Introduction
Cadmium (Cd) is a toxic heavy metal that is commonly
found at industrial worksites or in the environment (1). The vascular system is a critical
target of Cd toxicity and the action of Cd on the vascular system
may play important roles in mediating the pathophysiological
effects of Cd in specific target organs (1). Vascular endothelial cells are exposed
to Cd circulating in bloodstream and, if Cd is present at
sufficiently high concentrations, the endothelial cells are
injured. Endothelial dysfunction and damage may be attributable to
toxicity in parenchymal cells of various target organs, such as
kidney and liver (2). The most
commonly used therapeutic strategy against Cd toxicity is chelation
therapy to promote metal excretion. The chelating agent EDTA is
most widely used clinically. However, Cd chelators themselves
present a number of safety and efficacy concerns (1). In fact, CaNa2EDTA can cause renal
toxicity (in the proximal tubule in particular). Because of lack of
specificity, such essential metals as zinc, iron, and manganese are
excreted and depleted following CaNa2EDTA therapy (3). Therefore, the development of safe and
efficient strategies against Cd toxicity is required.
As a thiol-binding metal, free Cd primarily targets
highly abundant cellular glutathione (GSH), a reactive oxygen
species (ROS) scavenger (4).
Depletion of the GSH pool leads to poor scavenging of Cd, which
results in the disturbance of cellular redox balance leading to
oxidative stress. Cd interferes with not only antioxidant defense
systems but also the mitochondrial electron transport chain.
Although the complete pathology evoked by Cd toxicity is unknown,
the ability of Cd to elicit an oxidative stress response seems
apparent. Aiba et al (5)
revealed a novel pathway for the GSH-mediated reduction of Cd
toxicity in mammalian cells, namely, elevated GSH levels can
downregulate Cd uptake through the downregulation of Cd transporter
Zrt-, Irt-like protein 8 (ZIP8). Therefore, elevation of cellular
GSH levels is an important method for modulating Cd toxicity.
Nuclear factor erythroid 2-related factor 2 (Nrf2)
is a key transcription factor that plays a central role in
regulating the expression of glutamate cysteine ligase (GCL). GCL
is an enzyme that catalyzes the first and rate-limiting step in de
novo GSH synthesis. Sulforaphane, a compound found in broccoli
sprouts, is a potent Nrf2 activator (6). Recent studies have shown that
sulforaphane protects cells and tissues against Cd toxicity
(7,8). In vitro and in vivo
studies have demonstrated that Nrf2 activation protects against Cd
toxicity by increasing GSH levels (9). Interestingly, in vascular endothelial
cells after Cd exposure, Nrf2 partially contributes to the
expression of metallothionein (MT), the most potent protective
measure against Cd-induced toxicity (2).
Epalrestat (5-[(1Z,2E)-2-methyl-3-phenyl
propenylidene]-4-oxo-2-thioxo-3-thiazolidine acetic acid; EPS; Ono
Pharmaceuticals), which received approval for use in Japan in 1992,
is currently being used for the treatment of diabetic neuropathy.
EPS is an inhibitor of aldose reductase, a rate-limiting enzyme in
the polyol pathway. Under hyperglycemic conditions, EPS reduces
intracellular sorbitol accumulation, which is implicated in the
pathogenesis of diabetic complications (10). EPS is easily absorbed by neural
tissue and inhibits aldose reductase with minimum adverse effects
(11). Recently, we found that EPS
increased GSH levels in rat Schwann cells by upregulating GCL via
Nrf2 activation (12). In addition,
EPS increased GSH levels in bovine aortic endothelial cells (BAECs)
(13). The purpose of the present
study was to determine whether: i) EPS protects against Cd-induced
cytotoxicity in BAECs; ii) EPS affects GSH levels in cells exposed
to Cd; and iii) EPS has an effect on the intracellular levels of Cd
and MT.
Materials and methods
Cell culture and treatment with EPS
and Cd
BAECs were purchased from Dainippon Sumitomo Pharma
Co., Ltd. BAECs were grown to 80-90% confluence in DMEM containing
10% fetal bovine serum (FBS), L-glutamine (4 mM), penicillin (100
U/ml), and streptomycin (100 µg/ml) at 37˚C in a humidified
atmosphere of 5% CO2 and 95% air. Then, the cells were
passaged by trypsinization.
Before treating the cells with EPS (Wako Pure
Chemical Industries, Ltd.), the culture medium was replaced with
DMEM containing 2% FBS. EPS (10, 50 and 100 µM) was subsequently
added to the medium. After the treatment with EPS for 16 h, cells
were exposed to Cd chloride (25 and 50 µM).
Cell viability
Viability of BAECs was assessed by measuring lactate
dehydrogenase (LDH) release. After treatment of BAECs in 12-well
plates with EPS and Cd, aliquots of the medium were taken to
measure the activity of LDH released from cells. The remaining
intracellular LDH was released by adding 0.1% Triton X-100 in
phosphate-buffered saline (PBS) at pH 7.4. LDH activity was
measured spectrophotometrically on the basis of the increase in
absorbance at 340 nm with 60 mM lithium lactate in 0.3 M
diethanolamine buffer (pH 9.0), after the reaction was initiated by
adding 3 mM (final concentration) NAD+. Released LDH
activity was expressed as percentage of total LDH activity
(activities of LDH in the medium and in the remaining cells).
Treatment with N-acetylcysteine and
buthionine sulfoximine
BAECs were exposed to Cd at 25 µM for 24 h in the
presence or absence of N-acetylcysteine (NAC) (Sigma-Aldrich; Merck
KGaA) or GSH (free) (Wako Pure Chemical Industries, Ltd.) at 1 mM.
BAECs were pretreated with 100 µM buthionine sulfoximine (BSO)
(Sigma-Aldrich; Merck KGaA) for 16 h. Subsequently, the untreated
or BSO-treated cells were exposed to Cd at 25 µM for 24 h. The
effects of NAC, GSH, and BSO on cell viability was estimated by
measuring LDH release as described above.
