Introduction
Ischemic heart disease remains the most common cause
of death worldwide, accounting for up to 23% of global deaths
(1). To minimize the myocardial
damage following acute ischemia, early reperfusion is of crucial
importance (2). Unfortunately,
reperfusion itself is deleterious for the heart and causes ischemia
reperfusion (I/R) injury (2).
Cardioprotective interventions, such as ischemic and
pharmacological preconditioning, are powerful measures to confer
cardioprotection against myocardial I/R injury (3). Ischemic preconditioning (IPC) is
performed through the exposure to short cycles of ischemia followed
by reperfusion directly at the target organ (4). However, to date, the translation of
IPC into the clinic has failed due to the invasiveness of the
procedure. Nonetheless, this limitation was discovered to be
circumvented through using non-invasive remote IPC (RIPC), which
works by applying short cycles of I/R, for example at a limb, via a
blood pressure cuff (5). Although
promising experimental results have been published following years
of research on RIPC, the exact underlying mechanism of RIPC-induced
cardioprotection is still not fully understood (6). The proposed mechanism includes the
generation of humoral factors in the remote organ, which can then
enter the blood stream to activate and induce protective signaling
cascades in the heart (5).
It was previously demonstrated that conditioning
strategies protected the myocardium by activating the reperfusion
injury salvage kinases (RISK) pathway (7); this pathway mediates cell survival
through various signaling cascades, including the activation of
endothelial nitric oxide synthase, protein kinase G, PI3K and AKT.
Li et al (8) reported that
members of the RISK pathway were involved in RIPC-induced
cardioprotection, while the PI3K inhibitor wortmannin abolished the
cardioprotective effect in a combined in vivo/in
vitro model. In addition, increased AKT phosphorylation levels
were observed in tissues 15 min after RIPC treatment compared with
the control group. However, to the best of our knowledge, whether
RIPC-induced AKT phosphorylation is merely transient or maintained
during I/R remains unclear.
Furthermore, previous research has demonstrated that
conditioning with various stimuli protects the heart from I/R
injury via activation of the survivor activating factor enhancement
pathway, which involves TNFα, TNF receptor type 2 (TNFR2), Janus
kinase (JAK) and STAT3(9). The
tyrosine phosphorylation of STATs by JAKs leads to the dimerization
and subsequent translocation of the respective STATs into the
nucleus or mitochondria to regulate the expression levels of its
target genes, which are primarily involved in energy metabolism
(10). Heusch et al
(11) identified that the
phosphorylation levels of RISK pathway members or STAT3 were
increased in both RIPC-treated and control patients before
undergoing myocardial intervention, whereas the phosphorylation
levels of STAT5 were only upregulated in the RIPC group. While the
results from one previous study suggested the involvement STAT5 in
RIPC-induced cardioprotection (12), other previous studies have indicated
that the phosphorylation of STAT5 may not be involved (13,14).
Therefore, the role of STAT5 in RIPC-induced cardioprotection
remains unexplained.
As AKT and STAT5 are considered to serve an
important role in the signaling pathways of different
cardioprotective strategies, the present study aimed to investigate
the phosphorylation levels of these key mediators in RIPC-induced
cardioprotection in rat hearts in vivo. The current study
hypothesized that RIPC may induce the phosphorylation of AKT and/or
STAT5 at both time-points (TPs), that is, immediately after RIPC
and/or after I/R.
Materials and methods
Ethics approval
The current study was conducted in accordance with
the Guide for the Care and Use of Laboratory Animals published by
the National Institutes of Health (publication number 85-23,
revised 1996) and the experiments were performed after obtaining
ethical approval from the State Agency for Nature, Environment and
Consumer Protection, North-Rhine Westphalia, Germany (approval no.
8.87-50.10.37.09.148), and this study comply with the ARRIVE
guidelines (15). The animals used
were obtained from the breeding facility at the Central Animal
Research Facility of the Heinrich-Heine-University Duesseldorf.
