Introduction
Neutropenia is defined by a reduced absolute
neutrophil count (ANC). In Caucasian infants, aged between 2 weeks
and 1 year, the lower limit of the ANC is 1,000/µl, whereas for
children above the age of 1 year, the lower limit of the ANC is
1,500/µl. Frequently encountered in children, the disorder may be
responsible for various clinical representations, from benign and
transient forms to sometimes overwhelming disease (1,2). Mild
neutropenia is defined by an ANC between 1,000-1,500/µl, moderate
by 500-1,000/µl and the severe form by ANC <500/µl (3). Autoimmune neutropenia (AIN), a special
form of the disorder, is due to autoantibodies against
neutrophil-specific cell surface antigens, such as the human
neutrophil antigen (HNA)-1a, HNA-1b and HNA-2. Antibody-coated
neutrophils are destroyed in the peripheral blood, sometimes
augmented by deposition of C3 complement fraction. The
anti-neutrophil antibodies may also interfere with myelopoiesis in
the bone marrow (4). Historically,
its prevalence is considered to be 1/100,000 in children under the
age of 10, consequently belonging to a group of rare disorders. It
is part of the large and heterogeneous group of neutropenias, some
of intrinsic origin (mainly congenital, mostly a result of altered
myelopoiesis) and various others with extrinsic determination
(acquired, primary or secondary to altered myelopoiesis,
malignancy, drugs, infections). AIN itself can have a heterogeneous
background, as an isolated disorder (primary AIN) or occurring
within the frame of other autoimmune diseases and even as a
complication of infections, drugs, malignancy or vaccination
(secondary AIN). The identification of primary AIN, also named
chronic benign neutropenia of infancy and childhood, within the
large and heterogeneous group of neutropenias, is of utmost
importance, if we consider the risk of sometimes overwhelming
life-threatening infections on the one hand and the expensive
therapies (prophylactic G-CSF replacement, antibiotic prophylaxis)
on the other hand, which could and should be avoided. Primary AIN
of infancy and childhood usually has a harmless and self-limited
clinical course, with low frequency and severity of infections,
easily managed in an outpatient setting; although some cases,
mainly young infants, may suffer from severe infection (1,3,5). Our
objectives were to review the clinical and biological features, as
well as the outcome of primary AIN in a cohort of pediatric
patients and to assess the role of anti-neutrophil antibody
screening in confirming and decision-making of the therapeutic and
follow-up approach.
Patients and methods
This retrospective, cross-sectional cohort study was
performed on 18 patients with primary AIN, selected out of 3,488
patients consecutively admitted to the ‘Louis Țurcanu’ Emergency
Hospital for Children, with different diagnoses, but who also
developed neutropenia. The retrospective enrollment period was 3
years (January 2016 to December 2018). We had to exclude 224
patients from the analysis due to incomplete data. From the
remaining 3,264 patients, we focused on patients with primary AIN
of infancy. The approval of the local ethics committee, Ethics
Committee for Scientific Research of the Emergency Hospital for
Children ‘Louis Turcanu’ (approval no. 77/2020), was obtained prior
to starting the study. Patient's parental or caregiver's consents
were obtained where applicable.
Primary AIN of infancy and childhood represented
around 0.5% of all cases (18 patients). In order to be included in
the study, patients had to fulfill the criteria for neutropenia
(ANC <1,500/µl for children >1 year of age, ANC <1,000/µl
for infants aged over 2 weeks); and more than 3 months persistence
of neutropenia and granulocyte antibody screening performed.
Exclusion criteria were: underlying disorders that could render
neutropenia as secondary and patients with incomplete data or lost
to follow-up.
The granulocyte antibody screening consisted of the
granulocyte immunofluorescence test (GIFT), which detects
autoantibodies bound to patient neutrophils or in patient plasma
and the granulocyte agglutination test (GAT), which is used to
detect agglutination of control neutrophils in contact with
sensitized patient serum sample. For GIFT, paraformaldehyde-fixed
neutrophils are incubated with serum to allow neutrophil reactive
antibodies to bind to the antigenic epitopes and then washed and
incubated with a fluorescence-labeled anti-human globulin IgG, IgM
and IgA. Fluorescence microscopy is used for the analysis. In the
GAT, neutrophil-reactive antibodies bind to native antigens on
unfixed neutrophils, sensitizing the cells followed by a second
phase; sensitized neutrophils undergo chemotaxis and move towards
other neutrophils and agglutinate. The screening was performed by
means of both the GIFT test and the GAT test, in compliance with
the recommendations of the Second International Granulocyte
Serology Workshop (6).
