Introduction
Glioblastoma is the most common primary malignant
tumor in the central nervous system (CNS), accounting for 46.1% of
the total malignant tumors in the United States that are diagnosed
in the brain and spinal cord (1-3).
Glioblastoma has been indicated to constitute the majority (55.1%)
of gliomas that originate from glial cells or precursor cells
(1,4) and to be highly aggressive, which is
reflected by its classification as a grade IV neoplasm in CNS
tumors (5). The incidence of
glioblastoma has been estimated to be 3.2 per 100,000 individuals
worldwide (1). Current treatment
strategies for glioblastoma include the combination of surgical
resection with radiotherapy, systemic chemotherapy and local
treatment with carmustine wafer, immunotherapy with the
anti-angiogenic drug bevacizumab and electric field-based treatment
(6-10).
However, patients with glioblastoma undergoing the aforementioned
treatments have been indicated to exhibit a poor prognosis with a
5-year survival rate of ~5% and a median survival of ~15 months
(1,11,12).
The majority of patients has been revealed to ultimately succumb to
the disease due to local recurrence of the tumor (13), which results from the infiltrative
viability of glioblastoma that often hinders the complete surgical
removal of the initial tumors. Additionally, the blood-brain
barrier (BBB) has been indicated to prevent chemotherapy drugs from
entering the brain, and even when the drugs can be successfully
delivered to the tumor, glioblastomas have developed mechanisms of
drug resistance, which render chemotherapy less effective (8). Therefore, an urgent need still exists
to improve current treatment modalities and/or to develop novel
therapeutic strategies to ameliorate the clinical outcomes of
patients with glioblastoma.
Strategies focused on enhancing the delivery of
cytotoxic drugs or bioreactive agents into the glioblastoma tumor
cells with nanocarriers have been previously implemented in
preclinical studies and clinical trials (8,14-17).
The efficiency of targeted delivery for nanocarriers is determined
by their size, surface charge, surface hydration and targeting
moiety (15). An ideal nanocarrier
has been indicated to be able to cross the highly selective BBB to
reach the infiltrating glioblastoma cells, as well as to
extravasate via the disrupted blood-brain tumor barrier to enter
into the principal tumor core (15,18).
Moreover, the nanocarrier should be able to bypass the multidrug
resistance efflux transporters, such as P-glycoprotein (P-gp), the
multidrug resistance proteins (MRPs) and breast cancer resistance
protein, which are usually overexpressed in glioblastoma cells and
contribute to the tumor inherent resistance to multiple
chemotherapy drugs (19).
The anticancer drug paclitaxel (PTX) has been
indicated to inhibit glioblastoma viability in mice; however, this
antitumor effect was revealed to be reduced in endothelial and
glioblastoma cells where P-gp was overexpressed, thereby conferring
resistance to PTX, and preventing the entrance of PTX into tumor
cells (20,21). Therefore, the development of a
potent carrier to deliver PTX into glioblastoma cells is required.
The present study investigated whether previously reported
nanoparticle (20) may enhance the
antitumor activity of PTX in rat glioblastoma C6 cells in
vitro. This nanoparticle is composed of amphiphilic poly
(γ-glutamic
acid-maleimide-co-L-lactide)-1,2-dipalmitoylsn-glycero-3-phosphoethanolamine
(γ-PGA-MAL-PLA-DPPE) copolymer conjugated with a targeting moiety
transferrin (Tf) and exhibits a great potential to be an ideal
nanocarrier (20). To address these
issues, the present study aimed to develop an efficient
nano-delivery system for delivering therapeutic agents into
glioblastoma cells. The present study provided solid evidence for
subsequent investigation of PTX-Tf-NPs in animal models of
glioblastoma to better elucidate the antitumor potency of
PTX-Tf-NPs.
Materials and methods
MTT assay
Transferrin (Tf), 4-Nitro-phenyl chloroformate (pNP)
(97%), and paclitaxel (PTX) were obtained from Alfa Aesar,
Sigma-Aldrich (Merck KGaA) and Beijing HuaFeng Unite Co. Ltd.,
respectively, and were prepared as previously described (22). MTT assay was used to evaluate the
effect of Tf-NPs, PTX and PTX-Tf-NPs on cell viability in C6
(glioblastoma) and HT22 cells (mouse hippocampal neuron cell line,
purchased from American Type Culture Collection).
Cell culture
The cells were cultured in DMEM (Thermo Fisher
Scientific, Inc.) supplemented with 10% FBS (Thermo Fisher
Scientific, Inc.) and 1.2% penicillin/streptomycin for 24-48 h in a
humidified 5% CO2 atmosphere at 37˚C. Cells at the
logarithmic growth phase were washed twice with PBS, trypsinized at
37˚C incubator for 5 min and counted using a hemocytometer. A total
of 1x104 cells were seeded into each well of a 96-well
plate. After 24 h, the culture media were removed, and 100 µl DMEM
containing Tf-NPs at different concentrations (10.0000, 2.0000,
0.4000, 0.0800, 0.0160 or 0.0032 µg/ml) were added into the cells
in quadruplicate. The same concentrations of PTX or PTX-Tf-NPs were
also assayed on the cultured cells in quadruplicate. Cells without
treatment served as a blank control. After 48 h, the culture media
containing Tf-NPs, PTX or PTX-Tf-NPs were removed, and the cells
were washed with PBS. A standard MTT assay was subsequently
performed to measure the cytotoxicity of the drugs. The purple
formazan was dissolved in DMSO and the absorbance was measured at
570 nm. The experiment was repeated three times. The cell viability
curves with different concentrations of Tf-NPs, PTX or PTX-Tf-NPs
were plotted.
Flow cytometry
Flow cytometry was performed to analyze the cell
cycle distribution of C6 and HT22 cells treated with PTX or
PTX-Tf-NPs. C6 or HT22 cells at the logarithmic growth phase were
trypsinized at 37˚C incubator for 5 min and counted with a
hemocytometer. A total of 2x105 cells were seeded into
each well of a 6-well plate. When the cells entered the logarithmic
growth phase after 24 h, they were treated with PTX, PTX-Tf-NPs
(dissolved in cell culture media) at the concentrations of 0.0032,
0.016, 0.080 or 0.400 µg/ml (for C6 cells) or 0.06, 0.25 or 1.00
µg/ml (for HT22 cells) in duplicate. Cell culture medium was used
as a solvent control. After 48 h, the cells were collected and
centrifuged at 705 x g at 4˚C for 5 min. The cells were washed
twice with pre-cooled PBS and fixed with pre-cooled 70% ethanol at
4˚C for 30 min. Following fixation, the cells were centrifuged at
705 x g for 5 min, and 70% ethanol was removed. RNA was digested
with 100 µl of RNase A (0.1 mg/ml) at 37˚C for 30 min. The cells
were washed twice with PBS and subsequently stained with 200 µl
propidium iodide (PI, Beyotime Institute of Biotechnology) (0.05
mg/ml) containing 0.03% Triton X-100 at 4˚C for 30 min. The cell
suspension was filtered using a nylon mesh with 40 µm pores before
being analyzed using a FACSAria flow cytometer (Becton, Dickinson
and Company) and analyzed using FlowJo 7.6 (FlowJo LLC). The
percentages of cells at G0/G1, S and
G2/M phases were calculated. The experiment was repeated
three times.
Examination of the uptake and
subcellular localization of nanoparticles
A total of 4x105 C6 cells were seeded in
a 35 mm dish with an integrated cover glass bottom of a 0.1 mm
thickness and 14 mm diameter (MatTek Corporation). After 24 h, the
cells were washed twice with PBS and incubated at 37˚C with DMEM
containing 10 µg/ml FITC-conjugated PTX-Tf-NPs for 0, 0.5, 1, 2 and
4 h. The cells were washed with PBS before being examined under
FLUOVIEW FV1000 confocal laser scanning microscope (magnification,
x40; Olympus Corporation).