Surgical preparation
In part A of the present study, heart tissues were
used from rats exclusively treated during the present study. In
part B, the heart tissues were obtained from animals used in a
previous study by Brandenburger et al (16), which demonstrated a significant
reduction in infarct size following RIPC compared with the control
hearts.
Surgical preparation was performed as described
previously (16). Briefly, male
Wistar rats were anesthetized by intraperitoneal sodium
pentobarbital injection (60 mg/kg body weight). Following tracheal
intubation, mechanical ventilation was performed with 30%
oxygen/70% nitrogen and monitored by blood gas analysis throughout
the experiments to maintain the acid-base state within the
physiological limits. Anesthesia was maintained by continuous
infusion of pentobarbital (40 mg/kg/h) and the body temperature was
maintained at 37-37.5˚C using a heating pad. Lateral left-sided
thoracotomy was performed by looping a ligature (5-0 Prolene;
Ethicon, Inc.; Johnson & Johnson) around the left anterior
descending (LAD) coronary artery to tighten and occlude the
coronary artery to induce ischemia. Following successful
implementation of the surgical procedures, all animals were allowed
to stabilize for 10 min prior to the start of the respective
experimental protocol.
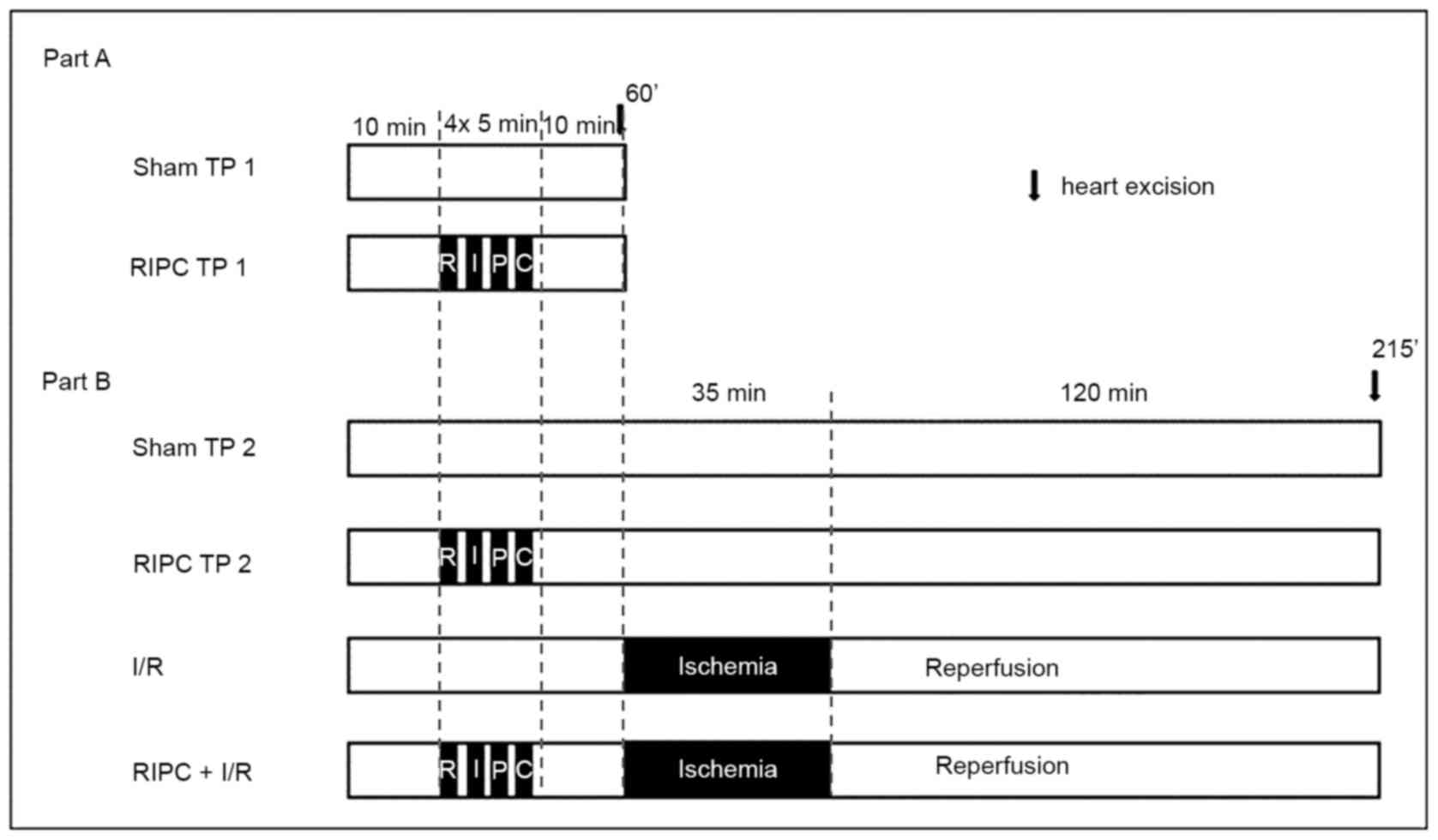
Experimental protocol Part A
Part A was performed to investigate the activation
of AKT and STAT5 immediately following RIPC. Therefore, the hearts
required for the analysis were harvested 10 min after sham or RIPC
treatment (TP 1:60 min). The rats (weight, 280±27 g) used for these
experiments were randomly assigned into one of two experimental
groups (n=6/group; Fig. 1): i) Sham
early TP (TP1) group, in which the rats received sham I treatment;
and ii) RIPC TP1 group, in which rats received RIPC treatment with
4 cycles of bilateral hind limb ischemia for 5 min interspersed
with 5 min of reperfusion. RIPC treatment was performed by
bilateral hind limb ischemia, which was induced by inflating
