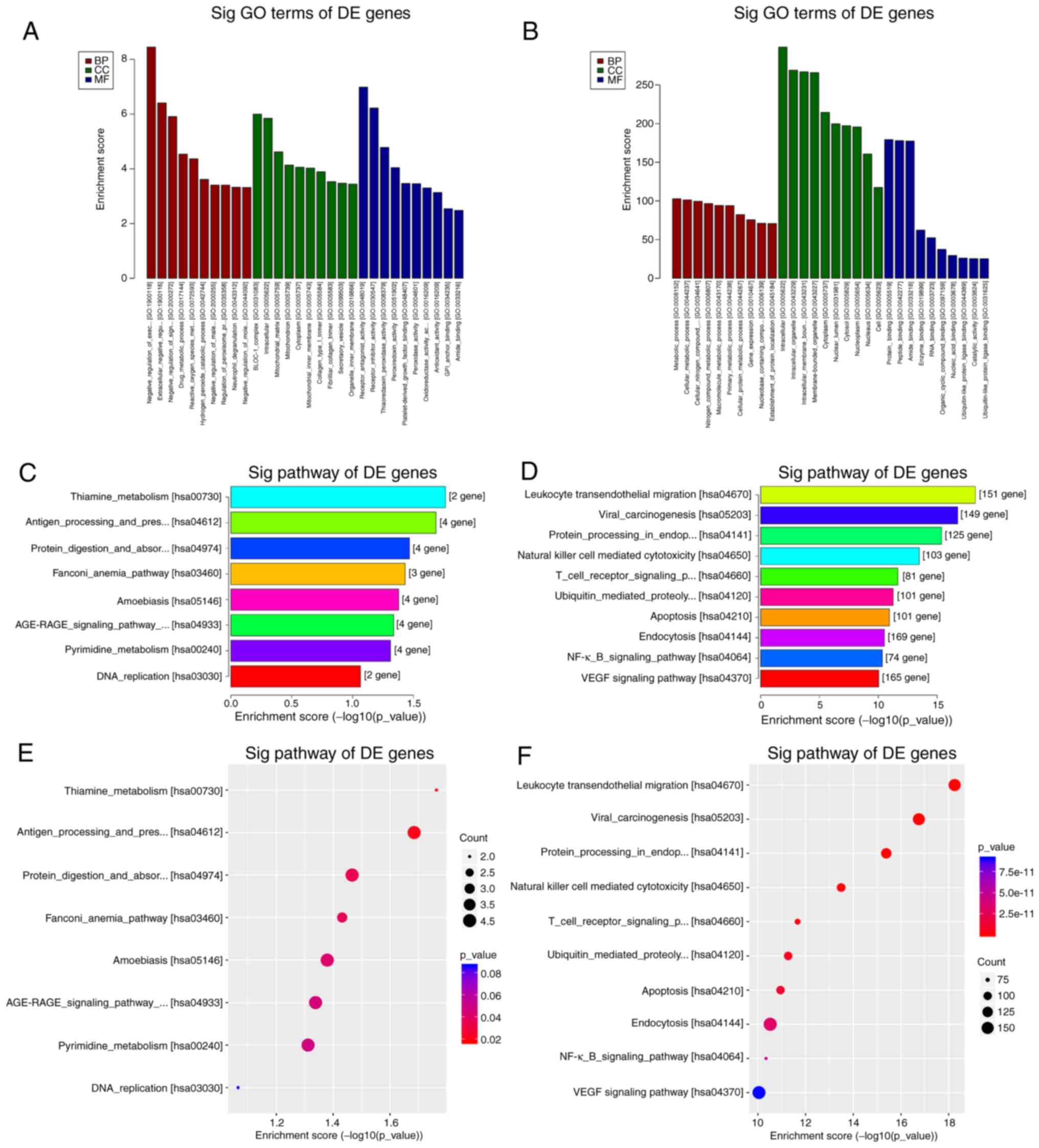
GO and KEGG pathway analyses
GO enrichment and KEGG pathway analysis were
performed with genes of a total of 3,328 differentially expressed
circRNAs (Fig. 3). The top 10 most
enriched GO terms by the upregulated and downregulated genes of
identified circRNAs in the categories biological process (BP),
cellular component and molecular function (MF) are presented in
Fig. 3A and B. The top 10 GO terms for the
downregulated genes of identified circRNAs in the category BP were
mainly involved in metabolic processes, while those in the category
MF mainly involved different types of binding functions. The KEGG
pathway analysis revealed that the upregulated genes of identified
circRNAs mapped to 8 pathways, including ‘thiamine metabolism’,
‘pyrimidine metabolism’ and ‘DNA replication’ (Fig. 3C and E). Furthermore, the downregulated genes of
identified circRNAs were mapped to 144 pathways, including
‘leukocyte transendothelial migration’ in Fig. 4A, ‘natural killer cell mediated
cytotoxicity’ in Fig. 4B, ‘T cell