Knockdown of Nrf2 in BAECs with small
interfering RNA (siRNA)
Oligonucleotides directed against bovine Nrf2
(Sigma-Aldrich; Merck KGaA) and control siRNA (Ambion) were
transfected into BAECs using Lipofectamine RNAiMAX (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. Briefly, both Nrf2 siRNA and control siRNA were diluted
with Opti-MEM medium and then, diluted Lipofectamine RNAiMAX was
added. The transfection mixture was incubated at room temperature
for 20 min. When BAECs reached 30-50% confluence, the culture
medium was replaced with DMEM (without FBS) and the transfection
mixture was added to each well. The final concentration of siRNA
was 20 nM.
Intracellular Cd concentrations
Intracellular Cd concentrations were measured by
graphite furnace atomic absorption spectrometry (GF-AAS; AA-7000
Atomic Absorption Spectrophotometer), according to the method
described by Luczak et al (14). After treatment of BAECs in 6-cm
dishes with EPS and Cd, Cd-containing media were removed. Attached
cells were washed twice with warm DPBS and the cells were harvested
with a cell scraper in DPBS, collected by centrifugation, and
washed twice with ice-cold DPBS. The cells were then suspended in
400 µl of ice-cold deionized water and this was followed by the
addition of 400 µl of 10% nitric acid. After the samples were
heated at 50˚C for 60 min, Cd-containing extracts were collected by
centrifugation. The supernatants were diluted with water to give 2%
nitric acid prior to Cd measurements by GF-AAS.
For protein measurements, which were necessary for
the normalization of Cd concentrations, Cd-extracted cell pellets
were washed with ice-cold 5% nitric acid, centrifuged, and
solubilized in 100 µl of 0.5 M NaOH by incubation at 37˚C for 30
min. The dissolved pellets were used for protein measurements.
ZIP8 and MT mRNA levels
RT-qPCR analysis was carried out to measure mRNA
levels. Total RNA from the treated cells was extracted with RNAspin
Mini (GE Healthcare) according to the manufacturer's protocol.
mRNAs were reverse-transcribed into cDNA with a High-Capacity cDNA
Reverse Transcription Kit (Applied Biosystems; Thermo Fisher
Scientific, Inc.). RT-qPCR was performed with a 7500 Fast Real-Time
PCR System (Applied Biosystems; Thermo Fisher Scientific, Inc.).
Primers for bovine ZIP8 (Bt04283914_m1), bovine MT (Bt03279283_m1),
and GAPDH (Bt03210913_g1) were purchased from Applied
Biosystems;Thermo Fisher Scientific, Inc. Data were analyzed using
the 2-ΔΔCq method (15) and normalized to the internal
reference gene GAPDH. Relative mRNA levels were compared and
expressed as percentage of control levels.
MT protein levels
MT protein levels were analyzed by western blotting.
A total of 20 µg of protein per well was resolved using sodium
dodecyl sulfate polyacrylamide gel electrophoresis and
electro-transferred to a PVDF membrane. After blocking and washing,
the membrane was incubated with the following primary antibody:
anti-mouse MT polyclonal antibody (Dako) or anti-mouse β-actin
(Sigma-Aldrich; Merck KGaA). Following primary antibody incubation,
the membrane was incubated with horseradish-peroxidase-conjugated
secondary antibodies. Chemiluminescence was detected with an ECL
Plus western blot detection kit (GE Healthcare). Protein expression
in each sample was determined by normalizing target band intensity
to β-actin band intensity. Band intensities were quantified using
ImageJ software.
Measurement of GSH
Intracellular GSH levels were measured by
spectrophotometric methods, as previously described (16). Untreated or 50 µM EPS-pretreated
cells for 16 h in 12-well plates were measured after exposure to 50
µM Cd for 4 h. Each sample for GSH measurement was mixed with 0.6
mM 5,5'-dithiobis (2-nitrobenzoic acid) (DTNB), 0.2 mM reduced
nicotinamide adenine dinucleotide phosphate (NADPH), and 5 mM
ethylenediaminetetraacetic acid (EDTA) in 0.1 M sodium phosphate
buffer (pH 7.5). The reaction was initiated by adding glutathione
reductase.
Other procedures
Protein concentrations were determined using the
Bradford method with bovine serum albumin (BSA) as the
standard.
Statistical analysis
All experiments were performed independently at
least three times. Data were combined and expressed as means ± SD.
Statistical significance was determined using the Student's t-test,
one-way ANOVA or two-way analysis of variance (ANOVA) with Tukey's
post-hoc test. A P-value of <0.01 was considered to be
significant.
Results
Effect of EPS on Cd toxicity in
BAECs
The vascular system is a critical target of Cd
toxicity (1). Endothelial
dysfunction and damage may be attributed to toxicity in parenchymal
cells of such target organs as kidney (1). In our previous work, we demonstrated
that EPS increased GSH levels in BAECs by upregulating GCL via Nrf2
activation (13). In the present
study, we first examined the effect of EPS on Cd-induced
cytotoxicity, using BAECs as an in vitro model of the
vascular endothelium. Fig. 1A shows
the protective ability of EPS against Cd-induced cytotoxicity in
BAECs, which was estimated by measuring LDH release, a frequently
used endpoint for cytotoxicity studies. Cd-induced release of LDH
was almost completely suppressed by pretreatment with EPS at 50 and
100 µM. EPS at 10 µM failed to suppress the LDH release. Our
previous study demonstrated that 50 and 100 µM EPS, but not 10 µM
EPS, increased GSH levels in BAECs (13). As shown in Fig. 1B, the addition of NAC or GSH (free)
reduced Cd-induced LDH release from BAECs, consistent with
published results (17).
Intracellular GSH depletion by BSO aggravated Cd-induced toxicity
in BAECs (Fig. 1C). These results
indicate that GSH plays a protective role against Cd-induced
cytotoxicity.