C6 cells were stained with 75 nM LysoTracker Red™
DND-99 at 37˚C for 1 h (Invitrogen; Thermo Fisher Scientific, Inc.)
following incubation at 37˚C with 10 µg/ml FITC-conjugated
PTX-Tf-NPs or Tf-NPs for 4 h. The cells were washed three times
with Dulbecco's PBS before being examined under FLUOVIEW FV1000
confocal laser scanning microscope (magnification, x40; FV10 ASW,
Olympus Corporation). The filter sets for FITC and LysoTracker Red
DND-99 were 488 nm (excitation)/510 nm (emission) and 488 nm
(excitation)/560 nm (emission), respectively.
Statistical analysis
The experiments were repeated three times. The data
are presented as the mean ± SD. SPSS 13.0 software (SPSS, Inc.) was
used for data analysis. One-way ANOVA followed by Turkey's post hoc
test was used for the comparison of multiple groups in Figs. 1 and 2. P<0.05 was considered to indicate a
statistically significant difference.
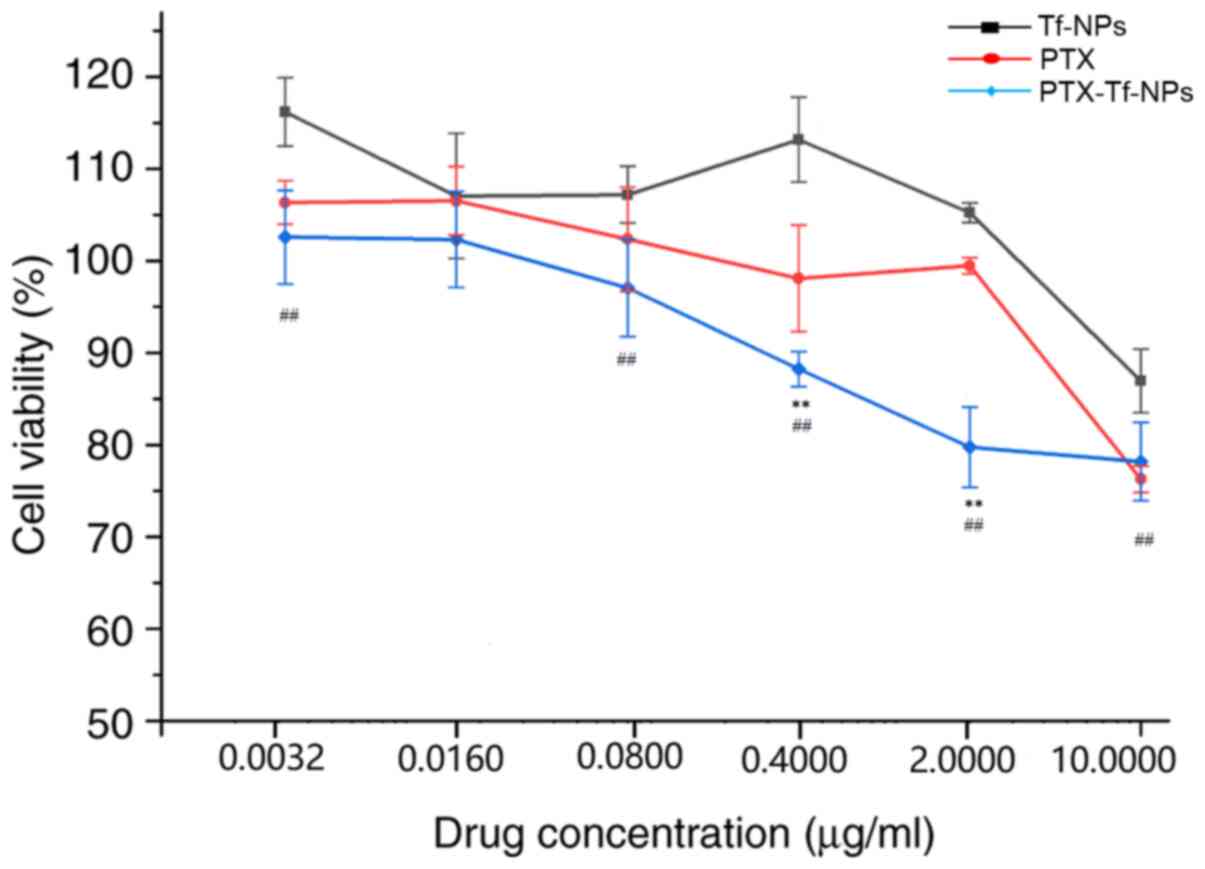
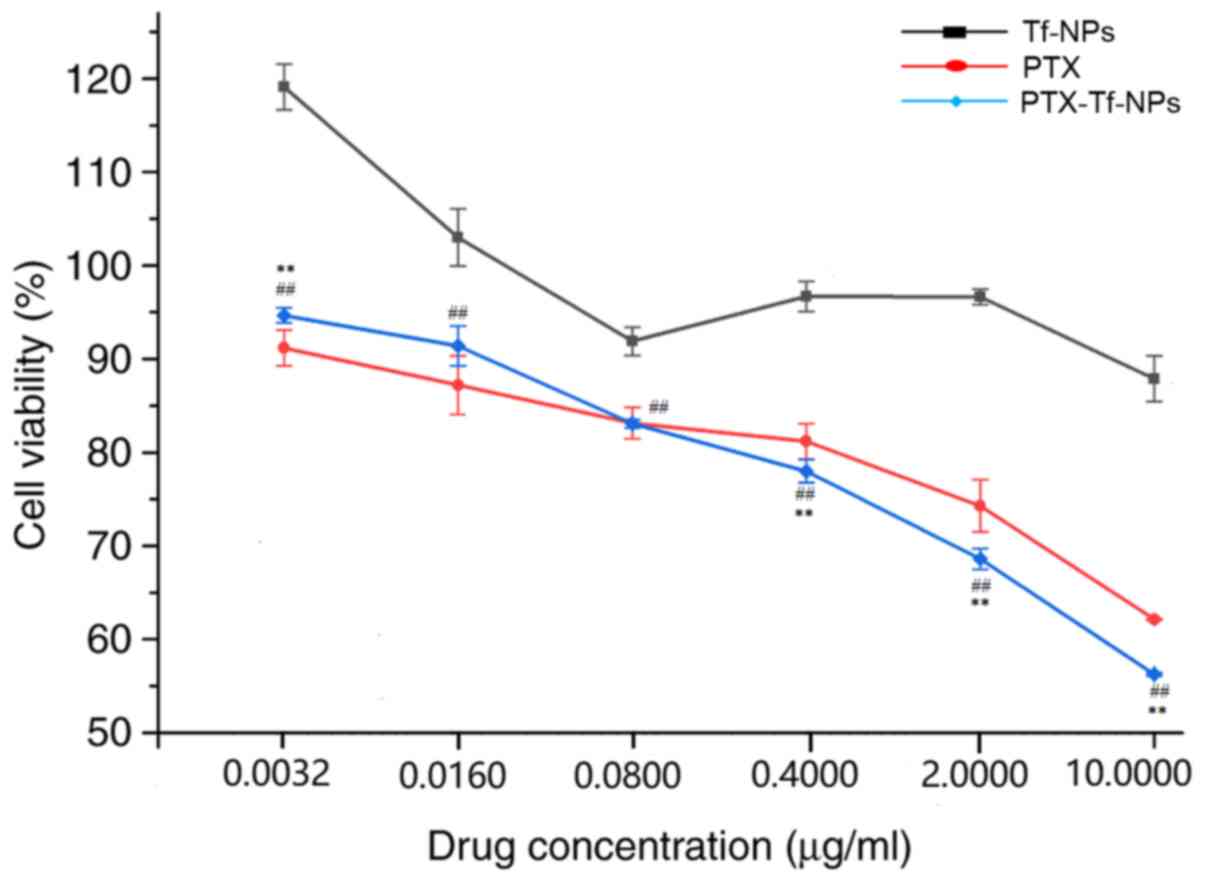 | Figure 1PTX or PTX-Tf-NP treatment reduces
the viability of rat glioblastoma C6 cells in a dose-dependent
manner, but PTX-Tf-NPs exhibit a stronger inhibitory effect at
higher concentrations. C6 cells were treated with Tf-NPs, PTX or
PTX-Tf-NPs at the concentrations of 0.0032, 0.016, 0.08, 0.4, 2 or
10 µg/ml for 48 h, followed by detection of the cell viability
using an MTT assay. The data are presented as the mean ± SD, and
ANOVA followed by Tukey's post hoc test was used for statistical
analysis. **P<0.01 vs. PTX; ##P<0.01
vs. Tf-NPs. PTX, paclitaxel; Tf-NPs, transferrin-nanoparticles. |
Results
PTX or PTX-Tf-NPs reduce the viability
of rat glioblastoma C6 cells in a dose-dependent manner, but
PTX-Tf-NPs exhibit a stronger inhibitory effect at higher
concentrations compared with PTX
Rat glioblastoma C6 cells were treated with Tf-NPs,
PTX or PTX-Tf-NPs for 48 h, and cell viability was detected using
the MTT assay. The percentages of cell viability are presented in
Fig. 1 and Table I. The results indicated that
treatment with Tf-NPs at concentrations of 0.0032 or 0.016 µg/ml
did not inhibit C6 cell viability, whereas Tf-NP treatment at
concentrations of 0.08, 0.4, 2 and 10 µg/ml resulted in a cell
viability of 92, 97, 97 and 88% in C6 cells, respectively, compared
with control cells, indicating that Tf-NPs alone cause a low
cytotoxicity in C6 cells. Both PTX and PTX-Tf-NPs exhibited a
dose-dependent effect on cell viability in C6 cells. Following PTX
treatment at concentrations of 0.0032, 0.016 and 0.08 µg/ml, C6
cell viability was 91, 87 and 83%, respectively, while following
PTX-Tf-NP treatment, cell viability was 95, 91 and 83%,
respectively. Statistical analysis revealed that at a concentration
of ≤0.08 µg/ml, no significant difference in cell viability by
treatment with either PTX or PTX-Tf-NPs was observed, indicating
that PTX and PTX-Tf-NPs exhibit similar cell viability inhibitory
effects at these concentrations. Nevertheless, at concentrations of
0.4, 2 and 10 µg/ml, C6 cells treated with PTX exhibited an average
viability of 81, 74 and 62%, respectively, but C6 cells treated
with PTX-Tf-NPs exhibited significantly lower viability compared
with cells treated with PTX (78, 69 and 56%, respectively). This
suggested that PTX-Tf-NPs were more potent compared with PTX in
reducing the viability of C6 glioblastoma cells at higher
concentrations.