receptor signaling pathway’ and ‘NF-κB signaling pathway’ (Fig. 3D and F). The 60 significantly differentially
expressed circRNAs were also analyzed through GO enrichment and
KEGG pathway analysis. The GO enrichment analysis was unsuccessful
due to the low number of circRNAs. Only five pathways were obtained
in the KEGG pathway analysis of the 60 host genes of the circRNAs
identified, namely ‘leukocyte transendothelial migration’, ‘natural
killer cell mediated cytotoxicity’, ‘VEGF signaling pathway’, ‘axon
guidance’ and ‘Wnt signaling pathway’.
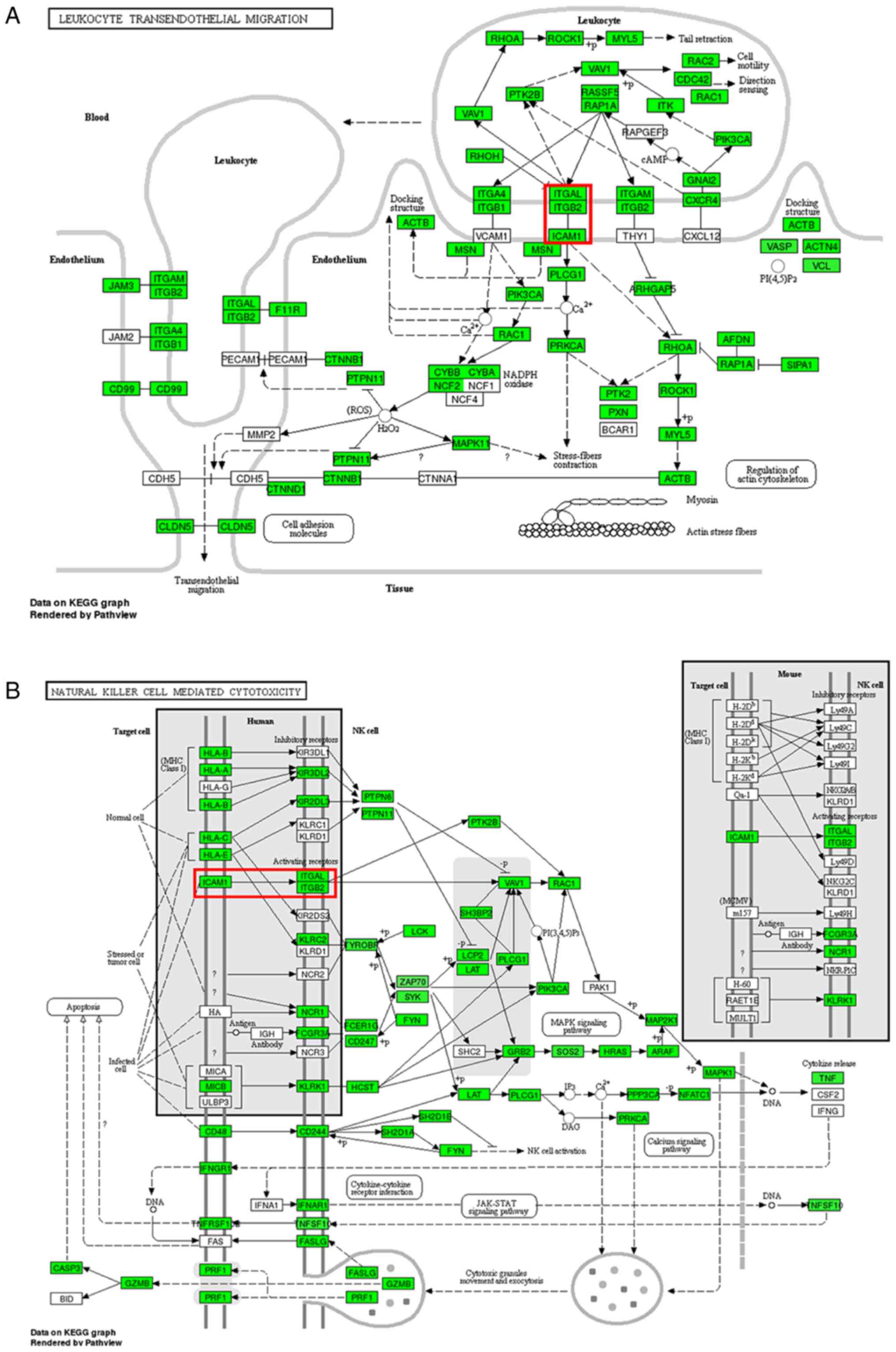 | Figure 4Pathways associated with the
formation of multiple intracranial aneurysms. (A) Leukocyte
transendothelial migration pathway. (B) Natural killer cell
mediated cytotoxicity pathway. Green nodes represent gene
enrichment of the pathway, while white nodes have no significance.
The red square represents adhesion between intercellular cell
adhesion molecule-1 and the complex of integrin subunit αL/integrin
subunit β2. KEGG, Kyoto Encyclopedia of Genes and Genomes; NK,
natural killer; ROS, reactive oxygen species;
H2O2, hydrogen peroxide; DNA,
deoxyribonucleic acid; cAMP, cyclic adenosine monophosphate; NADPH,
nicotinamide adenine dinucleotide phosphate; DAG, diacylglycerol;
IP3, inositol triphosphate. |
miRNA prediction and ceRNA network
construction
TargetScan, circBank and miRanda were utilized to
predict miRNAs targeted by the differentially expressed circRNAs.
The top 5 miRNAs predicted to be targeted by each of the three
selected significantly differentially expressed circRNAs, which
were all contained in the three databases, are presented in
Table IV. The miRNA-mRNA
regulatory relationships were further identified using miRTarBase
and TargetScan. The miRNAs targeted by the circRNAs associated with
‘leukocyte transendothelial migration’ and their regulated mRNAs
were further selected for ceRNA network construction. In this
network, an upregulated circRNA (hsa_circ_0135895) and two
downregulated circRNAs (hsa_circ_0000682 and hsa_circ_0000690)
regulate protein tyrosine kinase 2 (PTK2), protein kinase Cβ
(PRKCB) and integrin subunit αL (ITGAL), respectively, by sponging
a number of different miRNAs (Fig.