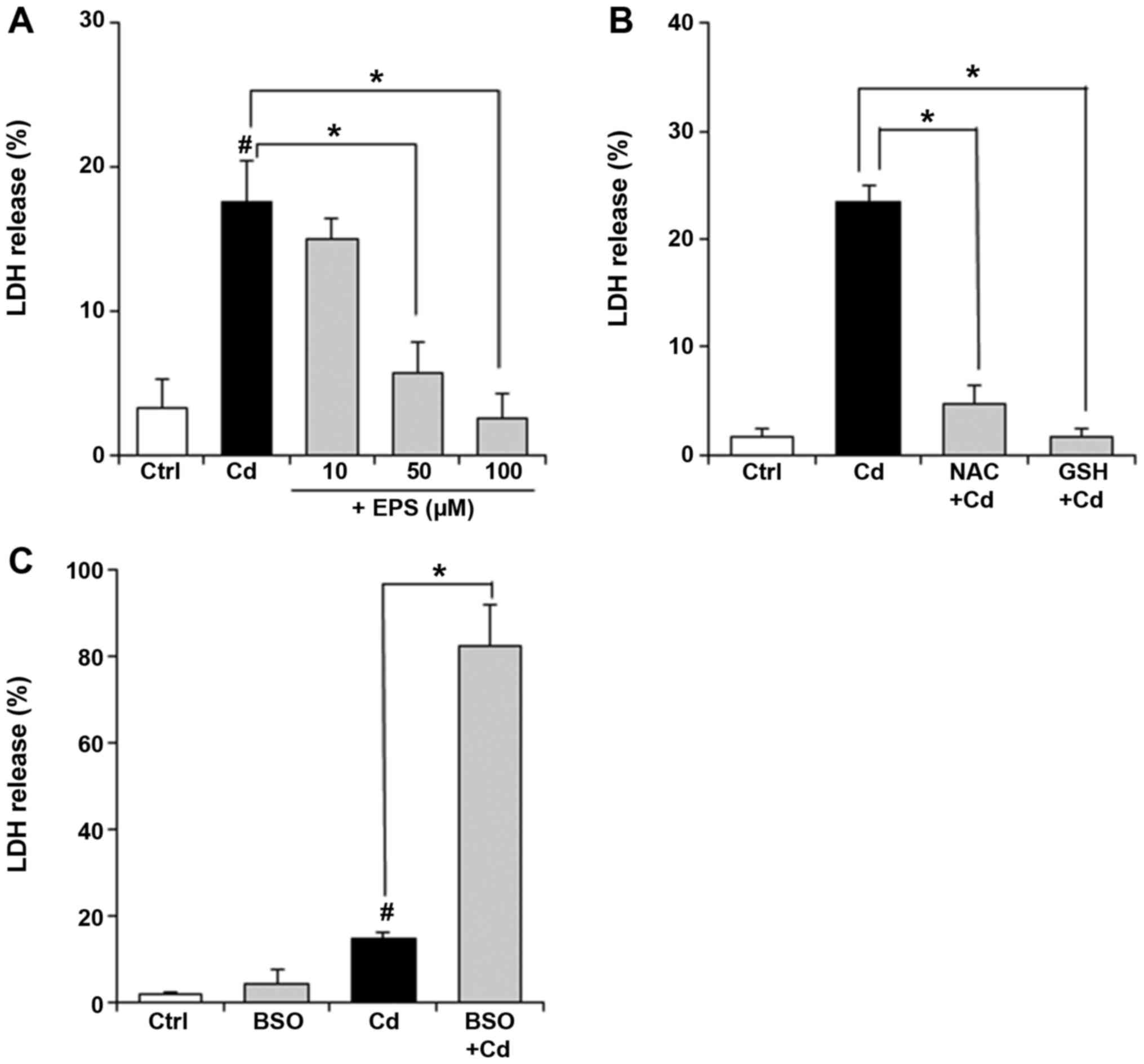 | Figure 1Effect of EPS on viability of BAECs
exposed to Cd. (A) BAECs were pretreated with EPS (10, 50 or 100
µM) for 16 h. Subsequently, the untreated or EPS-treated cells were
exposed to Cd at 25 µM for 24 h. Cell viability was estimated by
measuring LDH release. Values are means ± SD of three experiments.
(B) BAECs were exposed to Cd at 25 µM for 24 h in the presence or
absence of NAC or GSH at 1 mM. Cell viability was estimated by
measuring LDH release. Values are means ± SD of three experiments.
(C) BAECs were pretreated with 100 µM BSO for 16 h. Subsequently,
the untreated or BSO-treated cells were exposed to Cd at 25 µM for
24 h. Cell viability was estimated by measuring LDH release. Values
are means ± SD of three experiments. *P<0.01.