modified blood pressure cuffs to 200 mmHg, followed by the
initiation of reperfusion by deflating the cuffs. For the sham I
treatment, the cuffs were placed on the hind limbs, but not
inflated. Following 10 min of the respective treatments, the heart
was excised, frozen in liquid nitrogen and stored at -80˚C for
further analysis.
Part B
Part B experiments served to investigate the
activation of AKT and STAT5 induced by RIPC following I/R injury.
As previously described above, the surgeries were performed during
our previous study and only the heart tissue from the respective
animals was obtained for the present study (16). To facilitate the understanding of
the experimental protocol, the procedure is briefly outlined below.
The rats received sham I or RIPC treatment followed by I/R or a
time matched sham II treatment without any intervention (TP 2:215
min). After reperfusion, in the groups which received I/R
treatment, the LAD was reoccluded and 5 ml Evans blue solution was
injected intravenously. The non area of risk (non-AAR) was stained
blue, while the area of risk (AAR) remained unstained. The hearts
were removed, separated in non-AAR and ARR, respectively frozen in
liquid nitrogen and stored at -80˚C. For further analyses, only
tissue from the AAR was used. In the groups that did not receive
I/R treatment, the hearts were removed after the matched TP, frozen
in liquid nitrogen and stored at -80˚C. The experimental groups
were as follows (n=6/group): i) Sham late TP (TP2) group, in which
the rats received sham I and II treatment; ii) RIPC TP2 group, in
which rats received RIPC treatment with 4 cycles of 5 min bilateral
hind limb ischemia interspersed with 5 min of reperfusion, followed
by sham II treatment; iii) I/R group, in which rats received sham I
treatment and I/R treatment with 35 min of LAD occlusion and 120
min of reperfusion; and iv) RIPC + I/R group, in which the rats
received RIPC treatment with 4 cycles of 5 min bilateral hind limb
ischemia interspersed with 5 min of reperfusion and I/R treatment
with 35 min of ischemia (LAD occlusion) and 120 min of
reperfusion.