5). hsa-miR-4778-3p may combine with both hsa_circ_0135895 and
hsa_circ_0000690, while hsa-miR-543 may combine with both
hsa_circ_0135895 and hsa_circ_0000682. Several genes may combine
with >20 predicted miRNAs, including ITGAL, IL2 inducible T-cell
kinase, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic
subunit α, phospholipase C γ1, PRKCB, PTK2, protein tyrosine
phosphatase non-receptor type 11, RAP1A member of RAS oncogene
family, ras homolog family member H, rho associated coiled-coil
containing protein kinase 1 and signal-induced
proliferation-associated 1. Among the three circRNAs,
hsa_circ_0135895 sponged the most mRNAs.
| Table IVPredicted miRNAs for the selected
significantly differential circRNAs in multiple intracranial
aneurysm. |
Table IV
Predicted miRNAs for the selected
significantly differential circRNAs in multiple intracranial
aneurysm.
| CircRNA ID | miRNA |
|---|
|
hsa_circ_0135895 | hsa-miR-619-3p,
hsa-miR-4324, hsa-miR-5687, hsa-miR-3529-5p, hsa-miR-379-5p |
|
hsa_circ_0000682 | hsa-miR-448,
hsa-miR-1248, hsa-miR-302a-5p, hsa-miR-627-3p, hsa-miR-1248 |
|
hsa_circ_0000690 | hsa-miR-4726-3p,
hsa-miR-4520-3p, hsa-miR-4514, hsa-miR-4692, hsa-miR-6842-3p |
Discussion
MIA is an unpredictably and lethal disease. To the
best of our knowledge, the specific molecular biological mechanisms
of MIA have remained to be elucidated. IA, particularly MIA and
familial aneurysms, may be hereditary (19). Previous studies have focused on
genes associated with vascular smooth muscle cell dysfunction,
extracellular matrix components and vascular inflammation; however,
these genes may be associated with a change in risk of IA by <5%
(20,21). Gene expression is regulated by
various factors, which are relatively reversible. As non-coding
RNAs, a novel class of lncRNAs, circRNAs, are widely involved in
gene transcription, post-transcriptional regulation and other
biological processes. They have been identified and determined to
be involved in neurologic diseases (22), particularly cerebrovascular disease
(23).
In the present study, RNA-seq technology was used to
profile differentially expressed circRNAs in peripheral blood
mononuclear cells from patients with MIA. Samples from three pairs
of patients with MIA and matched healthy individuals were selected
to perform circRNA profiling. A total of 3,328 differentially
expressed circRNAs were identified and 60 of these circRNAs were
significantly differentially expressed, including 20 upregulated
and 40 downregulated circRNAs. The reliability of the results was
validated using RT-qPCR. The trends of all 10 selected circRNAs
were the same (Fig. 2A), but some
of them were not statistically significant (Fig. 2B). These results suggested that
expression changes of these circRNAs are associated with the
pathogenesis of MIA.
The GO enrichment and KEGG pathway analyses of the
present study revealed the functional roles of the target genes.
Since few pathways were identified for the upregulated genes, the
present study focused on the pathways associated with the
downregulated genes. It was revealed that most genes that were
downregulated in MIA were mainly involved in inflammation, which is
known to be critical for the pathogenesis of IA. This was similar
to the results of Hao et al (24) and Huang et al (25). Inflammation of the arterial wall is
one of the recognized mechanisms of IA (26). In addition, the aggregation of white
blood cells and their adherence to inflammatory tissues are the
basis of an inflammatory response (27). Inflammatory response and leukocyte
infiltration are the pathological basis for the development of IA
(28,29). Infiltration of inflammatory cells is
the primary pathological change during the early stages of IA
(30). circRNAs have been
demonstrated to serve important roles in inflammation, including
the regulation of inflammation-associated genes, recruitment of
macrophages and modulation of crucial signaling pathways (31). The present results suggested that
several inflammation-related biological mechanisms and pathways in
MIA are potentially regulated by circRNAs. The most affected
pathway was the ‘leukocyte transendothelial migration’ in Fig. 4A. Due to the blood-brain barrier,
there is almost no peripheral blood lymphocyte aggregation in
normal brain tissues. However, the adhesion between lymphocyte
function-associated antigen-1 (LFA-1)/intercellular cell adhesion
molecule-1 (ICAM-1) in the pathway ‘leukocyte transendothelial
migration’ serves an important role in the migration and
infiltration of lymphocytes into cerebrovascular wall tissues
(32). ‘Natural killer cell
mediated cytotoxicity’ in Fig. 4B
was the other signaling pathway associated with the downregulated
genes and was associated with MIA formation and development. In
addition, studies have demonstrated that natural killer cells
intensively infiltrate in abdominal aortic aneurysm and damage the
vessel by inducing cytotoxicity in arterial smooth muscle cells
(33).