#P<0.01 vs. control. EPS, epalrestat; BAECs, bovine
aortic endothelial cells; Cd, csadmium; LDH, lactate dehydrogenase;
SD, standard deviation; NAC, N-acetylcysteine; GSH, glutathione;
BSO, buthionine sulfoximine. |
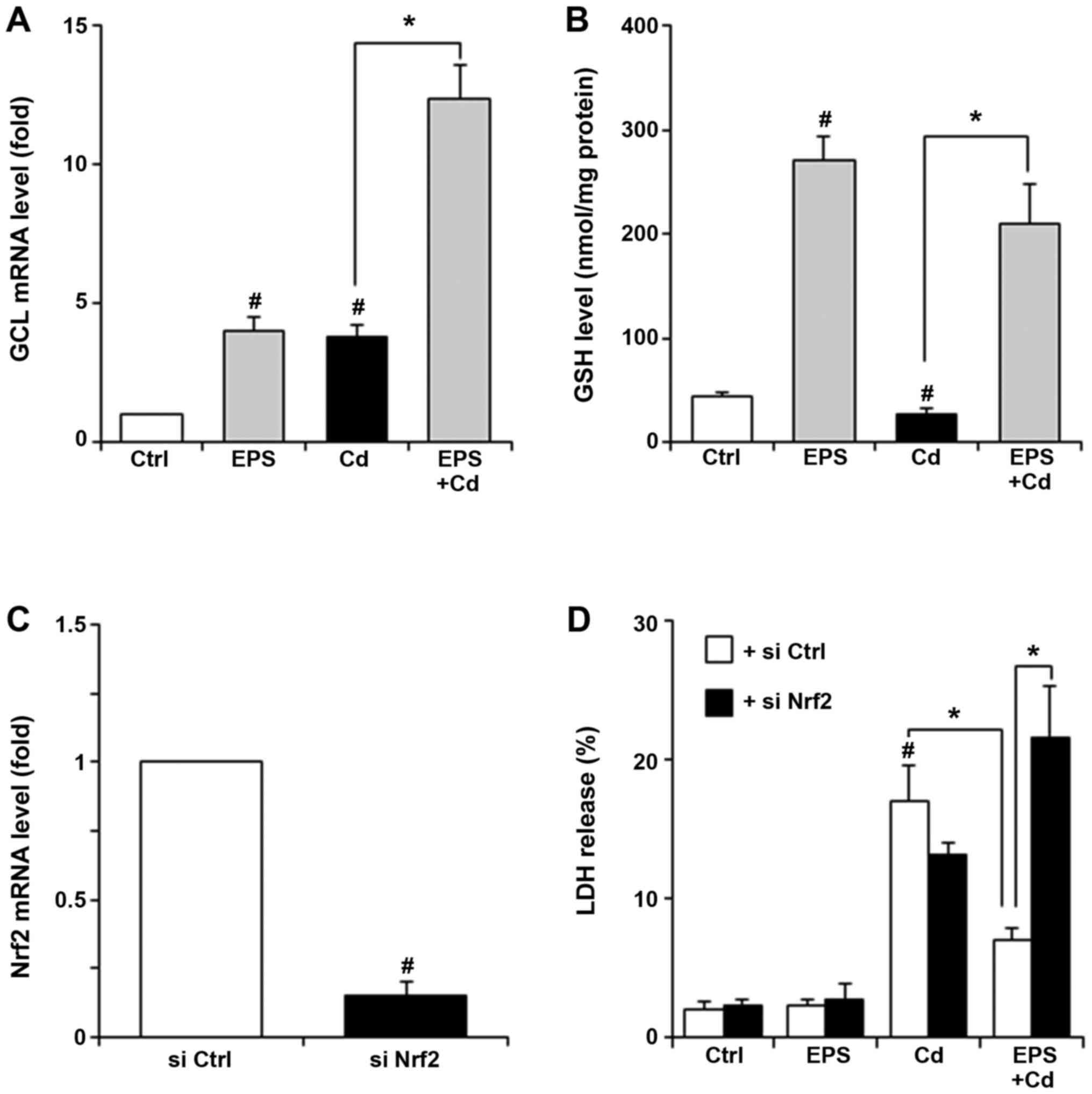
GCL is an enzyme that catalyzes the first and
rate-limiting step in de novo GSH synthesis (18). We measured GCL mRNA and GSH levels
in BAECs (Fig. 2A and B). EPS increased GCL mRNA levels as well
as intracellular GSH levels. Increases in GCL mRNA levels were also
observed in both untreated and EPS-treated cells after exposure to
Cd. Cd exposure significantly decreased GSH levels. These effects
of Cd on GCL and GSH levels are consistent with published results
(19), probably reflecting GSH
consumption as a result of Cd exposure. When EPS-pretreated BAECs
were exposed to Cd, GCL mRNA levels markedly increased by 3-fold
relative to that of EPS-pretreated cells without Cd exposure. In
the EPS-pretreated cells, high GSH level was observed after Cd
exposure, even though Cd exposure may result in the consumption of
excess GSH resulting from the upregulation of GCL. We examined
whether Nrf2 levels could alter the cell viability treated with Cd
and/or EPS, by means of Nrf2 knockdown in BAECs. BAECs were
transfected with control siRNA or Nrf2 siRNA. Nrf2 mRNA expression
levels in cells transfected with Nrf2 siRNA were reduced by 80%
relative to those in control siRNA transfected cells (Fig. 2C). Fig.
2D demonstrates that the protective ability of EPS against the
Cd-induced LDH release completely disappeared following Nrf2 siRNA
transfection.
Effect of EPS on Cd uptake in
BAECs
We next measured intracellular Cd levels in BAECs.
After untreated and EPS-pretreated BAECs were exposed to 50 µM Cd
for 4 h, intracellular Cd levels were determined by measuring Cd
content in the pellet suspended in HNO3 solution using GF-AAS. As
shown in Fig. 3A, pretreatment with
EPS at 50 µM reduced intracellular Cd levels in BAECs. Then, we
examined that the effect of EPS on ZIP8 levels in BAECs. When BAECs
were exposed to 25 µM Cd for 24 h, increases in ZIP8 mRNA levels
were confirmed (Fig. 3B). EPS
pretreatment inhibited the increases in ZIP8 mRNA levels induced by
Cd exposure. In other experiments in which NAC or GSH (free) was
added, a similar trend was observed; both NAC and GSH inhibited the
Cd-induced increases in ZIP8 mRNA levels (Fig. 3C).