Protein extraction and subcellular
fractioning
Tissue homogenization and protein extraction were
performed as previously described (16). Briefly, pulverized, frozen heart
tissue was dissolved in lysis buffer containing 5 mM Tris base, 2
mM EGTA, phosphatase inhibitors (50 mM NaF and 2 mM
Na3VO4), complete protease inhibitor mix
(Roche Diagnostics) and 5 mM DTT. The lysates were homogenized and
centrifuged at 600 x g at 4˚C for 10 min to discard the cell
debris.
Western blotting
Western blotting was performed as described
previously (16). Briefly, the
protein concentration was determined using the Lowry method and
proteins were dissolved in loading buffer containing
mercaptoethanol. Following the seperation of the proteins via 10%
SDS-PAGE, the proteins were subsquently transferred onto PVDF
membranes and blocked with 5% milk powder solution. The membranes
were then incubated with the following primary antibodies in 1%
milk powder solution overnight: Anti-GAPDH (1:5,000; cat. no.
ab8245; Abcam), anti-AKT (1:1,000; cat. no. 9272; Cell Signaling
Technology, Inc.), anti-phosphorylated (phospho)-AKT-serine
(Ser)473 (1:1,000; cat. no. 9271; Cell Signaling
Technology, Inc.), anti-phospho-STAT5-tyrosine694
(1:1,000; cat. no. 4322; Cell Signaling Technology, Inc.) and
anti-STAT5 (1:1,000; cat. no. 9363; Cell Signaling Technology,
Inc.). Following the primary antibody incubation, the membranes
were incuabted with HRP-conjugated secondary antibodies (Jackson
ImmunoResearch Laboratories, Inc.) for 2 h. Protein bands were
visualized using an enhanced chemiluminescence system (Santa Cruz
Biotechnology, Inc.), and captured using a CoolSNAP HQ2 camera
(Teledyne Photometrics) and a Gel-Pro analyzer 6.0.0.349 (Media
Cybernetics, Inc.). Densitometric analysis was performed using
Image Studio Lite version 5.2.5 software (LI-COR Biosciences). All
signal intensities were normalized to the respective signaling
intensity of the loading control, GAPDH. Signal intensities of the
phosphorylated proteins were normalized to the respective total
protein signal intensity.
Statistical analysis
The sample size used (n=6/group) was determined in
order to detect the mean difference of 25% with a standard
deviation of 15% in the phosphorylation levels of target proteins
with α<0.05 and a power of 0.8(17). To detect the statistical differences
in the phosphorylation levels between the two groups, a Student's
t-test was performed using GraphPad Prism 6 software (GraphPad
Software, Inc.). Data are expressed as the mean ± SD. P<0.05 was
considered to indicate a statistically significant difference.