Adhesion between LFA-1 and ICAM-1 is involved in
both ‘leukocyte transendothelial migration’ and ‘Natural killer
cell mediated cytotoxicity’ pathways in Fig. 4. LFA-1 is expressed on the surfaces
of lymphocytes, neutrophils, monocytes and macrophages. As a member
of the integrin β2 family, LFA-1 is composed of an α and a β
subunit. The α subunit, also known as CD11a antigen, is encoded by
ITGAL, and the β subunit, also known as CD18, is encoded by
integrin subunit β2(34). CD11a,
which may be transcriptionally regulated by hsa_circ_0000690, is
the binding site for ICAM-1, enabling stable immune adhesion
between LFA-1 and ICAM-1.
‘Leukocyte transendothelial migration’ may be a key
biological event in MIA. In the present study, the KEGG pathway
‘leukocyte transendothelial migration’ was enriched by 151 genes
targeted by differentially expressed circRNAs. Most circRNAs have
been identified to interact with miRNAs via fewer binding sites,
acting as miRNA sponges to regulate transcription. Therefore, the
ceRNA mechanism of certain circRNAs associated with ‘leukocyte
transendothelial migration’ was explored. For this, three circRNAs
that interact with certain miRNAs that transcriptionally regulate
migration were selected (hsa_circ_0135895, hsa_circ_0000682 and
hsa_circ_0000690) and annotated, since the validation experiment
confirmed that these three circRNAs were significantly different
between the cases and controls. Furthermore, a ceRNA network was
constructed based on the selected circRNAs. Subsequently,
circRNA-miRNA interactions were assessed to further reveal the
roles of circRNAs as miRNA sponges.
It is worth mentioning that the present results
demonstrated that the downregulated genes were over-represented in
pathways associated with inflammation. It is known that
inflammation is an important factor in the occurrence and
development of IAs. Therefore, the opposite results would be
expected. The observed negative association appeared to be the
opposite of the expected trend. The exact mechanisms responsible
for these results remain elusive and it was not possible to offer a
reasonable explanation based on the results of the present study.
The most recent studies have focused on upregulated circRNAs due to
the ceRNA mechanism (35-37).
circRNAs are widely involved in gene transcription,
post-transcriptional regulation and other biological processes with
complex and unknown mechanisms. The ceRNA mechanism is just one
classical mechanism. Only a small number of studies have
investigated downregulated genes; however, increasing attention is
being paid to these (38,39). Therefore, the relationships among
downregulated genes, inflammation and MIA, as well as the
mechanisms underlying their associations, deserve further
investigation.
In the present study, circRNA expression profiles
were linked to MIA development to predict a regulatory role of
circRNAs in MIA formation, thereby expanding the knowledge of MIA.
However, the present study had certain limitations. First, only
circRNAs associated with ‘leukocyte transendothelial migration’
were used to construct the ceRNA network, while circRNAs associated
with other pathways may also have an effect on transcriptional
regulation in MIA. Therefore, further investigation including more
circRNAs is required. In addition, the effects of circRNAs on
miRNAs and mRNAs were determined by computational analyses and
remain to be experimentally demonstrated. Further biological in
vitro and in vivo experiments are required, including
validation of the expression of target miRNAs. As another
limitation, both ruptured and unruptured IAs were included in the
research objective. Differences in circRNA expression in MIA may be
caused by not only IA itself but also subarachnoid hemorrhage.
Whether the results may apply to unruptured MIA or single IA with
ruptured status warrants further investigation. Finally, only
samples from female patients were used in the present study. There
may be certain significant differentially expressed circRNAs in the
Y chromosome that the present study was not able to assess.