 | Table ICell viability of C6 cells following
treatment with Tf-NPs, PTX or PTX-Tf-NPs. |
Table I
Cell viability of C6 cells following
treatment with Tf-NPs, PTX or PTX-Tf-NPs.
| | Concentration
(µg/ml) |
|---|
| Drug | 0.0032 | 0.0160 | 0.0800 | 0.4000 | 2.0000 | 10.0000 |
|---|
| Tf-NPs | 119.11±2.45 | 103.01±3.07 | 91.89±1.52 | 96.69±1.63 | 96.64±0.85 | 87.89±2.44 |
| PTX | 91.19±1.94 | 87.21±3.13 | 83.12±1.67 | 81.19±1.91 | 74.28±2.81 | 62.14±0.07 |
| PTX-Tf-NPs | 94.66±0.81 | 91.4±2.12 | 83.09±0.44 |
78.0±1.23a |
68.6±1.12a |
56.25±0.20a |
PTX and PTX-Tf-NPs exhibit a lower
inhibitory effect on the viability of mouse hippocampal neuronal
HT22 cells compared with that on rat glioblastoma C6 cells
Immortalized mouse hippocampal neuronal HT22 cells
were treated with Tf-NPs, PTX or PTX-Tf-NPs for 48 h, and cell
viability was measured using an MTT assay. The cell viability
measurements are presented in Fig.
2 and Table II. The results
indicated that at a high concentration of 10 µg/ml, Tf-NP treatment
reduced HT22 cell viability to 87%. At a concentration of ≤2 µg/ml,
Tf-NPs did not affect HT22 cell viability. These data indicated
that treatment with Tf-NPs alone results in low cytotoxicity in
HT22 cells. Interestingly, PTX alone did not inhibit cell viability
at a concentration of ≤2 µg/ml, but reduced HT22 cell viability to
76% relatively to that of control cells at a concentration of 10
µg/ml (Fig. 2), which was different
from that in C6 cells (Fig. 1).
PTX-Tf-NPs did not inhibit HT22 cell viability at a concentration
of ≤0.08 µg/ml, but reduced HT22 cell viability to 88, 80 and 78%
relatively to that of control cells at the concentrations of 0.4,
2.0 and 10 µg/ml, respectively (Fig.
2). These data indicated that PTX-Tf-NPs exhibited a lower
inhibitory effect on the viability of HT22 cells compared with that
on C6 cells.
| Table IICell viability of HT22 cells
following treatment with Tf-NPs, PTX or PTX-Tf-NPs. |
Table II
Cell viability of HT22 cells
following treatment with Tf-NPs, PTX or PTX-Tf-NPs.
| | Concentration
(µg/ml) |
|---|
| Drug | 0.0032 | 0.0160 | 0.0800 | 0.4000 | 2.0000 | 10.0000 |
|---|
| Tf-NPs | 116.2±3.70 | 107.05±6.80 | 107.2±3.09 | 113.19±4.63 | 105.24±1.08 | 86.96±3.46 |
| PTX | 106.35±2.35 | 106.53±3.70 | 102.36±5.66 | 98.1±5.77 | 99.47±0.88 | 76.27±1.43 |
| Tf-NPs-PTX | 102.57±5.10 | 102.32±5.23 | 97.07±5.28 |
88.25±1.90a |
79.74±4.35a |
78.19±4.26a |
PTX and PTX-Tf-NPs induce
G2/M arrest differentially in C6 cells
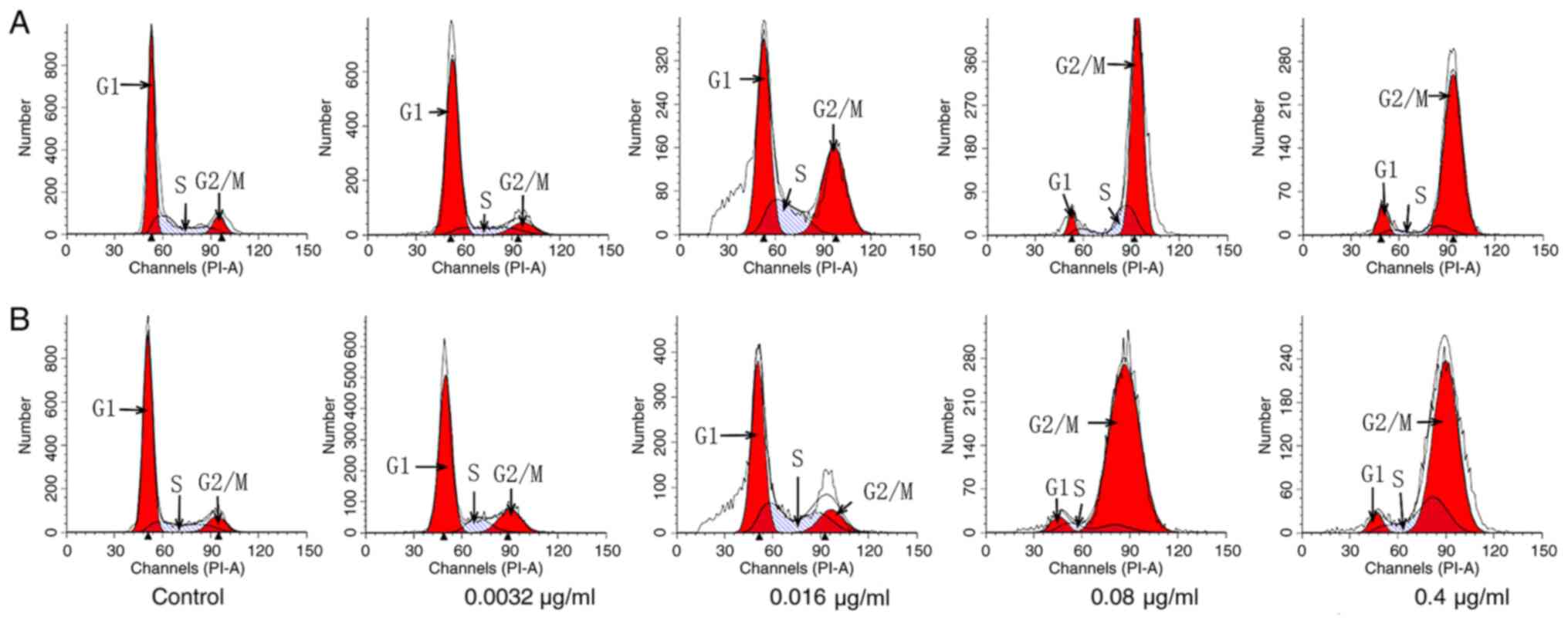
Following PTX treatment at the concentrations of
0.0032, 0.016, 0.08 or 0.4 µg/ml, C6 cells exhibited 9, 28, 73 and
86% G2/M distribution, respectively (Fig. 3A and Table III), whereas untreated cells
presented only 9%. These data suggested that PTX treatment at the
concentrations of 0.016, 0.08 and 0.4 µg/ml resulted in
G2/M arrest in C6 cells, and the percentage of
G2/M arrest was associated with the concentration of
PTX. On the other hand, following PTX-Tf-NP treatment at the
concentrations of 0.0032, 0.016, 0.08 and 0.4 µg/ml, C6 cells
exhibited 15, 17, 89 and 71% G2/M distribution,
respectively (Fig. 3B and Table III). These data indicated that
PTX-Tf-NPs induced G2/M cell cycle arrest in C6 cells
even at the lowest concentration of 0.0032 µg/ml. However, the
G2/M arrest effect of PTX-Tf-NPs peaked at a
concentration of 0.08 µg/ml, and this effect was reduced at a
concentration of 0.4 µg/ml. In addition, the aforementioned results
demonstrated that at the concentrations of 0.0032 and 0.08 µg/ml,
PTX-Tf-NPs resulted in a higher percentage of G2/M
arrest compared with PTX, but this difference was reversed at the
concentration of 0.4 µg/ml.