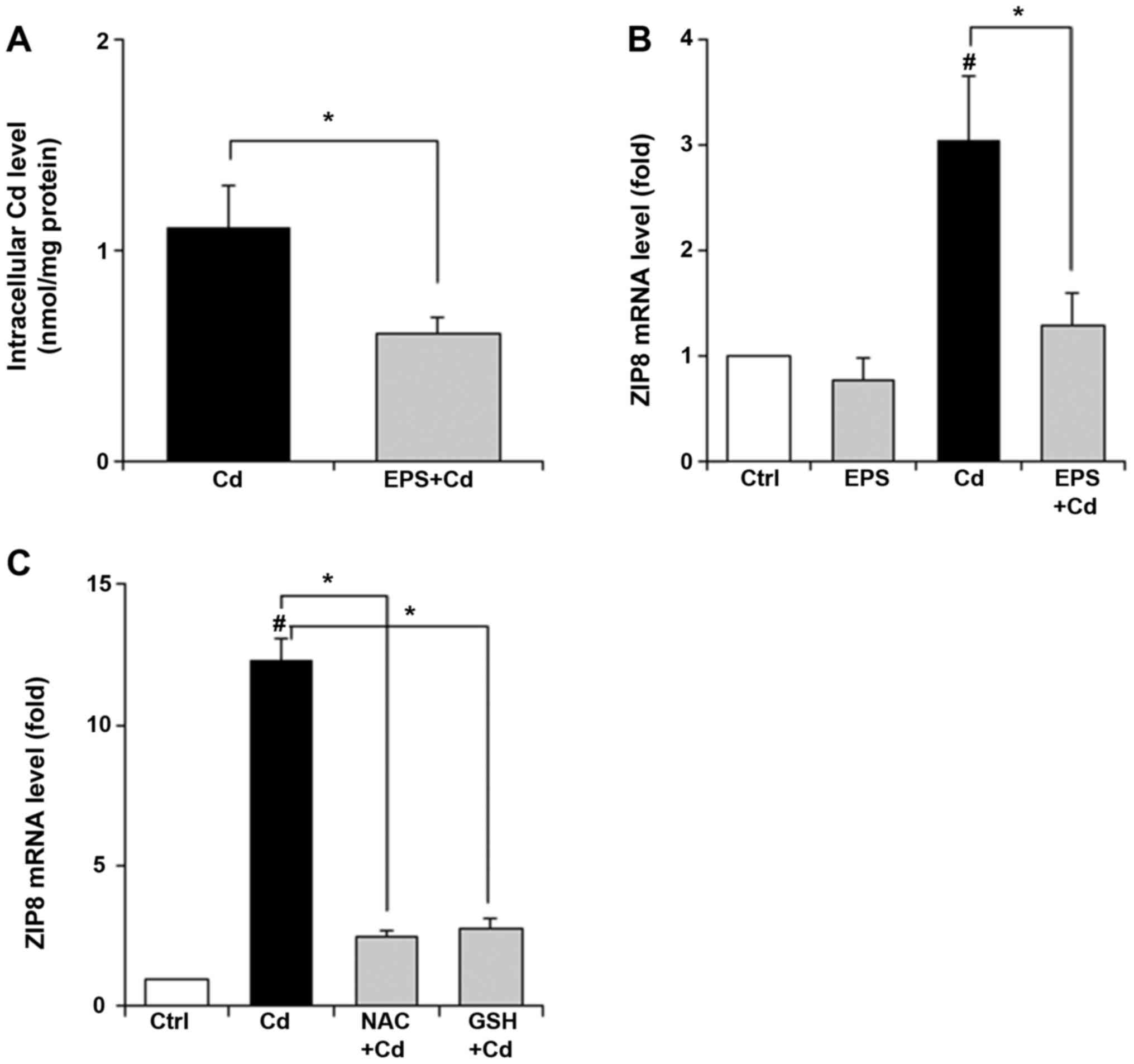 | Figure 3Effect of EPS on intracellular Cd
level and ZIP8 mRNA levels in BAECs. (A) BAECs were pretreated with
EPS (50 µM) for 16 h. After the cells were exposed to Cd at 50 µM
for 4 h, intracellular Cd accumulation was determined by measuring
Cd content in the pellet suspended in HNO3 solution by
GF-AAS. (B) BAECs were pretreated with EPS (50 µM) for 16 h. After
the cells were exposed to Cd at 25 µM for 24 h, ZIP8 mRNA levels
were measured. (C) BAECs were pretreated with NAC (1 mM) and GSH
(free, 1 mM) for 16 h. After the cells were exposed to Cd at 25 µM
for 24 h, ZIP8 mRNA levels were measured. Values are means ± SD of
three experiments. *P<0.01. #P<0.01 vs.
control. EPS, epalrestat; Cd, cadmium; ZIP8, Zrt-, Irt-like protein
8; BAECs, bovine aortic endothelial cells; GF-AAS, graphite furnace
atomic absorption spectrometry; NAC, N-acetylcysteine; GSH,
glutathione. |
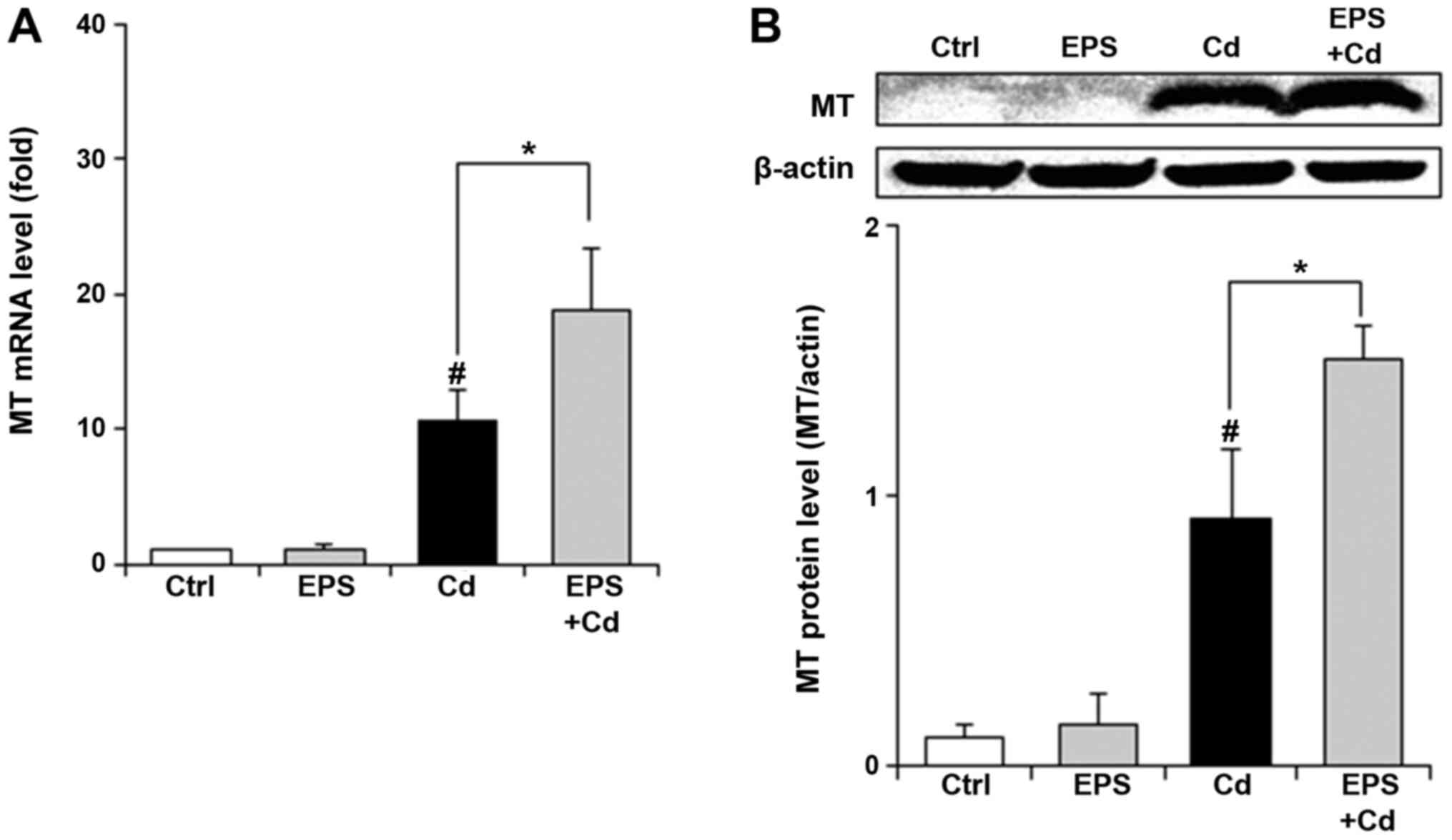
Effect of EPS on MT levels in
BAECs
Finally, we examined whether EPS affected MT in
BAECs. Exposure to Cd (25 µM) for 4 h resulted in elevated MT mRNA
and protein levels (Fig. 4A and
B). EPS pretreatment led to further
increases in the MT levels. In control cells, EPS alone had no
effect on the MT levels.
Discussion
Cd is an industrial and environmental pollutant that
targets the vascular endothelium (1). Recent advances in Cd toxicity research
have suggested an association between Cd and vascular diseases
(20). Although the molecular
targets of Cd toxicity are poorly understood, there are studies
indicating that Cd causes endothelial dysfunction and exhibits
cytotoxicity, indicating that functional damage to the endothelium
may be attributable to toxicity in parenchymal cells of various
target organs, such as kidney and liver (2). However, the mechanisms of Cd
implications in vascular diseases have yet to be explained.
EPS is the only aldose reductase inhibitor currently
available for the treatment of diabetic neuropathy. EPS is easily
absorbed by neural tissue and inhibits aldose reductase with
minimum adverse effects (11). The
usual dosage of EPS for oral use is 50 mg three times a day. The
plasma EPS concentration of 3.9 µg/ml (12 µM) was observed 1 h
after a single oral dose of 50 mg (21). In this study, we examined the
effects of EPS at near-plasma concentration on Cd-induced
cytotoxicity in BAECs. Our new findings are that: i) EPS protects
against Cd-induced cytotoxicity in BAECs; ii) EPS increases GSH
levels in cells exposed to Cd; and iii) EPS has an effect on the
intracellular levels of Cd, ZIP8, and MT.