For the anti-neutrophil antibody screen, 6 ml of
blood in EDTA vacutainers and 4 ml of non-anticoagulated blood were
drawn from each patient after prior approval by their parents or
caregivers. The workup of all patients also included: Complete
blood count (CBC) with differentials and reticulocyte count,
peripheral blood smear, biochemistry including C-reactive protein
(CRP), erythrocyte sedimentation rate (ESR), ferritin,
transaminases and lactate dehydrogenase (LDH), and immunological
screening with quantitative IgG, IgM and IgA measurement,
complement C3 and C4, anti-nuclear antibody screen, serological
markers for cytomegalovirus (CMV), Epstein-Barr virus (EBV),
hepatitis B, and C, human immunodeficiency virus (HIV) and
toxoplasmosis. Hematological investigations were performed with a
Sysmex XS800i analyzer (SYSMEX Corp., Kobe, Japan) using impedance
spectroscopy, flow cytometry, Hydro Dynamic Focusing (DC Detection
method). The biochemical investigations were performed with a Cobas
Integra 400 Plus analyzer (ROCHE Diagnostics GmbH, Germany). Bone
marrow (BM) smear examination was also performed in some patients.
Data were collected and analyzed from the patient files by means of
a database in Microsoft Office Excel software. Descriptive
statistics, correlations and Kaplan-Meier curves were performed
using IBM® SPSS® statistics version 25 (IBM
Corp.). A P-value of <0.05 was considered to be significant.
Results
The median age at onset of neutropenia of any type
was 3 months (range 0-9 months) and for children aged over 1 year,
3.5 years (range 1-17.9 years). The distribution of the types of
neutropenia are documented in Table
I.
 | Table IDistribution of the patients by type
of neutropenia (N=3,264). |
Table I
Distribution of the patients by type
of neutropenia (N=3,264).
| Type of
neutropenia | No. of patients
(%) |
|---|
| Autoimmune (primary
and secondary) | 31(1) |
| Congenital
neutropenia | 3 (0.1) |
| Neutropenia of
prematurity | 62 (1.9) |
| Bone marrow failure
syndromes | 8 (0.25) |
| Neutropenia in
malignancy and chemotherapy | 205 (6.3) |
| Transient
neutropenia | 2,955 (90.45) |
Autoimmune neutropenia (primary and secondary)
represented approximately 1% of the 3,264 patients enrolled in the
analysis, being found in 31 patients. From these patients, 18 were
diagnosed with primary AIN of infancy and childhood. The rest of
the patients (13) presented
secondary AIN (Table II). Our
study focused on the 18 patients with primary AIN.
| Table IITypes of secondary AIN (N=13). |
Table II
Types of secondary AIN (N=13).
| Type of secondary
AIN | No. of patients |
|---|
| Systemic lupus
erythematosus | 5 |
| Autoimmune
lymphoproliferative syndrome | 1 |
| Hemophagocytic
lymphohistiocytosis | 2 |
| Wiskott-Aldrich
syndrome | 1 |
| IgA deficiency | 3 |
| Fanconi anemia
associated with autoimmunity | 1 |
Most patients with primary AIN had their first
diagnosis during infancy, the median age at diagnosis being 7.5
months (range 1-20 months). The male/female ratio was 1.94.
Regarding the severity of neutropenia at first diagnosis, we found
that 10 patients (56%) presented severe neutropenia and 8 patients
(44%) moderate neutropenia. Median ANC at diagnosis was 315/µl
(range 0-800/µl). In contrast, during the course of the disease, 13
patients (72%) presented ANC counts below 500/µl, whereas only 5
patients stabilized at ANC between 500-1,000/µl. Table III presents the main
characteristics of the 18 study patients and the results of the
granulocyte antibody screening.
| Table IIICharacteristics of the study patients,
and results of the granulocyte antibody screening and outcome. |
Table III
Characteristics of the study patients,
and results of the granulocyte antibody screening and outcome.