To elucidate how circRNAs transcriptionally regulate
MIA formation and to identify biomarkers of MIA, research in this
field based on the present results may be performed.
In conclusion, to the best of our knowledge, the
present study was the first to identify differential expression
profiles of circRNAs in MIA. Most downregulated genes were mainly
involved in inflammation. The KEGG pathway ‘leukocyte
transendothelial migration’ may be critical for the pathogenesis of
MIA. circRNAs involved in this pathway, particularly
hsa_circ_0135895, hsa_circ_0000682 and hsa_circ_0000690, should be
further explored in the future.
Acknowledgements
The authors would like to thank Dr Yingying Lin
(Department of Neurosurgery, Ren Ji Hospital Affiliated to the
School of Medicine of Shanghai Jiao Tong University, Shanghai,
China), who supported the present study in terms of experimental
design and technology regarding RNA isolation and purification.
Funding
The present study was supported by the Basic
Research and Young Talent Programs Foundation of the Science and
Technology Department of Longyan City (grant no. 2018LYF8019).
Availability of data and materials
The datasets used or analyzed during the current
study are available from the corresponding author on reasonable
request. They are available from the Gene Expression Omnibus (GEO)
repository (series entry, GSE159631; (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE159631).
Authors' contributions
HC and YH participated in the design of the present
study and performed the statistical analysis. HC, JC and XL
performed the experiments and data acquisition. HC, JC and YH
confirmed the authenticity of the raw data. YH, TL, PQ and SQ
analyzed and interpreted the data. YH drafted the manuscript and
obtained funding. HC, JC, TL, PQ and SQ revised the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Scientific Research
Ethics Committee of Longyan First Hospital Affiliated to Fujian
Medical University (Longyan, China; Ethical Review Scientific
Research no. 003; 2018). Written informed consent was obtained from
the patients or their relatives for participation in this study,
including the RNA-seq cohort and validation cohort.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nieuwkamp DJ, Setz LE, Algra A, Linn FH,
de Rooij NK and Rinkel GJ: Changes in case fatality of aneurysmal
subarachnoid haemorrhage over time, according to age, sex, and
region: A meta-analysis. Lancet Neurol. 8:635–642. 2009.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Brown RD Jr, Huston J, Hornung R, Foroud
T, Kallmes DF, Kleindorfer D, Meissner I, Woo D, Sauerbeck L and
Broderick J: Screening for brain aneurysm in the Familial
Intracranial Aneurysm study: Frequency and predictors of lesion
detection. J Neurosurg. 108:1132–1138. 2008.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bo L, Wei B, Wang Z, Kong D, Gao Z and
Miao Z: Screening of Critical Genes and MicroRNAs in Blood Samples
of Patients with Ruptured Intracranial Aneurysms by Bioinformatic
Analysis of Gene Expression Data. Med Sci Monit. 23:4518–4525.
2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wei L, Wang Q, Zhang Y, Yang C, Guan H,
Chen Y and Sun Z: Identification of key genes, transcription
factors and microRNAs involved in intracranial aneurysm. Mol Med
Rep. 17:891–897. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Korostynski M, Morga R, Piechota M,
Hoinkis D, Golda S, Dziedzic T, Slowik A, Moskala M and Pera J:
Inflammatory Responses Induced by the Rupture of Intracranial
Aneurysms Are Modulated by miRNAs. Mol Neurobiol. 57:988–996.