Some antioxidants decrease the cytotoxic effects of
Cd (7,22,23).
Sulforaphane, which is a natural and highly effective antioxidant,
reduced Cd-induced cell death in lymphocytes and monocytes
(8). Sulforaphane is a potent Nrf2
activator that promotes the restoration of cellular GSH levels
(24). Sulforaphane prevents
testicular damage by Cd in association with the Nrf2 pathway
(8). Quercetin attenuates
Cd-induced oxidative damage and apoptosis in ovarian granulosa
cells (25). Both sulforaphane and
quercetin are Nrf2 activators (26). Sulforaphane and quercetin modulate
GSH homeostasis via Nrf2. In kidney cells, the activation of Nrf2
is an adaptive intracellular response to Cd-induced oxidative
stress, and that Nrf2 is protective against Cd-induced apoptosis
(27). Nrf2 has important roles in
suppression the carcinogenicity of Cd in terms of protection from
oxidative stress-induced DNA damage (28). Therefore, the therapeutic Nrf2
activator dose may be a new strategy against Cd-induced toxicity.
In our previous reports, we showed that EPS increases GSH levels in
rat Schwann cells and BAECs in association with the Nrf2 pathway
(12,13). EPS could be expected to protect
against Cd-induced toxicity in the same manner as sulforaphane and
quercetin. In fact, our present study indicates that EPS protects
against Cd-induced cytotoxicity in BAECs (Fig. 1A). Cd exposure to untreated cells
resulted in decreased GSH levels. GSH plays a protective role
against Cd-induced cytotoxicity (Fig.
1B and C). It is possible that
the EPS-induced protection against Cd-induced cytotoxicity is
mainly due to the increase in GSH levels. The mechanism seems to
involve the upregulation of GSH via Nrf2. When EPS-pretreated cells
were exposed to Cd, the increase in GSH levels prevented the loss
of viable cells induced by the exposure to Cd. EPS increased GCL
mRNA levels and an increase in GCL mRNA levels was also observed in
both untreated and EPS-treated cells after exposure to Cd in BAECs
(Fig. 2A). However, in the
EPS-pretreated BAECs, there were no significant differences in GSH
levels between EPS treatment alone and EPS treatment followed by Cd
exposure (Fig. 2B), implying that
Cd exposure may result in the consumption of GSH. Cd is tightly
bound to GSH for chelation (29).
In addition, the experiments using Nrf2 knockdown cells (Fig. 2D) indicated that Nrf2 may be
involved in the protective ability of EPS against Cd-induced
cytotoxicity. Together, these results suggest that EPS protects
against Cd-induced cytotoxicity in BAECs by increasing GSH levels.
Possibly, Nrf2 may be involved in the protection against Cd-induced
cytotoxicity.