Results
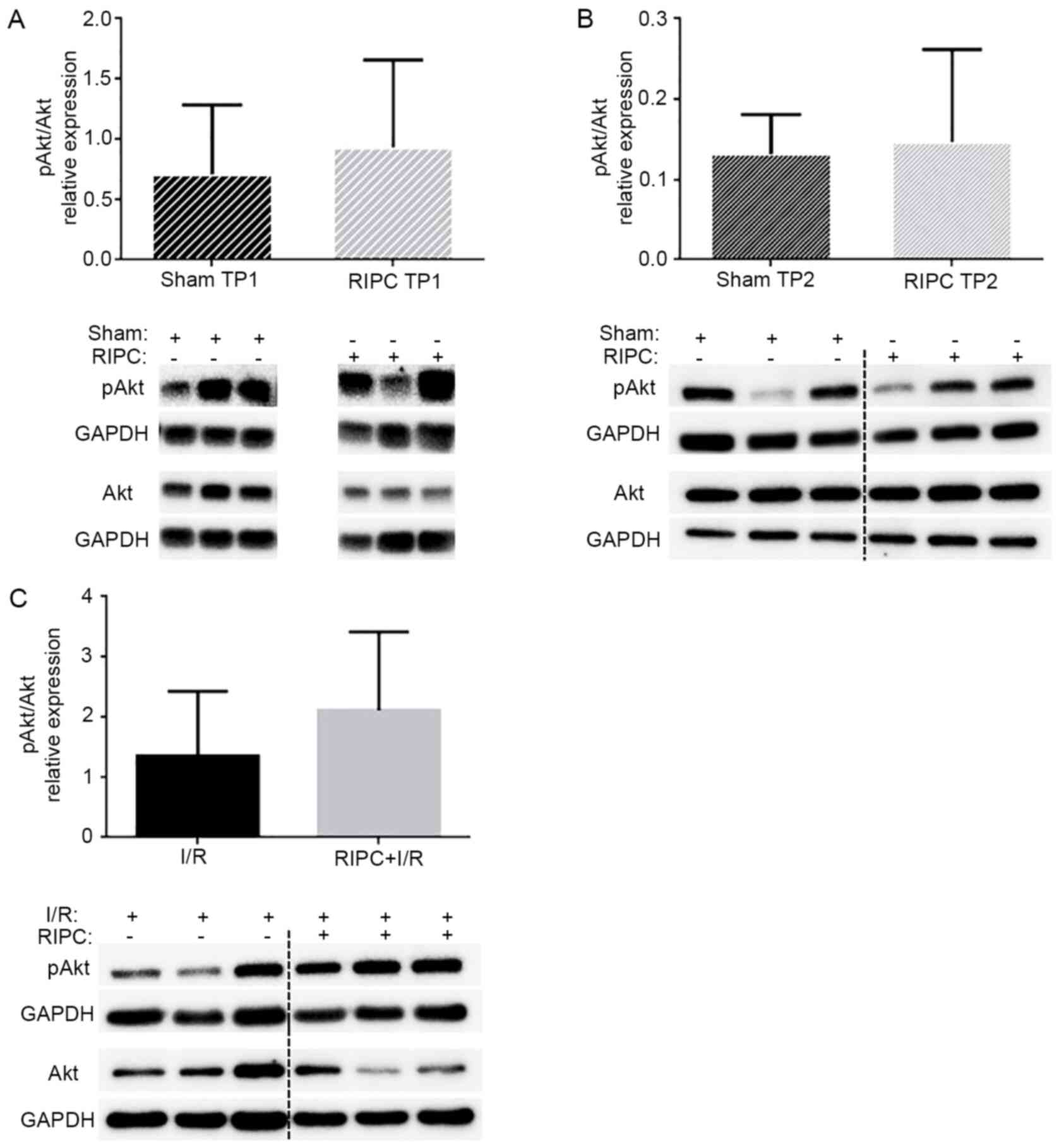
Analysis of AKT expression levels
A previous study revealed that RIPC treatment, as
used in the present study, significantly reduced the infarct size
(16). Therefore, to analyze the
role of AKT in the present study, the levels of AKT phosphorylation
at Ser473 were immediately determined following RIPC
treatment (TP1) or after a subsequent I/R phase (TP2). As shown in
Fig. 2, RIPC treatment did not
significantly alter the phosphorylation levels of AKT compared with
the sham-operated control animals at TP1 (0.93±0.73 vs. 0.71±0.57;
P=0.57; Fig. 2A) or TP2 (0.15±0.12
vs. 0.13±0.05; P=0.77; Fig. 2B).
Furthermore, no significant differences were identified in the AKT
phosphorylation levels between the I/R group (1.36±1.07) and RIPC +
I/R group (2.12±1.30) (P=0.300; Fig.
2C).