| Pt. no | Age at diagnosis
(months) | ANC/µl at
diagnosis | Severity of
neutropenia (minimal ANC during course of disease) | Anti-neutrophil
antibody screening | Outcome (time to
resolution) |
|---|
| 1 | 7 | 120 | Severe (min
0/µl) | GIFT positive with 2
out of 4 test cells/GAT weak positive with 4 out of 4 test
cells | 23 months |
| 2 | 3 | 730 | Severe (min
0/µl) | GIFT weak positive
with 3 out of 4 test cells/GAT negative with 4 out of 4 test
cells | >15 months |
| 3 | 9 | 70 | Severe (min
70/µl) | GIFT positive with 4
out of 4 test cells/GAT positive with 2 out of 3 test cells | 12 months |
| 4 | 11 | 100 | Severe (min
30/µl) | GIFT positive for
anti-IgG with 3 out of 4 test cells, GIFT negative for anti-IgM
with 2 out of 2 test cells/GAT positive at 30˚C with 4 out of 4
test cells, GAT negative at 37˚C with 4 out of 4 test cells | 18 months |
| 5 | 5 | 130 | Severe (min
0/µl) | GIFT negative with 4
out of 4 test cells/GAT negative with 4 out of 4 test cells | 26 months |
| 6 | 8 | 70 | Severe (min
0/µl) | GIFT positive with 3
out of 4 test cells/GAT negative with 2 out of 2 test cells | 13 months |
| 7 | 3 | 800 | Moderate (min
630/µl) | GIFT negative with 4
out of 4 test cells/GAT negative with 4 out of 4 test cells | 7 months |
| 8 | 8 | 200 | Severe (min
70/µl) | GIFT clearly positive
with 1 out of 3 test cells/GAT negative with 3 out of 3 test
cells | 5 months |
| 9 | 20 | 90 | Severe (min
40/µl) | GIFT strong positive
with 4 out of 4 test cells/GAT negative with 4 out of 4 test
cells/LIFT negative with 4 out of 4 test cells | 18 months |
| 10 | 10 | 0 | Severe (min
0/µl) | GIFT weak to strong
positive with 3 out of 4 test cells/GAT negative with 4 out of 4
test cells/LIFT negative with 4 out of 4 test cells/Glycoprotein
specific immune assay to CD11a, CD11b, CD16b, CD18, CD117,
HLA-class I antigen | 18 months |
| 11 | 8 | 0 | Severe (min
0/µl) | GIFT negative with 4
out of 4 test cells/GAT negative with 4 out of 4 test cells | 14 months |
| 12 | 1 | 700 | Moderate (min
700/µl) | GIFT negative with 4
out of 4 test cells/GAT negative with 4 out of 4 test cells | 9 months |
| 13 | 1 | 800 | Severe (min
430/µl) | GIFT weak positive
with 3 out of 4 test cells (anti-HNA1a)/GAT negative with 4 out of
4 test cells | 13 months |
| 14 | 3 | 620 | Severe (min
410/µl) | GIFT weak positive
with 4 out of 4 test cells/GAT negative with 4 out of 4 test
cells | 16 months |
| 15 | 11 | 430 | Severe (min
0/µl) | GIFT negative with 4
out of 4 test cells/GAT negative with 4 out of 4 test cells | 5 months |
| 16 | 6 | 640 | Severe (min
460/µl) | GIFT weak positive
with 3 out of 4 test cells/GAT negative with 4 out of 4 test
cells | 12 months |
| 17 | 7 | 780 | Severe (min
250/µl) | GIFT negative with 4
out of 4 test cells/GAT negative with 4 out of 4 test cells | 8 months |
| 18 | 15 | 540 | Moderate (min
540/µl) | GIFT negative with 4
out of 4 test cells/GAT negative with 4 out of 4 test cells | 8 months |
| | Median 7.5
months | Median 315/µl | Median 55/µl | | Median 13 months |
Regarding the clinical manifestations at diagnosis,
6 patients (33%) were asymptomatic, neutropenia being revealed by a
routine analysis. Table IV depicts
the reasons for presentation of the patients at initial
diagnosis.
| Table IVClinical manifestations and reasons
for examination at the time of diagnosis (N=18). |
Table IV
Clinical manifestations and reasons
for examination at the time of diagnosis (N=18).
| Reason for
presentation at diagnosis | No. of patients
(%) |
|---|
| Evaluation for
anemia | 3 (16.9) |
| Rhinopharyngitis | 3 (16.9) |
| Ethmoiditis | 1 (5.5) |
| Enterocolitis | 2 (11.2) |
| Urinary tract
infection | 1 (5.5) |
| Pneumonia | 1 (5.5) |
| Neonatal
omphalitis | 1 (5.5) |
| Asymptomatic
(routine analysis) | 6(33) |
Bone marrow (BM) examination was performed in 8
patients (45%). In 6 cases, the marrow smears revealed a bone
marrow sample with rich cellularity and moderate myeloid
hyperplasia. In patient no. 2 (Table
III), the smear showed hypoplasia of the myeloid series with
delayed maturation and hypoplasia of the erythroblastic series and
moderate lymphocytosis. The patient presented a positive GIFT test
and did not recover yet, after 15 months of follow-up. Patient no.