Accumulating evidence indicates that oxidative
stress could be partially responsible for some cases of Cd-induced
cytotoxicity (30). We examined
whether ROS was generated by Cd in BAECs using the ROS probe and
found that ROS production was not induced by Cd in our present
study (data not shown). Cd is not able to produce radicals via
Fenton-type chemistry. Nonetheless, it induces oxidative stress
through a multifaceted mechanism, including the reduction of
antioxidative defense and ROS production as a result of
mitochondrial damage (9). The use
of antioxidants, the induction of antioxidant enzymes, and the
complexation of Cd with GSH and MT are the most potent protective
measures against Cd-induced oxidative stress.
Cd-induced cytotoxicity is dependent on the amount
of intracellular Cd (9). Fig. 3A shows that the pretreatment with
EPS reduced the intracellular accumulation of Cd in BAECs. This
reduction of the intracellular Cd accumulation may be involved in
the suppression of Cd-induced cytotoxicity by EPS. ZIP8 plays an
important role in Cd uptake in mammalian cells (31). One report has indicated that the
absence of ZIP8 expression in vascular endothelial cells is
associated with resistance to Cd-induced testicular toxicity
(32). That finding suggests that
ZIP8 expression in endothelial cells is crucial for the Cd-induced
cytotoxicity. Our results in Fig.
3B indicate that the effect of EPS on ZIP8 mRNA levels may
contribute to the suppression of Cd-induced cytotoxicity, albeit in
a limited data. Further studies might be needed to clarify the
effect of EPS on ZIP8 expression levels. Fig. 3C shows that NAC or GSH inhibited
Cd-induced ZIP8 mRNA levels. One study has revealed that elevated
GSH levels can downregulate Cd uptake by downregulating the Cd
transporter ZIP8(5). EPS inhibited
Cd-induced ZIP8 mRNA levels (Fig.
3B). Therefore, we attribute this inhibitory effect to the
increase in GSH levels by EPS.
MT as well as GSH is the most potent protective
measure against Cd-induced oxidative stress (9). Exposure to such heavy metals as Cd
triggers the induction of MT, which confers cells with resistance
to heavy-metal-induced toxicities (33). Exposure to Cd elevated MT mRNA and
protein levels and EPS pretreatment further increased the levels of
MT (Fig. 4A and B), indicating that EPS contributed to the
expression of MT in BAECs treated with Cd. Metal-responsive
transcription factor 1 (MTF-1) is considered to be a major
activator for MT gene expression (34,35).
On the other hand, Nrf2 partially contributes to MT expression in
vascular endothelial cells after Cd exposure (2). Our results indicate that the effect of
EPS on MT levels may contribute to mediate Nrf2 involved in the
protective ability of EPS partially regulates to MT levels in BAECs
treated with Cd. Further studies might be needed to clarify the
effect of EPS on MT levels.
In summary, we demonstrated for the first time that
EPS suppresses Cd-induced cytotoxicity in BAECs. The upregulation
of GSH may be involved in the suppression of Cd-induced
cytotoxicity by EPS. Our findings have led us to propose that
targeting the regulation of GSH, ZIP8 and MT by EPS is a promising
therapeutic approach in Cd poisoning.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by JSPS KAKENHI (grant no. JP
20K10434).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Author's contributions
RT and KS conceived and designed the experiments and
wrote the manuscript. YM and KY performed the experiments and
analyzed and interpreted the data. YY and SO performed the
experiments. YT analyzed and interpreted the data. All authors read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Prozialeck WC, Edwards JR, Nebert DW,
Woods JM, Barchowsky A and Atchison WD: The vascular system as a
target of metal toxicity. Toxicol Sci. 102:207–218. 2008.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Shinkai Y, Kimura T, Itagaki A, Yamamoto
C, Taguchi K, Yamamoto M, Kumagai Y and Kaji T: Partial
contribution of the Keap1-Nrf2 system to cadmium-mediated
metallothionein expression in vascular endothelial cells. Toxicol
Appl Pharmacol. 295:37–46. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Zhai Q, Narbad A and Chen W: Dietary
strategies for the treatment of cadmium and lead toxicity.
Nutrients. 7:552–571. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Nair AR, Degheselle O, Smeets K, Van
Kerkhove E and Cuypers A: Cadmium-induced pathologies: Where is the
oxidative balance lost (or Not)? Int J Mol Sci. 14:6116–6143.
2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Aiba I, Hossain A and Kuo MT: Elevated GSH
level increases cadmium resistance through down-regulation of
Sp1-dependent expression of the cadmium transporter ZIP8. Mol
Pharmacol. 74:823–833. 2008.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Sun Z, Niu Z, Wu S and Shan S: Protective
mechanism of sulforaphane in Nrf2 and anti-lung injury in ARDS
rabbits. Exp Ther Med. 15:4911–4915. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yang SH, Long M, Yu LH, Li L, Li P, Zhang
Y, Guo Y, Gao F, Liu MD and He JB: Sulforaphane prevents testicular
damage in Kunming mice exposed to cadmium via activation of
Nrf2/ARE signaling pathways. Int J Mol Sci.
17(E1703)2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Alkharashi NAO, Periasamy VS,
Athinarayanan J and Alshatwi AA: Sulforaphane mitigates
cadmium-induced toxicity pattern in human peripheral blood
lymphocytes and monocytes. Environ Toxicol Pharmacol. 55:223–239.
2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Sandbichler AM and Höckner M: Cadmium
protection strategies--a hidden trade-off? Int J Mol Sci.
17(E139)2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Steele JW, Faulds D and Goa KL:
Epalrestat. A review of its pharmacology, and therapeutic potential
in late-onset complications of diabetes mellitus. Drugs Aging.