2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Li H, Wang W, Zhang L, Lan Q, Wang J, Cao
Y and Zhao J: Identification of a Long Noncoding RNA-Associated
Competing Endogenous RNA Network in Intracranial Aneurysm. World
Neurosurg. 97:684–692.e4. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wu C, Song H, Wang Y, Gao L, Cai Y, Cheng
Q, Chen Y, Zheng Z, Liao Y, Lin J, et al: Long non-coding RNA
TCONS_00000200 as a non-invasive biomarker in patients with
intracranial aneurysm. Biosci Rep. 39(39)2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kulcheski FR, Christoff AP and Margis R:
Circular RNAs are miRNA sponges and can be used as a new class of
biomarker. J Biotechnol. 238:42–51. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
't Hoen PA, Ariyurek Y, Thygesen HH,
Vreugdenhil E, Vossen RH, de Menezes RX, Boer JM, van Ommen GJ and
den Dunnen JT: Deep sequencing-based expression analysis shows
major advances in robustness, resolution and inter-lab portability
over five microarray platforms. Nucleic Acids Res.
36(e141)2008.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Khan S and Kaihara KA: Single-Cell
RNA-Sequencing of Peripheral Blood Mononuclear Cells with ddSEQ.
Methods Mol Biol. 1979:155–176. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Jeck WR and Sharpless NE: Detecting and
characterizing circular RNAs. Nat Biotechnol. 32:453–461.
2014.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Li HM, Dai YW, Yu JY, Duan P, Ma XL, Dong
WW, Li N and Li HG: Comprehensive circRNA/miRNA/mRNA analysis
reveals circRNAs protect against toxicity induced by BPA in GC-2
cells. Epigenomics. 11:935–949. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wang Y, Wang Y, Li Y, Wang B, Miao Z, Liu
X and Ma Y: Decreased expression of circ_0020397 in intracranial
aneurysms may be contributing to decreased vascular smooth muscle
cell proliferation via increased expression of miR-138 and
subsequent decreased KDR expression. Cell Adhes Migr. 13:220–228.
2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
eLife. 4(4)2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Glažar P, Papavasileiou P and Rajewsky N:
circBase: A database for circular RNAs. RNA. 20:1666–1670.
2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Chou CH, Shrestha S, Yang CD, Chang NW,
Lin YL, Liao KW, Huang WC, Sun TH, Tu SJ, Lee WH, et al: miRTarBase
update 2018: A resource for experimentally validated
microRNA-target interactions. Nucleic Acids Res. 46D:D296–D302.
2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Maass PG, Glažar P, Memczak S, Dittmar G,
Hollfinger I, Schreyer L, Sauer AV, Toka O, Aiuti A, Luft FC, et
al: A map of human circular RNAs in clinically relevant tissues. J
Mol Med (Berl). 95:1179–1189. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Caranci F, Briganti F, Cirillo L, Leonardi
M and Muto M: Epidemiology and genetics of intracranial aneurysms.
Eur J Radiol. 82:1598–1605. 2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Alg VS, Sofat R, Houlden H and Werring DJ:
Genetic risk factors for intracranial aneurysms: A meta-analysis in
more than 116,000 individuals. Neurology. 80:2154–2165.
2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Hussain I, Duffis EJ, Gandhi CD and
Prestigiacomo CJ: Genome-wide association studies of intracranial
aneurysms: An update. Stroke. 44:2670–2675. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Shao Y and Chen Y: Roles of Circular RNAs
in Neurologic Disease. Front Mol Neurosci. 9(25)2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
van Rossum D, Verheijen BM and Pasterkamp
RJ: Circular RNAs: Novel Regulators of Neuronal Development. Front
Mol Neurosci. 9(74)2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Hao Z, Li Y, Yu N, Zhao Y, Hu S, Liu Z and
Li M: Analysis of differentially expressed circular RNAs in
endothelial cells under impinging flow. Mol Cell Probes.