11 (Table III), whose BM
examination also revealed a hypoplastic marrow, especially of the
myeloid series, with stimulation of the cellular immunocompetent
system and moderate stimulation of the erythropoiesis, had a
negative antibody screening, but recovered spontaneously at 14
months after diagnosis.
Granulocyte antibody screening was performed in all
18 patients. In 8 patients (45%), GIFT was positive, but GAT was
negative. In 3 patients (16%), both GIFT and GAT yielded positive
results, whereas in 7 patients (39%) both GIFT and GAT revealed
negative results. Table III
depicts the results of each patient. In all patients with positive
GAT test, results were confirmed by the GIFT test.
During the course of the disease, within the
follow-up period, 16 patients (89%) presented upper respiratory
tract infections, the majority of which were uncomplicated
rhinopharyngitis. Otitis media occurred in 3 patients, one of which
presented recurrent otitis. Two patients presented pneumonia and
other 2 urinary tract infections (UTI). Three patients developed
enterocolitis, one of which was due to rotavirus infection. One
patient presented omphalitis with E. coli, this being the
initial clinical finding at admission. The clinical follow-up of
the patients showed that in 3 cases (17%), the clinical course of
the disease was severe with the need for aggressive antibiotic
therapy, the patients presenting pneumonia and ethmoiditis.
Nevertheless, 50% of the patients required hospitalization for
neutropenic fever and/or proven bacterial infection, but no patient
needed antibiotic prophylaxis and the outcome of infections was in