| Table IIICell cycle analysis of C6 cells
following treatment with PTX or PTX-Tf-NPs. |
Table III
Cell cycle analysis of C6 cells
following treatment with PTX or PTX-Tf-NPs.
| Treatment | Concentration
(µg/ml) |
G0/G1 phase (%) | S phase (%) | G2/M
phase (%) |
|---|
| Control | - | 62.26±1.68 | 28.28±2.21 | 9.47±0.54 |
| PTX | | | | |
| | 0.0032 | 68.07±8.19 | 22.49±8.61 | 9.45±0.41 |
| | 0.0160 | 49.08±5.38 | 22.59±2.35 | 28.34±7.73 |
| | 0.0800 | 5.57±0.44 | 21.01±1.53 | 73.42±1.97 |
| | 0.4000 | 7.14±1.30 | 6.57±2.70 | 86.3±4.02 |
| PTX-Tf-NPs | 0.0032 |
70.68±1.46a | 14.74±2.39 |
14.58±3.84a |
| | 0.0160 |
57.73±3.70a | 25.7±6.42 |
16.58±2.70a |
| | 0.0800 |
5.62±1.22a | 5.77±1.88 |
88.61±0.66a |
| | 0.4000 |
5.50±1.13a | 23.68±2.23 |
70.83±3.36a |
PTX and PTX-Tf-NPs induce
G2/M arrest differentially in HT22 cells
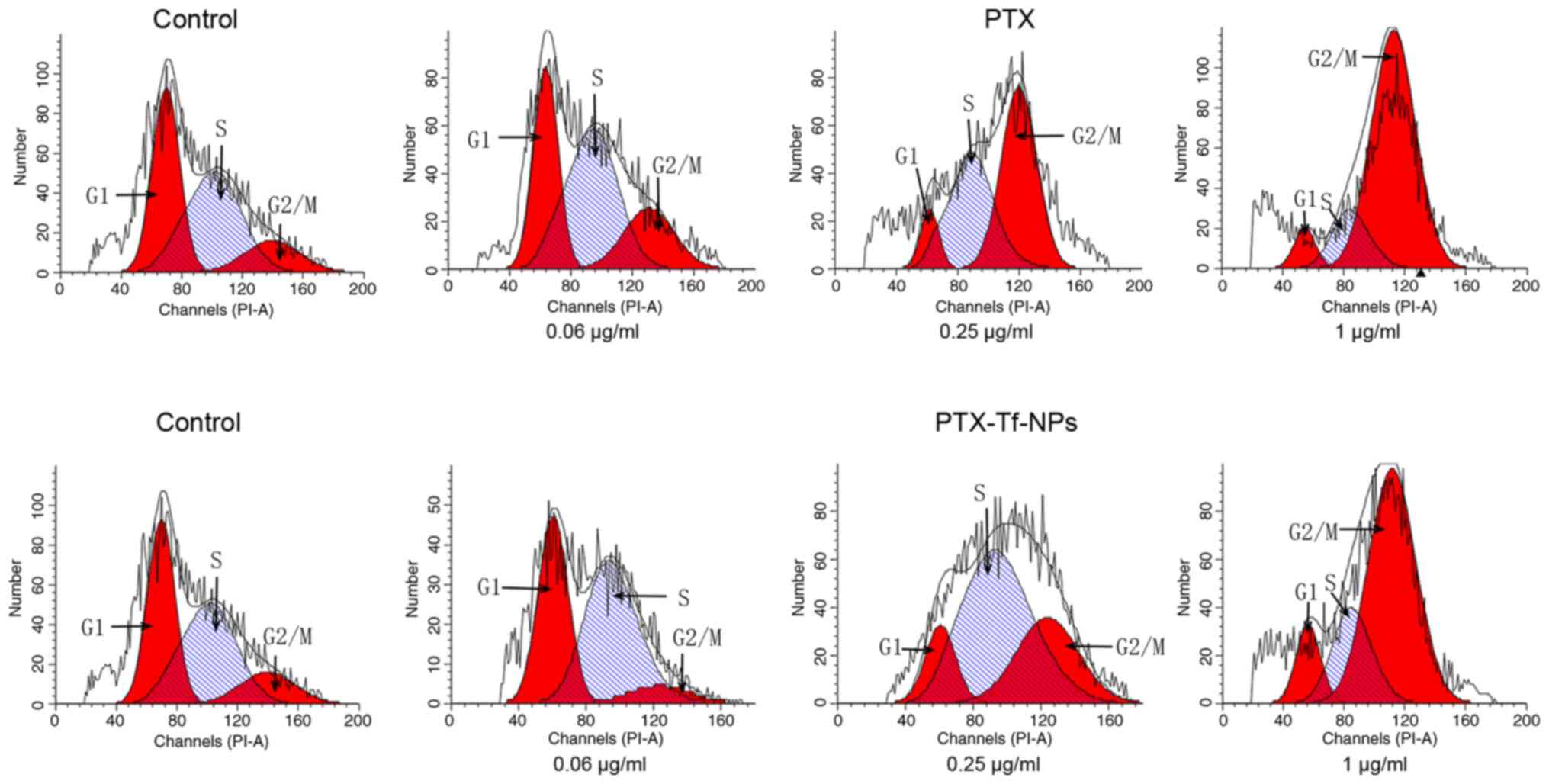
Following PTX treatment at the concentrations of
0.06, 0.25 or 1 µg/ml, HT22 cells exhibited 20, 52 and 73%
G2/M distribution, respectively, whereas the untreated
cells exhibited only 14% (Fig. 4,
row 1; Table IV), indicating that
G2/M cell cycle arrest in HT22 cells was associated with
the concentration of PTX. On the contrary, HT22 cells treated with
0.06, 0.25 or 1 µg/ml PTX-Tf-NPs exhibited 4, 29 or 66%
G2/M phase distribution, respectively (Fig. 4, row 2; Table IV). These data suggested that
PTX-Tf-NPs induced G2/M cell cycle arrest in HT22 cells
at the concentrations of 0.25 and 1 µg/ml, but not 0.06 µg/ml.