51(101539)2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Huang Q, Huang QY, Sun Y and Wu S:
High-Throughput Data Reveals Novel Circular RNAs via Competitive
Endogenous RNA Networks Associated with Human Intracranial
Aneurysms. Med Sci Monit. 25:4819–4830. 2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Signorelli F, Sela S, Gesualdo L, Chevrel
S, Tollet F, Pailler-Mattei C, Tacconi L, Turjman F, Vacca A and
Schul DB: Hemodynamic Stress, Inflammation, and Intracranial
Aneurysm Development and Rupture: A Systematic Review. World
Neurosurg. 115:234–244. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Chidlow JH Jr, Glawe JD, Alexander JS and
Kevil CG: VEGF164 differentially regulates neutrophil and T cell
adhesion through ItgaL- and ItgaM-dependent mechanisms. Am J
Physiol Gastrointest Liver Physiol. 299:G1361–G1367.
2010.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Hoh BL, Rojas K, Lin L, Fazal HZ, Hourani
S, Nowicki KW, Schneider MB and Hosaka K: Estrogen Deficiency
Promotes Cerebral Aneurysm Rupture by Upregulation of Th17 Cells
and Interleukin-17A Which Downregulates E-Cadherin. J Am Heart
Assoc. 7(7)2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Sawyer DM, Pace LA, Pascale CL, Kutchin
AC, O'Neill BE, Starke RM and Dumont AS: Lymphocytes influence
intracranial aneurysm formation and rupture: Role of extracellular
matrix remodeling and phenotypic modulation of vascular smooth
muscle cells. J Neuroinflammation. 13(185)2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Liu Y, Zhang Y, Dai D and Xu Z: Expression
of NF-kappaB, MCP-1 and MMP-9 in a Cerebral Aneurysm Rabbit Model.
Can J Neurol Sci. 41:200–205. 2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Dou C, Cao Z, Yang B, Ding N, Hou T, Luo
F, Kang F, Li J, Yang X, Jiang H, et al: Changing expression
profiles of lncRNAs, mRNAs, circRNAs and miRNAs during
osteoclastogenesis. Sci Rep. 6(21499)2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Faveeuw C, Di Mauro ME, Price AA and Ager
A: Roles of alpha(4) integrins/VCAM-1 and LFA-1/ICAM-1 in the
binding and transendothelial migration of T lymphocytes and T
lymphoblasts across high endothelial venules. Int Immunol.
12:241–251. 2000.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Hinterseher I, Schworer CM, Lillvis JH,
Stahl E, Erdman R, Gatalica Z, Tromp G and Kuivaniemi H:
Immunohistochemical analysis of the natural killer cell
cytotoxicity pathway in human abdominal aortic aneurysms. Int J Mol
Sci. 16:11196–11212. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Yang H, Graham LC, Reagan AM, Grabowska
WA, Schott WH and Howell GR: Transcriptome profiling of brain
myeloid cells revealed activation of Itgal, Trem1, and Spp1 in
western diet-induced obesity. J Neuroinflammation.
16(169)2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhu Z, Li Y, Liu W, He J, Zhang L, Li H,
Li P and Lv L: Comprehensive circRNA expression profile and
construction of circRNA-associated ceRNA network in fur skin. Exp
Dermatol. 27:251–257. 2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wu J, Liu S, Xiang Y, Qu X, Xie Y and
Zhang X: Bioinformatic Analysis of Circular RNA-Associated ceRNA
Network Associated with Hepatocellular Carcinoma. BioMed Res Int.
2019(8308694)2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Cao M, Zhang L, Wang JH, Zeng H, Peng Y,
Zou J, Shi J, Zhang L, Li Y, Yoshida S, et al: Identifying
circRNA-associated-ceRNA networks in retinal neovascularization in
mice. Int J Med Sci. 16:1356–1365. 2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Li X, Ding J, Wang X, Cheng Z and Zhu Q:
NUDT21 regulates circRNA cyclization and ceRNA crosstalk in
hepatocellular carcinoma. Oncogene. 39:891–904. 2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Li C, Li M and Xue Y: Downregulation of
CircRNA CDR1as specifically triggered low-dose Diosbulbin-B induced
gastric cancer cell death by regulating miR-7-5p/REGγ axis. Biomed
Pharmacother. 120(109462)2019.PubMed/NCBI View Article : Google Scholar
|