all cases favorable. None of the patients developed systemic or
localized fungal infections, except for oral thrush, which was
treated topically. Eleven patients (61%) required antibiotic
therapy during the course of the disease or during the follow-up
period and in 4 patients (22%), a short course of low-dose G-CSF
was used during infectious episodes. Intravenous immunoglobulin
(IvIg) was administered in one patient and it was very efficient,
inducing permanent remission of the AIN (Table V).
| Table VCharacteristics of infections during
the course of disease. |
Table V
Characteristics of infections during
the course of disease.
| Parameter | No. of patients
(%) | Observation |
|---|
| Severe
infections | 3(17) | Pneumonia,
ethmoiditis |
| Need for
infection-driven hospitalization | 9(50) | Fever and/or proven
bacterial infection |
| Fungal
infections | 0 (0) | Exception: Oral
thrush topical treatment |
| Favorable
outcome | 18(100) | |
| Antibiotic
prophylaxis | 0 (0) | |
| Antibiotic use
during hospitalization or home treatment | 11(61) | |
| Use of G-CSF
(on-demand) | 4(22) | Short
course/low-dose |
| Use of IvIg | 1 (5.5) | Induced permanent
remission |
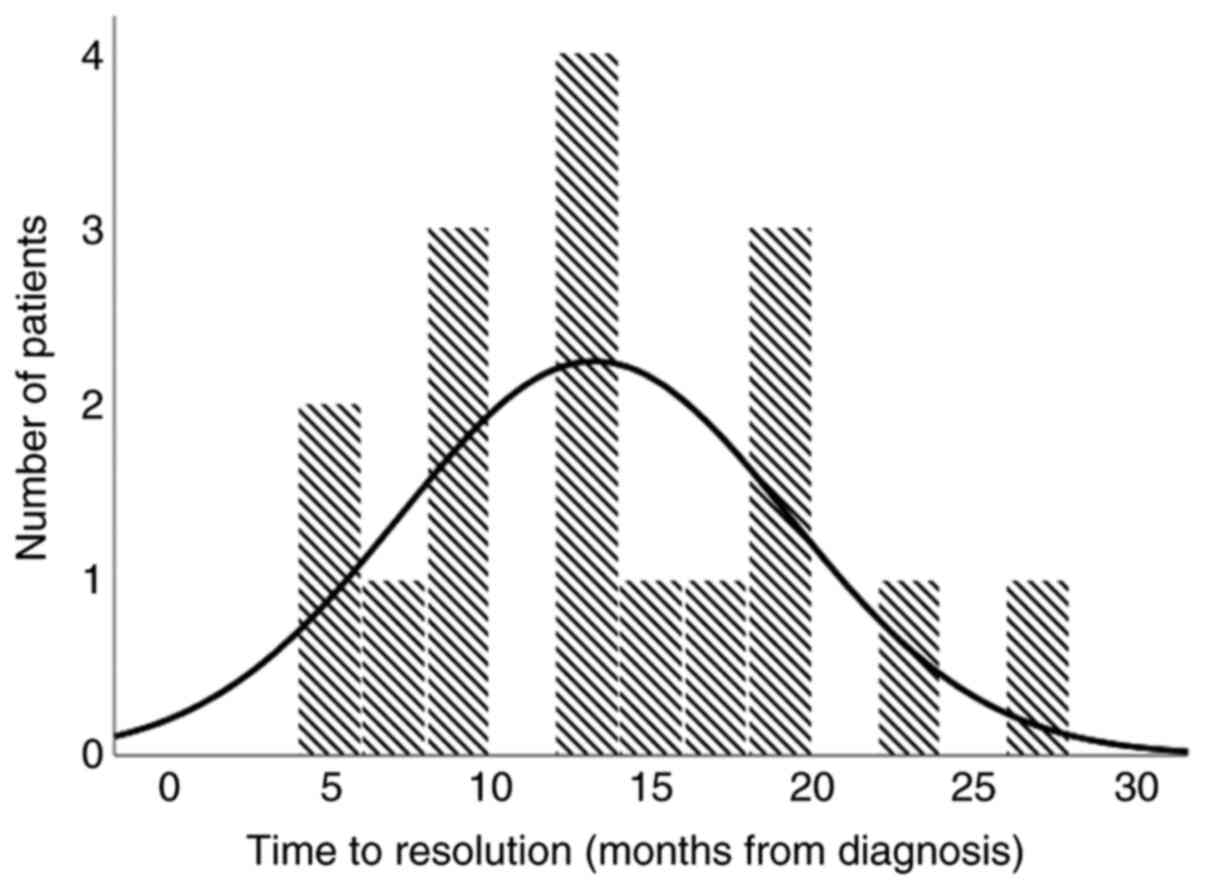
The median time to recovery of neutropenia was 13
months (range 5-26 months), as shown in Fig. 1. In 89% of the patients, the time to
recovery from neutropenia was within 20 months after diagnosis.
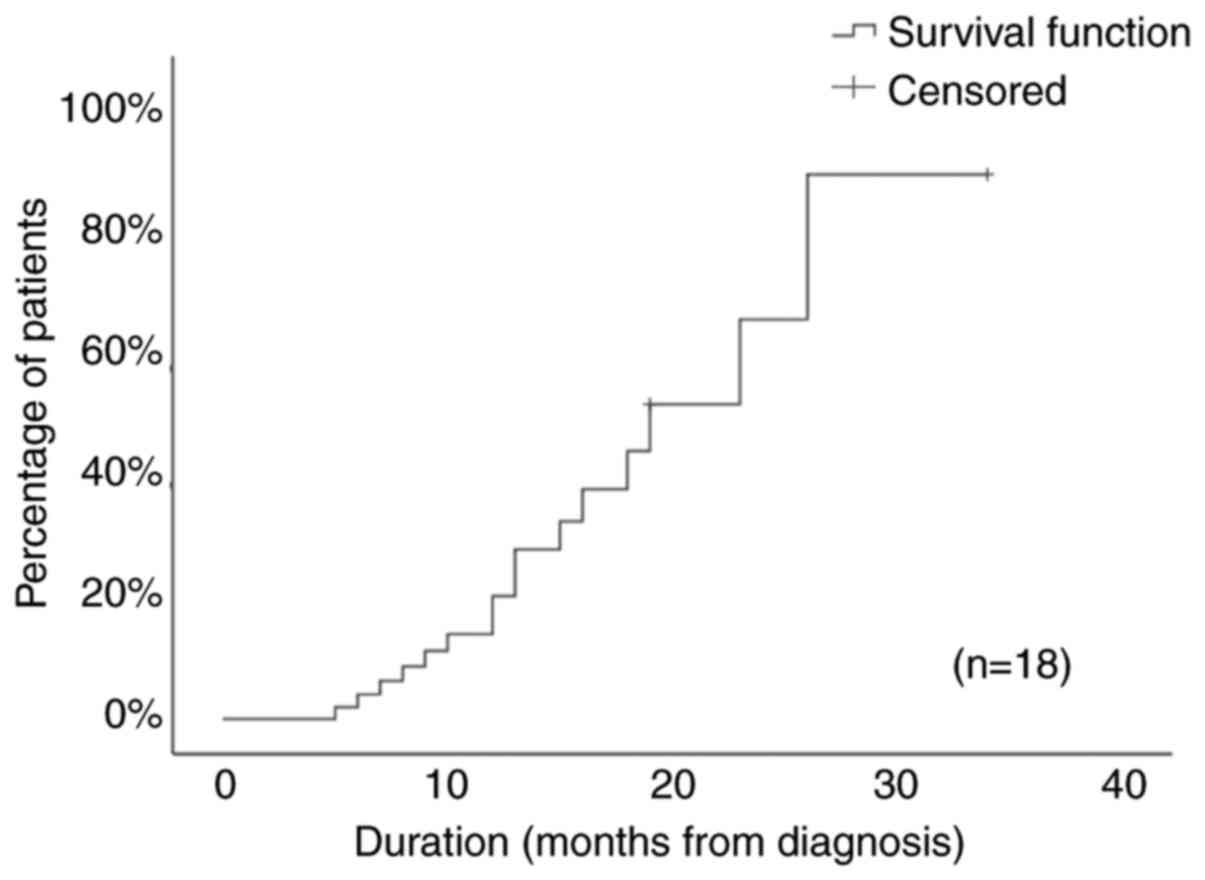
Fig. 2 depicts the Kaplan-Meier
recovery curve for the study cohort. The pattern of neutrophil
recovery was intermittent, with alternating phases of normalization
and relapse in 5 patients (28%) and continuous in the other 13
(72%).