Moreover, at these concentrations, PTX-Tf-NPs resulted in a lower
percentage of G2/M arrest compared with PTX.
| Table IVCell cycle analysis of HT22
cells. |
Table IV
Cell cycle analysis of HT22
cells.
| Treatment | Concentration
(µg/ml) |
G0/G1 phase (%) | S phase (%) | G2/M
phase (%) |
|---|
| Control | - | 40.83±2.76 | 45.03±3.71 | 14.14±0.56 |
| PTX | 0.06 | 29.52±3.06 | 50.85±3.54 | 19.64±0.48 |
| | 0.25 | 7.89±0.78 | 40.39±0.15 | 51.72±0.92 |
| | 1.00 | 8.86±2.39 | 18.54±2.53 | 72.61±4.92 |
| PTX-Tf-NPs | 0.06 |
39.38±0.08a | 56.17±3.46 |
4.46±3.38a |
| | 0.25 |
12.82±2.19a | 58.5±5.41 |
28.68±3.00a |
| | 1.00 | 11.41±1.17a | 22.23±1.60 |
66.36±5.52a |
PTX-Tf-NP endocytosis in C6 cells
increases in a time-dependent manner
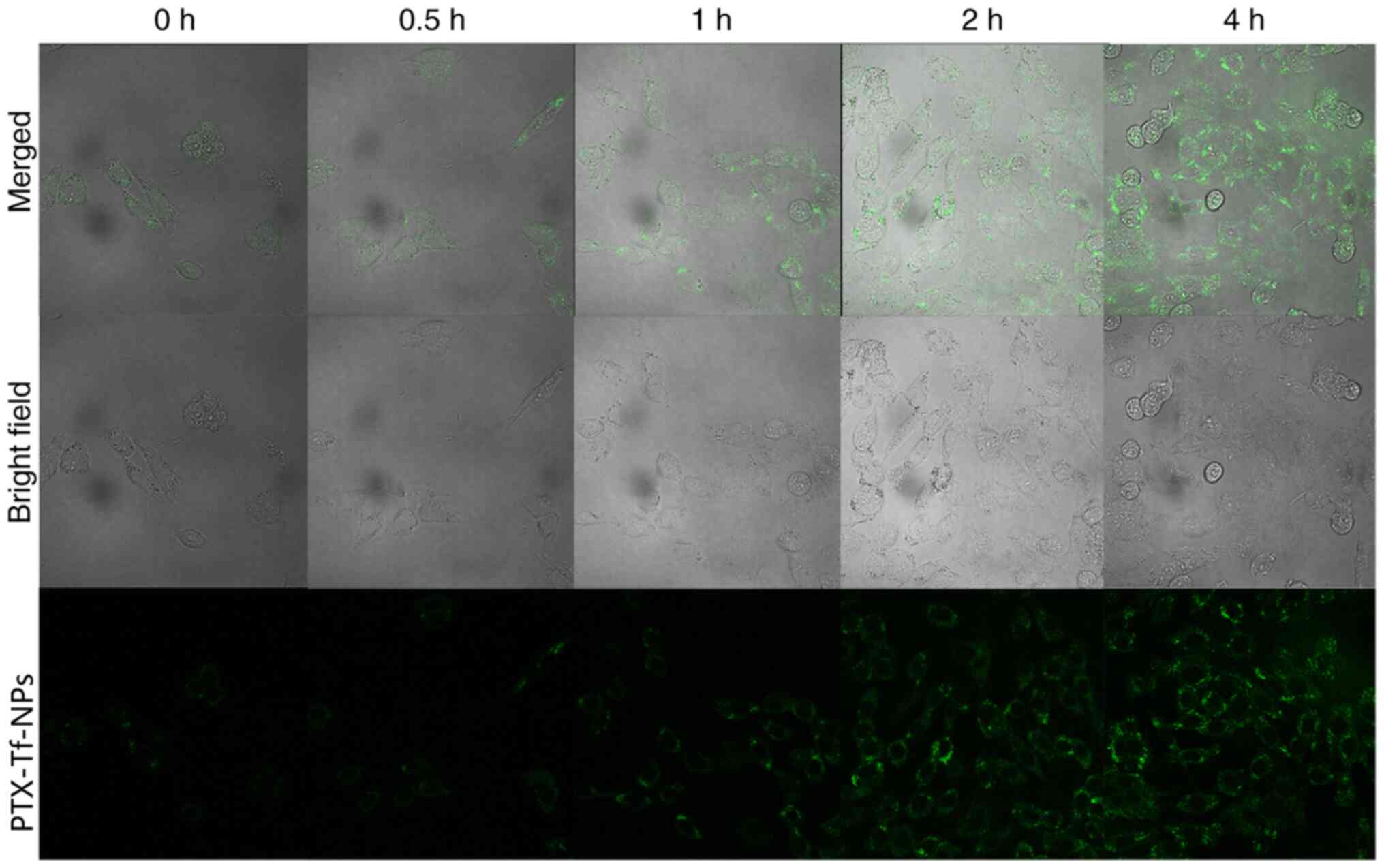
To visualize the distribution of PTX-Tf-NPs in C6
cells, a high concentration of the particles (10 µg/ml) was used,
as at this concentration an increased amount of particles is more
likely to enter the cells. The cells were incubated with
FITC-labeled PTX-Tf-NPs and the green fluorescence signal was
detected under confocal laser microscope. Following treatment for
0.5 h, a small amount of fluorescence signal was detected in the
cytosol of C6 cells, indicating that PTX-Tf-NPs had entered into C6
cells (Fig. 5). With increased
incubation time, an increased number of PTX-Tf-NPs entered C6
cells. Following treatment for 4 h, a strong fluorescence signal
was visible in nearly all C6 cells (Fig. 5), indicating that the majority of
PTX-Tf-NPs were successfully endocytosed by C6 cells.
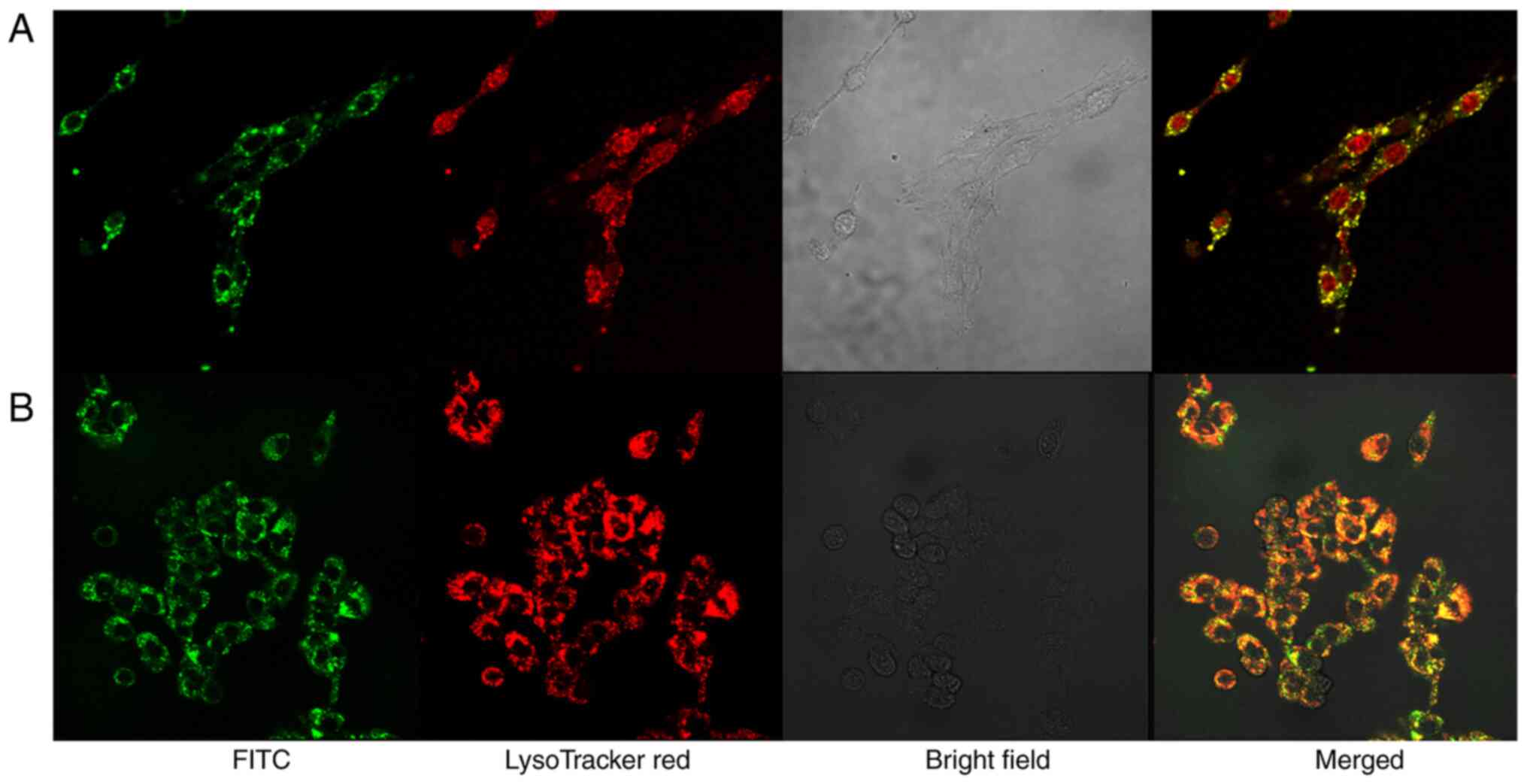
Intracellular PTX-Tf-NPs or Tf-NPs are
co-localized with lysosomes
C6 cells were treated with FITC-labeled PTX-Tf-NPs
or Tf-NPs and additionally incubated with LysoTracker Red DND-99.