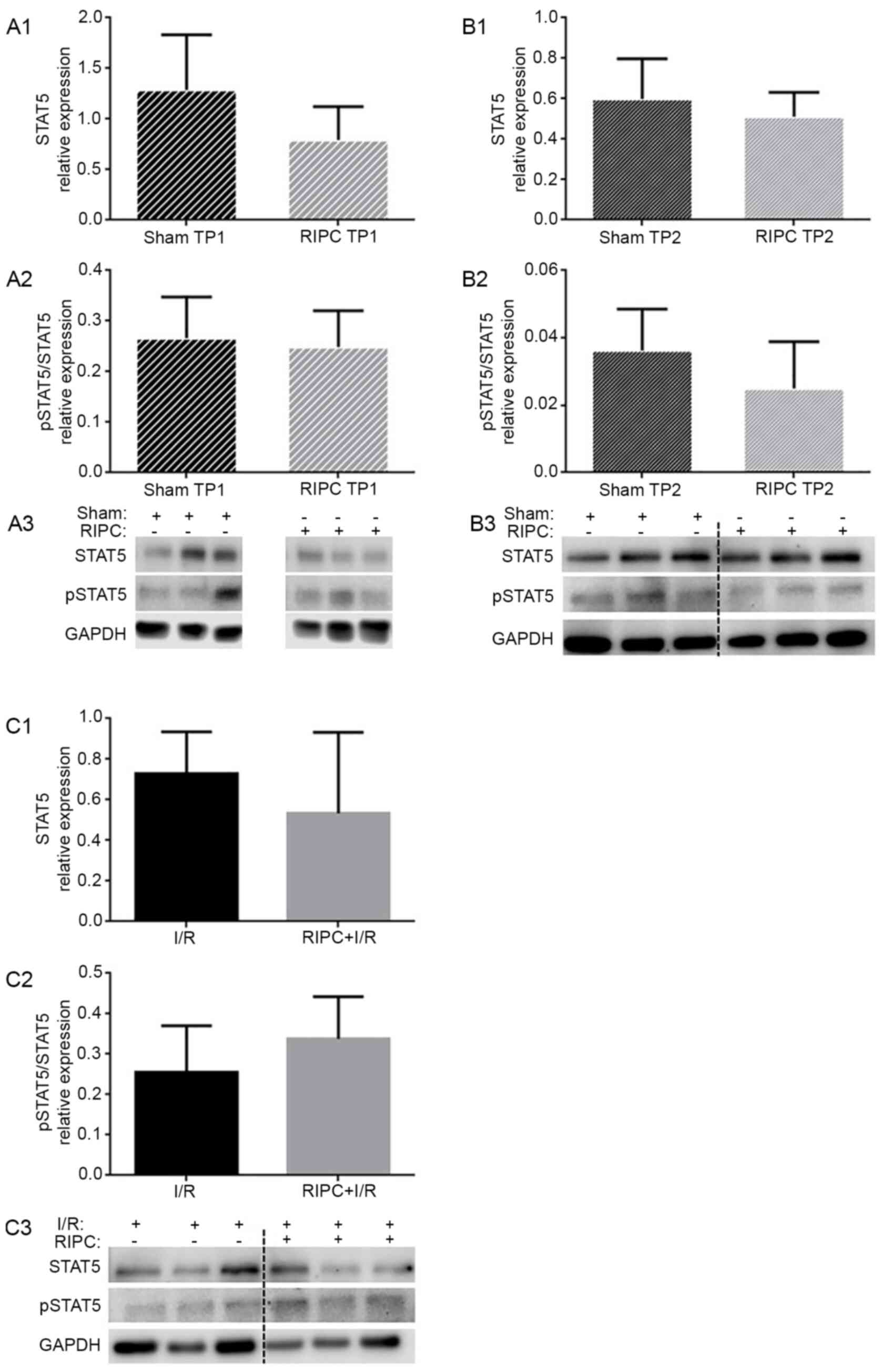
Analysis of STAT5 expression
levels
To determine the involvement of STAT5 in
RIPC-induced cardioprotection, the expression levels of STAT5 were
determined to verify a possible translocation from the cytosol into
the nuclei or mitochondria. At the TP1, STAT5 expression levels
were not significantly altered following RIPC treatment compared
with the sham-treated group (0.79±0.33 vs. 1.29±0.54; P=0.09;
Fig. 3A1). At the TP2, RIPC also
did not influence STAT5 expression levels compared with the sham
group (0.51±0.12 vs. 0.60±0.20; P=0.38; Fig. 3B1). Furthermore, RIPC followed by
I/R did not alter the STAT5 expression levels compared with the I/R
group (0.53±0.40 vs. 0.73±0.21; P=0.31; Fig. 3C1).