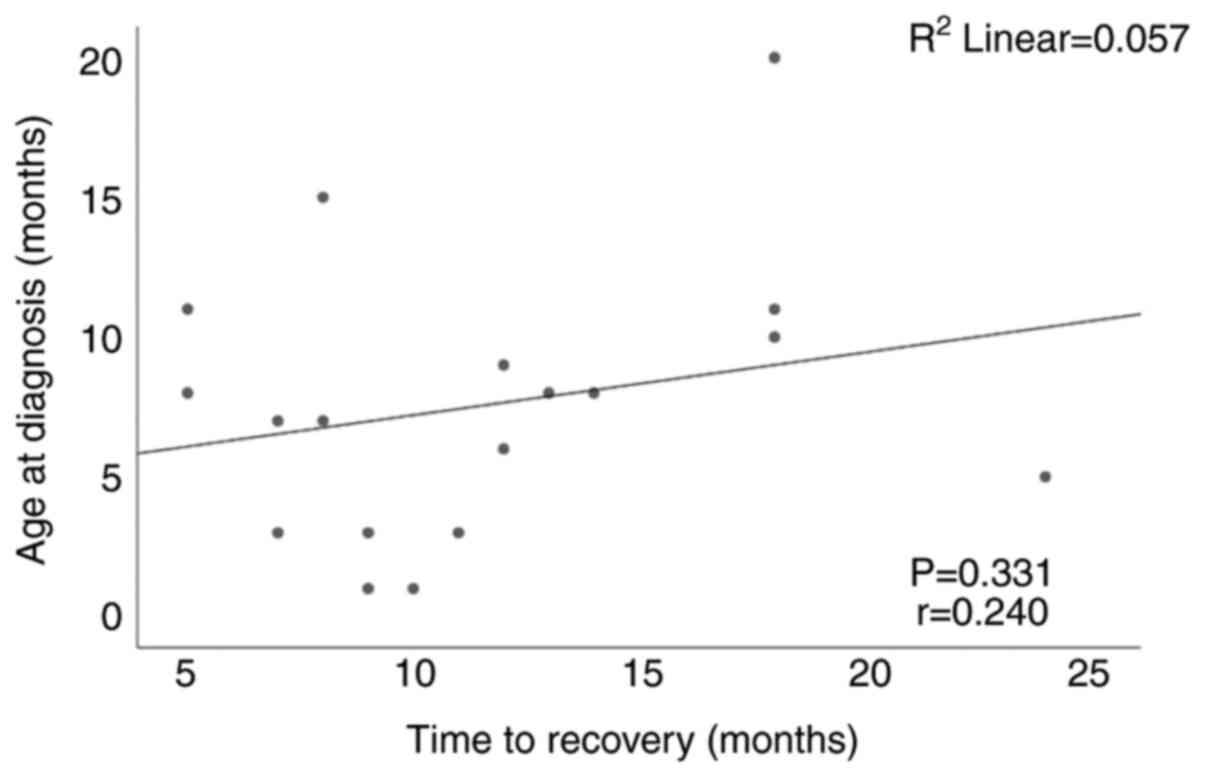
One of the objectives of the study was to establish
if age at diagnosis correlates with the time to recovery. Fig. 3 shows that there was no
statistically significant correlation (r=0.240, P=0.331) between
the two variables.