3:532–555. 1993.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Hotta N, Akanuma Y, Kawamori R, Matsuoka
K, Oka Y, Shichiri M, Toyota T, Nakashima M, Yoshimura I, Sakamoto
N, et al: Long-term clinical effects of epalrestat, an aldose
reductase inhibitor, on diabetic peripheral neuropathy: The 3-year,
multicenter, comparative Aldose Reductase Inhibitor-Diabetes
Complications Trial. Diabetes Care. 29:1538–1544. 2006.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Sato K, Yama K, Murao Y, Tatsunami R and
Tampo Y: Epalrestat increases intracellular glutathione levels in
Schwann cells through transcription regulation. Redox Biol.
2:15–21. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yama K, Sato K, Abe N, Murao Y, Tatsunami
R and Tampo Y: Epalrestat increases glutathione, thioredoxin, and
heme oxygenase-1 by stimulating Nrf2 pathway in endothelial cells.
Redox Biol. 4:87–96. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Luczak MW and Zhitkovich A: Role of direct
reactivity with metals in chemoprotection by N-acetylcysteine
against chromium(VI), cadmium(II), and cobalt(II). Free Radic Biol
Med. 65:262–269. 2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-ΔΔ C(T)) Method. Methods. 25:402–408. 2001.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Tsukamoto M, Tampo Y, Sawada M and Yonaha
M: Paraquat-induced oxidative stress and dysfunction of the
glutathione redox cycle in pulmonary microvascular endothelial
cells. Toxicol Appl Pharmacol. 178:82–92. 2002.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhou YJ, Zhang SP, Liu CW and Cai YQ: The
protection of selenium on ROS mediated-apoptosis by mitochondria
dysfunction in cadmium-induced LLC-PK(1) cells. Toxicol In Vitro.
23:288–294. 2009.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Meister A: Selective modification of
glutathione metabolism. Science. 220:472–477. 1983.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Nair PMG, Park SY, Chung JW and Choi J:
Transcriptional regulation of glutathione biosynthesis genes,
γ-glutamyl-cysteine ligase and glutathione synthetase in response
to cadmium and nonylphenol in Chironomus riparius. Environ Toxicol
Pharmacol. 36:265–273. 2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Kolluru GK, Tamilarasan KP, Geetha Priya
S, Durgha NP and Chatterjee S: Cadmium induced endothelial
dysfunction: Consequence of defective migratory pattern of
endothelial cells in association with poor nitric oxide
availability under cadmium challenge. Cell Biol Int. 30:427–438.
2006.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ono Pharmaceutical Co., Ltd.,
Kinedak® Tablets 50 mg. Epalrestat, Package Insert,
Osaka, Japan, 2009.
|
|
22
|
Harabawy AS and Mosleh YY: The role of
vitamins A, C, E and selenium as antioxidants against genotoxicity
and cytotoxicity of cadmium, copper, lead and zinc on erythrocytes
of Nile tilapia, Oreochromis niloticus. Ecotoxicol Environ Saf.
104:28–35. 2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
El-Boshy ME, Risha EF, Abdelhamid FM,
Mubarak MS and Hadda TB: Protective effects of selenium against
cadmium induced hematological disturbances, immunosuppressive,
oxidative stress and hepatorenal damage in rats. J Trace Elem Med
Biol. 29:104–110. 2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Mizuno K, Kume T, Muto C, Takada-Takatori
Y, Izumi Y, Sugimoto H and Akaike A: Glutathione biosynthesis via
activation of the nuclear factor E2-related factor 2
(Nrf2)--antioxidant-response element (ARE) pathway is essential for
neuroprotective effects of sulforaphane and 6-(methylsulfinyl)
hexyl isothiocyanate. J Pharmacol Sci. 115:320–328. 2011.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Jia Y, Lin J, Mi Y and Zhang C: Quercetin
attenuates cadmium-induced oxidative damage and apoptosis in
granulosa cells from chicken ovarian follicles. Reprod Toxicol.
31:477–485. 2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Bahar E, Kim JY and Yoon H: Quercetin
attenuates manganese-induced neuroinflammation by alleviating
oxidative stress through regulation of apoptosis, iNOS/NF-κB and
HO-1/Nrf2 pathways. Int J Mol Sci. 18(E1989)2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Chen J and Shaikh ZA: Activation of Nrf2
by cadmium and its role in protection against cadmium-induced
apoptosis in rat kidney cells. Toxicol Appl Pharmacol. 241:81–89.
2009.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Park JY and Seo YR: The protective role of
Nrf2 in cadmium-induced DNA damage. Mol Cell Toxicol. 7:61–66.
2011. View Article : Google Scholar
|
|
29
|
Leverrier P, Montigny C, Garrigos M and
Champeil P: Metal binding to ligands: Cadmium complexes with
glutathione revisited. Anal Biochem. 371:215–228. 2007.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Thévenod F and Lee WK: Cadmium and
cellular signaling cascades: Interactions between cell death and
survival pathways. Arch Toxicol. 87:1743–1786. 2013.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Himeno S, Yanagiya T and Fujishiro H: The
role of zinc transporters in cadmium and manganese transport in
mammalian cells. Biochimie. 91:1218–1222. 2009.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Dalton TP, He L, Wang B, Miller ML, Jin L,
Stringer KF, Chang X, Baxter CS and Nebert DW: Identification of
mouse SLC39A8 as the transporter responsible for cadmium-induced
toxicity in the testis. Proc Natl Acad Sci USA. 102:3401–3406.
2005.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Chen L, Ma L, Bai Q, Zhu X, Zhang J, Wei
Q, Li D, Gao C, Li J, Zhang Z, et al: Heavy metal-induced
metallothionein expression is regulated by specific protein
phosphatase 2A complexes. J Biol Chem. 289:22413–22426.
2014.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Murphy BJ, Andrews GK, Bittel D, Discher
DJ, McCue J, Green CJ, Yanovsky M, Giaccia A, Sutherland RM,
Laderoute KR, et al: Activation of metallothionein gene expression
by hypoxia involves metal response elements and metal transcription
factor-1. Cancer Res. 59:1315–1322. 1999.PubMed/NCBI
|
|
35
|
Andrews GK: Regulation of metallothionein
gene expression by oxidative stress and metal ions. Biochem
Pharmacol. 59:95–104. 2000.PubMed/NCBI View Article : Google Scholar
|