To investigate the role of STAT5 activation by RIPC,
the phosphorylation levels of STAT5 were determined. At the TP1, no
significant differences were reported in the phosphorylation levels
of STAT5 between the sham and RIPC groups (0.27±0.08 vs. 0.25±0.07;
P=0.70; Fig. 3A2). At the TP2,
neither RIPC treatment alone (0.03±0.01; P=0.17; Fig. 3B2) nor RIPC followed by I/R (I/R,
0.26±0.12 vs. RIPC + I/R, 0.34±0.10; P=0.22; Fig. 3C2) influenced the phosphorylation
levels of STAT5.
Discussion
The present study aimed to elucidate whether the
activation of AKT and/or STAT5 served a role in RIPC at the
investigated TPs. The results of the current study indicated that
neither the activation of AKT nor STAT5 were modulated by RIPC with
or without subsequent I/R, suggesting that the activation of the
respective targets at the measured TPs may not be involved in
RIPC-induced cardioprotection.
Focusing on the activation of AKT, the present
findings revealed that the cardioprotective signaling induced by
RIPC was not mediated through the phosphorylation of AKT, at least
at the chosen TPs of measurement in the present study. At the TP1,
in contrast to the findings of the present study, previous studies
have described the activation of AKT by RIPC. For example, in an
in vivo mouse model, increased levels of phosphorylated AKT
were detected 15 min after non-invasive RIPC treatment (8), while invasive RIPC via femoral artery
occlusion induced the phosphorylation of AKT immediately following
intervention in a rat model (18).
These controversial findings suggested that the phosphorylation of
AKT requires >10 min or a more pronounced stimulus, such as
invasive RIPC, to possibly reduce this time span following
intervention. In addition, species-specific differences may also be
responsible for the different results. In pigs, Skyschally et
al (19) reported no
differences in the phosphorylation levels of AKT between the
control and RIPC group following 1 h, indicating that the
activation of AKT may only be transient.
With regards to the TP2, similar to the results of
the present study, Hildebrandt et al (14) detected no changes in the
phosphorylation levels of AKT 2 h after reperfusion in a
translational protocol transferring human RIPC plasma, collected
from 5 min to 6 h after treatment, onto in vitro mouse
hearts prior to I/R. These results supported the findings from the
current study regarding the TP2 (following 2 h of reperfusion),
indicating that the activation of AKT does not occur at this TP.
Nonetheless, the selected TP2 during reperfusion in the current
experimental protocol may have been too late for detecting AKT
phosphorylation levels. In accordance with this hypothesis, Ma
et al (20) observed an
increase in the phosphorylated levels of AKT by RIPC 5 min after
the onset of reperfusion. Taking into account the fact that
increased levels of AKT phosphorylation are detectable immediately
at the beginning of reperfusion may suggest that the selected TP (2
h after the begining of reperfusion) may be too late to succesfully
investigate AKT phosphorylation levels. Interestingly,
postconditionig with RIPC or transfering RIPC plasma between
species (for example, pig plasma into rat hearts) enabled the
detection of AKT during the late periods of reperfusion. In
addition, inducing a postconditioning stimulus with RIPC led to the
activation of AKT, with even higher levels of phosphorylated AKT
observed 3 h after the onset compared with at the beginning of
reperfusion (21). Elevated Akt
phosphorylation levels were also detected following 2 h of
reperfusion through using a translational cardioprotective strategy
of RIPC plasma transfer (19). In
summary, the activation of AKT by RIPC seems to be dependent on the
TPs chosen for measurements, the species and the stimulus.