Discussion
In our observational study, we present the clinical
and biological features of 18 patients with primary autoimmune
neutropenia (AIN) starting from the premise that all types of
neutropenia are frequent in infancy and early childhood but the
persistent, chronic forms are rare. Within the frame of multiple
etiologies and mechanisms involved in the pathological background
of neutropenia, the diagnosis of primary AIN of infancy remains one
of exclusion. The diagnosis is often suggested by the benign course
of the disease, rarely severe, despite the impressively low
absolute neutrophil count (ANC). Its main differential diagnoses
are alloimmune neutropenia (AN), cyclic neutropenia (CN) and severe
congenital neutropenia (SCN), which are to be excluded from the
beginning (7,8). In 33% of our patients, no sign of
infection could be identified at the time of diagnosis despite the
strikingly low ANC. Due to this discrepancy between the low ANC,
which can pose the risk of potentially life-threatening infection,
clinicians have to proceed immediately with an algorithmic work-up
in order to exclude other etiologies of the neutropenia, after
confirmation of its persistence, in order to rule out laboratory
error (3). There is still debate
whether or not to perform anti-neutrophil antibody screening.
Walkovich and Boxer suggest that anti-neutrophil antibody screening
should start in an infant who remains asymptomatic despite the
persistence of low ANC (9). Bone
marrow investigation may also be postponed until the result of the
anti-neutrophil antibody screening is negative (8). Several methods for the anti-neutrophil
antibody screening have been employed and developed, with different
limitations from the point of view of their sensitivities and
specificities. The International Granulocyte Immunology Workshop
recommends using both the GIFT and GAT tests for diagnostic studies
(4,5,8-10).
It has been shown that GIFT is more sensitive than GAT.
Nevertheless, there are certain alloantibody specificities, such as
anti-5b, anti-NB2 and anti-9a and also autoantibodies, which are
easier to be detected by the GAT. In addition, antibody titers are
usually low, raising the need for the tests to be repeated. Testing
for specific anti-neutrophil antibodies may be difficult to perform
as well, considering the need for a sufficient number of isolated
granulocytes and to the unspecific binding of IgG immune complexes
to the Fcγ receptors II and IIIb of the activated neutrophils.
Conversely, it has been shown that if cells from patients are
activated by inflammation and if their plasma contains large
amounts of immune complexes, the elevated granulocyte-associated
IgG levels may lead to false-positive results (4,8,11,12).
Bux et al reported the detection of granulocyte-specific
antibodies in 74% of the investigated cases from a cohort of 240
tested children within the first investigation, but there was a
need to repeat testing for the remainder 26% of the patients
(8). Bruin et al (13) reported a rate of positivity of 80%.
Farruggia and Dufour found a 62% positivity rate with a single
assay, but repeated assay increased the rate of positivity to 82%
(5). Our findings are in
accordance, showing a positivity rate of 61% at the initial
testing. The limitations of the two methods can be overcome by the
MAIGA (monoclonal antibody-specific immobilization of granulocyte
antigen) assay (13). This
technique is not routinely available and is used for the location
of autoantigens (4,11,12).
Repeating the combined GIFT and GAT test seems to be useful,
especially in challenging cases in which a bone marrow
investigation may also be warranted. It has been recommended to use
bone marrow examination for diagnosis in cases with severe
infections, severe stomatitis or recurrent high fever or in the
case of findings that may suggest leukemia, myelodysplastic
syndromes or bone marrow failure syndromes (3). The usual finding in primary AIN is
that of a reactive marrow with no morphological abnormalities and
an increased myeloid to erythroid ratio. The arrest of maturation
of the myeloid series has also been observed. In a minority of
patients, a hypoplastic bone marrow can also be encountered,
reflecting the presence of autoantibodies reactive to myeloid
precursors (4,5,14). In
2 of our patients, BM examination revealed a hypoplastic marrow. We
found an incidence of 17% severe infections and a hospitalization
rate of 50%. In a retrospective study, Farruggia et al found
a rate of 44.2% hospitalizations and only 9.6% severe infections in
157 patients (15). The use of
granulocyte-colony stimulating factor (G-CSF) was necessary in 22%
of the cases and was administered on-demand, as short course and in
low doses. No corticosteroids or prophylactic antibiotics were
used. IvIg was used in one single patient and the response was
rapid and induced complete neutrophil recovery. All cohort studies
have reported the use of G-CSF in primary AIN, with a rapid
response rate. The effect of IvIg is reported to be good, but short
lasting (8,13). The time to recovery of neutrophils
in different reports was between 1 and 4 years from diagnosis in
the majority of patients (3,5). Two
patterns of recovery, continuous and intermittent, have been
described, but no parameter could be identified to predict this
pattern (15). The recovery curve
in our study cohort showed that ~90% of the patients recovered
within 20 months after diagnosis. There was no statistically
significant correlation between the age at diagnosis and the time
to recovery; however, this could be attributed to the small number
of patients in our study.