LysoTracker Red DND-99 can specifically accumulate in acidic
organelles, such as lysosomes (21). The fluorescence signals were
examined using a confocal laser microscope. As demonstrated in
Fig. 6, FITC-labeled PTX-Tf-NPs or
Tf-NPs co-localized with LysoTracker Red DND-99. These data
suggested that both PTX-Tf-NPs and TF-NPs are localized in
lysosomes upon endocytosis.
Discussion
The present study demonstrated that PTX-Tf-NP
treatment resulted in a higher cytotoxicity in glioblastoma C6
cells at the higher concentrations tested (0.4, 2 and 10 µg/ml)
compared with PTX alone. Additionally, it was observed that
PTX-Tf-NPs induced higher G2/M arrest at the lower
concentrations tested (0.0032 and 0.0800 µg/ml) in C6 cells
compared with PTX alone. Moreover, PTX-Tf-NP treatment resulted in
a higher cytotoxicity in C6 cells compared with that on mouse
hippocampal neuronal HT22 cells. Furthermore, Tf-NPs alone
exhibited a low cytotoxicity in both glioblastoma C6 and
hippocampal neuronal HT22 cells, only slightly inhibiting the
viability of these cells at a high concentration of 10 µg/ml, which
indicated that Tf-NPs can be safe for therapeutic applications.
Therefore, the results of the present study suggested that Tf-NPs
exhibit a great potential to be an excellent nanocarrier for
glioblastoma drug targeting. Based on the Tf-NP structure, there
are two key features that may contribute to the successful
targeting of glioblastoma cells by PTX-Tf-NPs in vivo,
including the targeting moiety Tf (22) and the backbone of Tf-NPs, which is
composed of an amphiphilic γ-PGA-MAL-PLA-DPPE copolymer (20).
Under normal physiological conditions, Tf functions
as an iron carrier in plasma and transfers iron to various types of
tissues, such as the brain (23).
Currently, how iron molecules cross the BBB remains unclear. The
models of Tf receptor (TfR)-mediated endocytosis and transcytosis
have been proposed to demonstrate the movement of iron from
endothelial cells in the BBB to the brain parenchyma. Generally,
iron-loaded holo-Tf has been indicated to bind to TfR, which is
localized on the luminal cell surface of endothelial cells in the
brain (24). The complex of holo-Tf
with TfR is internalized into the cell via the clathrin-coated pit
and wrapped into the clathrin-coated vesicle (24). Subsequently, the vesicle becomes
uncoated and fuses with the early endosome. The iron transport via
TfR-mediated endocytosis or transcytosis has been revealed to
depend on whether holo-Tf can be transferred to brain parenchyma,
which is dependent upon the ability of Tf to cross the abluminal
membrane together with iron (24).
In TfR-mediated endocytosis, the acidified environment inside the
endosome has been reported to induce the separation of iron from
holo-Tf, and iron has been indicated to be subsequently pumped out
of the endosome by divalent metal transporter 1 into the cytosol,
followed by the transportation of iron to the abluminal membrane of
endothelial cells by ferroportin (24). The majority of TfR andiron-free
apo-Tf complexes recycle back to the respective cell surface and
plasma, whereas only a small amount of the TfR-Tf complex undergoes
lysosomal degradation (25). On the
other hand, a certain amount of iron in the form of holo-Tf has
been indicated to be transported as a complex across the abluminal
membrane of the brain's endothelial cells to reach the brain
parenchyma in TfR-mediated transcytosis (24).
As a targeting moiety, Tf may serve vital roles in
the transportation of PTX-Tf-NPs across the BBB. TfR1 is highly
expressed in endothelial cells from the BBB (26), which makes it a feasible target for
the delivery of therapeutic agents into the brain parenchyma via
TfR-mediated transcytosis. Several groups have reported that
Tf-conjugated nanoparticles enter the brain in greater amounts
in vivo compared with those without a Tf targeting moiety
(25,27-29).
Sonali et al (29)
demonstrated that Tf-conjugated gold nanoparticles crossed the BBB
in a manner dependent on the avidity of the nanoparticles to TfR,
which was determined by the size of the nanoparticles and the
amount of Tf conjugated to the surface of the nanoparticles. For
example, nanoparticles with a diameter of 45 nm and 30 Tf/particle
have been indicated to reach the mouse brain parenchyma in larger
amounts compared with same-sized nanoparticles with either 20 or
100 Tf/particle (29).
Nanoparticles with a diameter of 80 nm and 20 Tf/particle have been
reported to enter the mouse brain at a higher number compared with
same-sized nanoparticles with 200 Tf/particle (27). Tf-conjugated nanoparticles with a
moderate avidity to Tf have been demonstrated to be more likely
transcytosed to the brain parenchyma in comparison to nanoparticles
with a high avidity to Tf (27). To
further increase the amount of transcytosed Tf-conjugated
nanoparticles, the same research group incorporated an
acid-cleavable linkage between Tf and the nanoparticle core
(28). It has been revealed that
this acid-cleavable linkage increased the mobility of high-avidity
Tf-containing nanoparticles (80 nm in diameter; 200 Tf/particle),
but not low-avidity Tf-containing nanoparticles (80 nm in diameter;
20 Tf/particle). High-avidity Tf-containing nanoparticles with the
acid-cleavable linkage have been demonstrated to reach the mouse
brain parenchyma more efficiently compared with low-avidity
Tf-containing nanoparticles without the acid-cleavable linkage
(28). This may be attributed to
the fact that the Tf-containing nanoparticles with high avidity to
TfR may be endocytosed by endothelial cells in the BBB to a greater
extent compared with their low-avidity counterparts, whereas the
acid-cleavable linkage may induce an endosomal separation of the
nanoparticle core from both Tf and TfR and facilitate the ability
of the nanoparticle core to enter the transcytotic pathway.
However, despite the ability of the acid-cleavable linkage to
increase the amount of Tf-containing nanoparticles entering the
mouse brain, the transcytosed nanoparticles without the Tf
targeting moiety would have lost their targeting ability to
glioblastoma cells, which exhibit a high expression of TfR
(30,31).
There are two major advantages for
glioblastoma-targeted delivery of drug-loaded Tf-modified
nanocarriers via TfR-mediated endocytosis. As aforementioned, one
advantage is the achievement of an efficient targeted delivery of
anticancer agents via binding of a Tf-modified nanocarrier to TfR,
which has been indicated to be upregulated owing to the increased
uptake of iron by glioblastoma cells (32,33).