In conclusion, the results of our study support the
use of the granulocyte antibody screening to confirm the diagnosis
of primary AIN in order to apply a rather non-aggressive management
to these patients, avoiding unnecessary use of antifungals and
antibiotics and most importantly of hospitalization during febrile
episodes in confirmed patients. It may also provide confidence and
reassurance for the families of these children.
Acknowledgements
We would like to thank Professor Dr U. Sachs and
Professor Dr G. Bein from the Granulocyte Laboratory of the Zentrum
fur Transfusionsmedizin und Hamotherapie of the Uniklinikum Giessen
und Marburg (UKGM), Giessen, Germany for performing the granulocyte
antibody screening. Professional editing, linguistic and technical
assistance were performed by Irina Radu.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Author's contributions
CJ, MS and SA conceived and designed the study. AM
and EU performed the acquisition of data. CJ, AM and EU were
involved in the analysis and interpretation of collected data. CJ,
MS, AM and EU wrote the original draft. MS, CJ and SA were
responsible for the critical revision of the manuscript's content.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The approval of the local ethics committee (Ethics
Committee for Scientific Research of the Emergency Hospital for
Children ‘Louis Turcanu’) (approval no. 77/2020) was obtained prior
to starting the study. Patient's parental or caregiver's consents
were obtained where applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Thomas AE and Simpson LA: A step-by-step
approach to paediatric neutropenia. Paediatrics and Child Health.
27:511–516. 2017.
|
|
2
|
Celkan T and Koç BŞ: Approach to the
patient with neutropenia in childhood. Turk Pediatri Ars.
50:136–144. 2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Newburger PE: Autoimmune and other
acquired neutropenias. Hematology Am Soc Hematol Educ Program.
2016:38–42. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Capsoni F, Sarzi-Puttini P and Zanella A:
Primary and secondary autoimmune neutropenia. Arthritis Res Ther.
7:208–214. 2005.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Farruggia P and Dufour C: Diagnosis and
management of primary autoimmune neutropenia in children: Insights
for clinicians. Ther Adv Hematol. 6:15–24. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Bux J and Chapman J: Report on the second
international granulocyte serology workshop. Transfusion.
37:977–983. 1997.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Donadieu J, Fenneteau O, Beaupain B,
Mahlaoui N and Chantelot CB: Congenital neutropenia: Diagnosis,
molecular bases and patient management. Orphanet J Rare Dis.
6(26)2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bux J, Behrens G, Jaeger G and Welte K:
Diagnosis and clinical course of autoimmune neutropenia in infancy:
Analysis of 240 cases. Blood. 91:181–186. 1998.PubMed/NCBI
|
|
9
|
Walkovich K and Boxer LA: How to approach
neutropenia in childhood. Pediatr Rev. 34:173–184. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Dale DC: How I manage children with
neutropenia. Br J Haematol. 178:351–363. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Autrel-Moignet A and Lamy T: Autoimmune
neutropenia. Presse Med. 43(4 Pt 2):e105–118. 2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Heinzl MW, Schönbacher M, Dauber EM,
Panzer S, Mayr WR and Körmöczi GF: Detection of
granulocyte-reactive antibodies: A comparison of different methods.
Vox Sang. 108:287–293. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Bruin MC, von dem Borne AE, Tamminga RY,
Kleijer M, Buddelmeijer L and de Haas M: Neutrophil antibody
specificity in different types of childhood autoimmune neutropenia.
Blood. 94:1797–1802. 1999.PubMed/NCBI
|
|
14
|
Lyall EG, Lucas GF and Eden B: Autoimmune
neutropenia of infancy. J Clin Pathol. 45:431–434. 1992.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Farruggia P, Fioredda F, Puccio G,
Porretti L, Lanza T, Ramenghi U, Ferro F, Macaluso A, Barone A,
Bonanomi S, et al: Autoimmune neutropenia of infancy: Data from the
Italian Neutropenia Registry. Am J Hematol. 90:E221–E222.
2015.PubMed/NCBI View Article : Google Scholar
|