Another advantage is bypassing the multi-drug resistance developed
by glioblastoma cells due to their increased expression of efflux
transporters, such as MRP1(32).
Inhibition of MRP1 or downregulation of MRP1 has been demonstrated
to improve the sensitivity of glioblastoma cells to chemotherapy
drugs in vitro (19,33,34).
By taking into account the advantages of a Tf-modified nanocarrier
that may facilitate the transport of anticancer agents into
glioblastoma cells, the present study using in vitro
methods, as well as previous studies using in vivo methods,
demonstrated that PTX-Tf-NPs in addition to other drug-loaded
Tf-containing nanoparticles (Tf-PO-DOX, G4-DOX-PEG-Tf-TAM,
Tf-NP-DOX, NPs-ZOL-Tf) (35-38)
inhibited the viability of glioblastoma cells in comparison to
anticancer drugs or a drug-loaded nanocarrier without Tf
modification. Liu et al (36) prepared doxorubicin (Dox)-loaded
Tf-conjugated polyethylene glycol-polylactic acid NPs (Tf-NP-Dox)
and observed a 2-fold increase in the intracellular drug
concentration in C6 cells compared with NP-Dox alone. Pang et
al (35) further revealed that
Tf-conjugated biodegradable polymersomes (Tf-PO) enhanced the
delivery of Dox into the brain and tumors in glioblastoma-bearing
rats in comparison to PO alone. The present study for the first
time, to the best of our knowledge, demonstrated that drug-loaded
Tf-NP treatment resulted in a higher cytotoxicity in glioblastoma
cells (C6 cells) compared with normal neural cells (HT22 cells),
which supported the use of Tf in specifically targeting
glioblastoma cells. Moreover, the current study indicated that
PTX-Tf-NPs were localized in the lysosomes upon endocytosis in
these glioblastoma cells.
In addition to the Tf targeting moiety, the backbone
of Tf-NPs that was used in the present study, which was composed of
amphiphilic γ-PGA-MAL-PLA-DPPE copolymer, has been indicated to
exhibit several ideal characteristics for the systemic delivery of
anticancer drugs into the brain. Firstly, γ-PGA-MAL-PLA-DPPE
copolymer has been revealed to be highly biodegradable, as
polypeptide γ-PGA was made from carboxy-linked glutamate residues
(39), and PLA (40) and phospholipid DPPE (41) have all been reported to exhibit
excellent biodegradability and biocompatibility. The results of the
present study also indicated that Tf-NPs alone were biocompatible,
as they exhibited very low cytotoxicity in C6 and HT22 cells.
Secondly, a controlled and sustained release of PTX from PTX-Tf-NPs
has been observed at both pH 7.4 and pH 5.0, which simulated the pH
values at physiological conditions and in the lysosomal
environment, respectively (20).
Lastly, the PTX-Tf-NPs used in the present study have previously
been indicated to exhibit desirable near-neutral zeta potentials
(20), which may prevent the
aggregation of these nanoparticles and their uptake by the
mononuclear phagocyte system, thereby achieving a prolonged
circulation time in the bloodstream (42).
There is a limitation to the present study. Although
it was revealed that the nanoparticles were efficiently transported
into cells derived from the BBB, no in vivo evidence was
included in the current study to support that these particles can
cross the BBB. Additional in vivo assays are required to
confirm this effect.
The results of the present study indicated that
PTX-Tf-NPs were more potent compared with PTX alone in inhibiting
cell viability and inducing G2/M cell cycle arrest in
rat glioblastoma C6 cells. PTX-Tf-NPs were successfully endocytosed
by C6 cells, and were localized in lysosomes. These data indicated
that Tf-NPs can facilitate the entrance of PTX into C6 cells via
transferrin-mediated endocytosis and as a result, improve the
efficacy of PTX. Therefore, the present study laid the foundation
for subsequent investigation of Tf-NPs as a potential nanocarrier
in animal models of glioblastoma.
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant no. 81472850), Strategic
Priority Research Program of the Chinese Academy of Sciences (grant
no. XDA09030301), Heilong Jiang Natural Science Foundation (grant
no. H2015077), Graduate Science & Technology Research Project
of Jiamusi University (grant no. Sjz2012-16) and Graduate Science
& Technology Innovation Project of Jiamusi University (grant
no. LZR2014_016).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
YW and JL conceived the study; LW designed the
experiments and wrote the manuscript; LW, CL, FQ and ML performed
the experiments; HX and NC conducted statistics analysis; YW and JL
reviewed the experimental data and revised the manuscript
critically. YW and JL approved the final manuscript. YW and JL are
accountable for all aspects of the work in this study. All authors
read and approved the final version of the manuscript. LW, YW and
JL confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ostrom QT, Gittleman H, Fulop J, Liu M,
Blanda R, Kromer C, Wolinsky Y, Kruchko C and Barnholtz-Sloan JS:
CBTRUS Statistical Report: Primary Brain and Central Nervous System
Tumors Diagnosed in the United States in 2008-2012. Neuro-oncol. 17
(Suppl 4):iv1–iv62. 2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Zhang X, Zhang W, Cao WD, Cheng G and
Zhang YQ: Glioblastoma multiforme: Molecular characterization and
current treatment strategy (Review). Exp Ther Med. 3:9–14.
2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Magrath JW and Kim Y: Salinomycin's
potential to eliminate glioblastoma stem cells and treat
glioblastoma multiforme (Review). Int J Oncol. 51:753–759.
2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Jovčevska I, Kočevar N and Komel R: Glioma
and glioblastoma - how much do we (not) know? Mol Clin Oncol.
1:935–941. 2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Stupp R, Taillibert S, Kanner AA, Kesari
S, Steinberg DM, Toms SA, Taylor LP, Lieberman F, Silvani A, Fink
KL, et al: Maintenance Therapy With Tumor-Treating Fields Plus
Temozolomide vs Temozolomide Alone for Glioblastoma: A Randomized
Clinical Trial. JAMA. 314:2535–2543. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Toms SA and Tapinos N: Recent Advances in
the Treatment of Gliomas - Comprehensive Brain Tumor Center. R I
Med J (2013). 100:43–46. 2017.PubMed/NCBI
|
|
8
|
Chakroun RW, Zhang P, Lin R, Schiapparelli
P, Quinones-Hinojosa A and Cui H: Nanotherapeutic systems for local
treatment of brain tumors. Wiley Interdiscip Rev Nanomed
Nanobiotechnol. 10(e1479)2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Bryukhovetskiy IS, Dyuizen IV, Shevchenko
VE, Bryukhovetskiy AS, Mischenko PV, Milkina EV and Khotimchenko
YS: Hematopoietic stem cells as a tool for the treatment of
glioblastoma multiforme. Mol Med Rep. 14:4511–4520. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Bryukhovetskiy I, Bryukhovetsky A,
Khotimchenko Y, Mischenko P, Tolok E and Khotimchenko R:
Combination of the multipotent mesenchymal stromal cell
transplantation with administration of temozolomide increases
survival of rats with experimental glioblastoma. Mol Med Rep.
12:2828–2834. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Preusser M, de Ribaupierre S, Wöhrer A,
Erridge SC, Hegi M, Weller M and Stupp R: Current concepts and
management of glioblastoma. Ann Neurol. 70:9–21. 2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Bryukhovetskiy I, Ponomarenko A, Lyakhova
I, Zaitsev S, Zayats Y, Korneyko M, Eliseikina M, Mischenko P,
Shevchenko V, Shanker Sharma H, et al: Personalized regulation of
glioblastoma cancer stem cells based on biomedical technologies:
From theory to experiment (Review). Int J Mol Med. 42:691–702.
2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hundsberger T, Reardon DA and Wen PY:
Angiogenesis inhibitors in tackling recurrent glioblastoma. Expert
Rev Anticancer Ther. 17:507–515. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kim SS, Harford JB, Pirollo KF and Chang
EH: Effective treatment of glioblastoma requires crossing the
blood-brain barrier and targeting tumors including cancer stem
cells: The promise of nanomedicine. Biochem Biophys Res Commun.
468:485–489. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Karim R, Palazzo C, Evrard B and Piel G:
Nanocarriers for the treatment of glioblastoma multiforme: Current
state-of-the-art. J Controll Release. 227:23–37. 2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Glaser T, Han I, Wu L and Zeng X: Targeted
Nanotechnology in Glioblastoma Multiforme. Front Pharmacol.
8(166)2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Fang K, Liu P, Dong S, Guo Y, Cui X, Zhu
X, Li X, Jiang L, Liu T and Wu Y: Magnetofection based on
superparamagnetic iron oxide nanoparticle-mediated low lncRNA
HOTAIR expression decreases the proliferation and invasion of
glioma stem cells. Int J Oncol. 49:509–518. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Lee JK, Nam DH and Lee J: Repurposing
antipsychotics as glioblastoma therapeutics: Potentials and
challenges. Oncol Lett. 11:1281–1286. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Tivnan A, Zakaria Z, O'Leary C, Kögel D,
Pokorny JL, Sarkaria JN and Prehn JH: Inhibition of multidrug
resistance protein 1 (MRP1) improves chemotherapy drug response in
primary and recurrent glioblastoma multiforme. Front Neurosci.
9(218)2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhao C, Liu X, Liu J, Yang Z, Rong X, Li
M, Liang X and Wu Y: Transferrin conjugated poly (γ-glutamic
acid-maleimide-co-L-lactide)-1,2-dipalmitoylsn-glycero-3-phosphoethanolamine
copolymer nanoparticles for targeting drug delivery. Colloids Surf
B Biointerfaces. 123:787–796. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Pierzyńska-Mach A, Janowski PA and
Dobrucki JW: Evaluation of acridine orange, LysoTracker Red, and
quinacrine as fluorescent probes for long-term tracking of acidic
vesicles. Cytometry A. 85:729–737. 2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Choudhury H, Pandey M, Chin PX, Phang YL,
Cheah JY, Ooi SC, Mak KK, Pichika MR, Kesharwani P, Hussain Z, et
al: Transferrin receptors-targeting nanocarriers for efficient
targeted delivery and transcytosis of drugs into the brain tumors:
A review of recent advancements and emerging trends. Drug Deliv
Transl Res. 8:1545–1563. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Gkouvatsos K, Papanikolaou G and
Pantopoulos K: Regulation of iron transport and the role of
transferrin. Biochim Biophys Acta. 1820:188–202. 2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Skjørringe T, Burkhart A, Johnsen KB and
Moos T: Divalent metal transporter 1 (DMT1) in the brain:
Implications for a role in iron transport at the blood-brain
barrier, and neuronal and glial pathology. Front Mol Neurosci.
8(19)2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Yan F, Wang Y, He S, Ku S, Gu W and Ye L:
Transferrin-conjugated, fluorescein-loaded magnetic nanoparticles
for targeted delivery across the blood-brain barrier. J Mater Sci
Mater Med. 24:2371–2379. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Uchida Y, Ohtsuki S, Katsukura Y, Ikeda C,
Suzuki T, Kamiie J and Terasaki T: Quantitative targeted absolute
proteomics of human blood-brain barrier transporters and receptors.
J Neurochem. 117:333–345. 2011.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wiley DT, Webster P, Gale A and Davis ME:
Transcytosis and brain uptake of transferrin-containing
nanoparticles by tuning avidity to transferrin receptor. Proc Natl
Acad Sci USA. 110:8662–8667. 2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Clark AJ and Davis ME: Increased brain
uptake of targeted nanoparticles by adding an acid-cleavable
linkage between transferrin and the nanoparticle core. Proc Natl
Acad Sci USA. 112:12486–12491. 2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Sonali Singh RP, Singh N, Sharma G,
Vijayakumar MR, Koch B, Singh S, Singh U, Dash D, Pandey BL, et al:
Transferrin liposomes of docetaxel for brain-targeted cancer
applications: Formulation and brain theranostics. Drug Deliv.
23:1261–1271. 2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Recht L, Torres CO, Smith TW, Raso V and
Griffin TW: Transferrin receptor in normal and neoplastic brain
tissue: Implications for brain-tumor immunotherapy. J Neurosurg.
72:941–945. 1990.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Voth B, Nagasawa DT, Pelargos PE, Chung
LK, Ung N, Gopen Q, Tenn S, Kamei DT and Yang I: Transferrin
receptors and glioblastoma multiforme: Current findings and
potential for treatment. J Clin Neurosci. 22:1071–1076.
2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Abe T, Hasegawa S, Taniguchi K, Yokomizo
A, Kuwano T, Ono M, Mori T, Hori S, Kohno K and Kuwano M: Possible
involvement of multidrug-resistance-associated protein (MRP) gene
expression in spontaneous drug resistance to vincristine, etoposide
and adriamycin in human glioma cells. Int J Cancer. 58:860–864.
1994.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Garrido W, Muñoz M, San Martín R and
Quezada C: FK506 confers chemosensitivity to anticancer drugs in
glioblastoma multiforme cells by decreasing the expression of the
multiple resistance-associated protein-1. Biochem Biophys Res
Commun. 411:62–68. 2011.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Peigñan L, Garrido W, Segura R, Melo R,
Rojas D, Cárcamo JG, San Martín R and Quezada C: Combined use of
anticancer drugs and an inhibitor of multiple drug
resistance-associated protein-1 increases sensitivity and decreases
survival of glioblastoma multiforme cells in vitro. Neurochem Res.
36:1397–1406. 2011.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Pang Z, Gao H, Yu Y, Guo L, Chen J, Pan S,
Ren J, Wen Z and Jiang X: Enhanced intracellular delivery and
chemotherapy for glioma rats by transferrin-conjugated
biodegradable polymersomes loaded with doxorubicin. Bioconjug Chem.
22:1171–1180. 2011.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Liu G, Mao J, Jiang Z, Sun T, Hu Y, Jiang
Z, Zhang C, Dong J, Huang Q and Lan Q: Transferrin-modified
Doxorubicin-loaded biodegradable nanoparticles exhibit enhanced
efficacy in treating brain glioma-bearing rats. Cancer Biother
Radiopharm. 28:691–696. 2013.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Porru M, Zappavigna S, Salzano G, Luce A,
Stoppacciaro A, Balestrieri ML, Artuso S, Lusa S, De Rosa G,
Leonetti C, et al: Medical treatment of orthotopic glioblastoma
with transferrin-conjugated nanoparticles encapsulating zoledronic
acid. Oncotarget. 5:10446–10459. 2014.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Li Y, He H, Jia X, Lu WL, Lou J and Wei Y:
A dual-targeting nanocarrier based on poly(amidoamine) dendrimers
conjugated with transferrin and tamoxifen for treating brain
gliomas. Biomaterials. 33:3899–3908. 2012.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Khalil IR, Burns AT, Radecka I, Kowalczuk
M, Khalaf T, Adamus G, Johnston B and Khechara MP:
Bacterial-Derived Polymer Poly-y-Glutamic Acid (y-PGA)-Based
Micro/Nanoparticles as a Delivery System for Antimicrobials and
Other Biomedical Applications. Int J Mol Sci.
18(313)2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Pavot V, Berthet M, Rességuier J, Legaz S,
Handké N, Gilbert SC, Paul S and Verrier B: Poly(lactic acid) and
poly(lactic-co-glycolic acid) particles as versatile carrier
platforms for vaccine delivery. Nanomedicine (Lond). 9:2703–2718.
2014.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Navarro G, Essex S, Sawant RR, Biswas S,
Nagesha D, Sridhar S, de ILarduya CT and Torchilin VP:
Phospholipid-modified polyethylenimine-based nanopreparations for
siRNA-mediated gene silencing: Implications for transfection and
the role of lipid components. Nanomedicine (Lond). 10:411–419.
2014.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Xiao K, Li Y, Luo J, Lee JS, Xiao W, Gonik
AM, Agarwal RG and Lam KS: The effect of surface charge on in vivo
biodistribution of PEG-oligocholic acid based micellar
nanoparticles. Biomaterials. 32:3435–3446. 2011.PubMed/NCBI View Article : Google Scholar
|