The present study also detected no significant
differences in the phosphorylation levels of STAT5 at the measured
TPs. Previous studies have demonstrated comparable results; for
example, the transfer of human RIPC plasma to mouse hearts or from
pigs to rat hearts in the respective models of I/R injury
identified no significant differences in the phosphorylation levels
of STAT5 compared with the control plasma (14,19).
In contrast, RIPC-induced decreased levels of phosphorylated STAT5
were detected 10 min after the onset of reperfusion compared with
in the pre-ischemia in pigs (19).
Despite these controversial results, it is hypothesized that the
activation of STAT5 may serve a role in RIPC-induced
cardioprotection. The induction of RIPC 24 h before I/R injury was
discovered to be cardioprotective and induce the phosphorylation of
STAT5; however, both effects were abolished in a
cardiomyocyte-specific STAT5 knockout mouse (12). Contrary to these findings, it seems
that the activation of STAT5 serves a rather minor role in the
experimental setting of cardioprotection in animal models, in
contrast to its importance in the clinical setting for patients.
For example, Heusch et al (11) suggested that in human hearts,
cardioprotection was associated with STAT5, but not STAT3, whereas
STAT3 activation and possibly STAT5 inhibition were associated with
protection in animals (11). Recent
clinical studies have also supported these findings, demonstrating
that the phosphorylation of STAT5 was enhanced by RIPC shortly
after reperfusion (10,22). Therefore, the inability to detect
the activation of STAT5 in the current rat model may have been be
due to species-dependent differences in signal transcription
factors.
It is widely accepted that RIPC treatment induces a
signal (e.g. humoral factors) release at the conditioned organ or
tissue into the blood stream (5).
After reaching the target organ (heart), these humoral factors
confer cardioprotective properties through activating signaling
cascades (5). Thus, a whole
organism is essential to study these mechanisms induced by RIPC
treatment and therefore, we selected an in vivo rat model of
I/R injury for the present study.
There are several limitations to the present study.
Firstly, the applied protocol for I/R, more specifically,
concerning the time intervals of I/R, has been successfully
established for analyzing the effects on infarct size and
hemodynamics; however, it may not be sufficient for detecting the
phosphorylation levels of AKT and STAT5. Therefore, a possible
activation of the respective targets at different TPs following
RIPC with or without I/R is conceivable and cannot be clarified
with the present experimental settings. Furthermore, the possible
activation of AKT via its second phosphorylation site, threonine
(Thr)308, was not analyzed. However, all other previous
studies discussed in this manuscript also only analyzed the
phosphorylation levels at Ser473. Therefore, the
additional detection of AKT phosphorylation levels at
Thr308 should be analyzed in future studies. In
addition, due to the study design, an analysis of the time effects
within groups was not possible. Finally, a possible translocation
of phosphorylated STAT5 into the nucleus or mitochondria was not
investigated in the present study, but it seems to be of great
interest with regards to the modulation of energy metabolism during
cardioprotection.
In conclusion, the findings of the present study
using an in vivo rat model revealed that neither the
phosphorylation of AKT nor STAT5 mediated the cardioprotective
effects of RIPC at the measured TPs, that is, 10 min after RIPC
treatment or 2 h after reperfusion. Due to the controversial
findings in the previous studies, the association between the
possible transient activation of phosphorylated AKT/STAT5 and the
cardioprotective properties needs to be investigated
comprehensively in future studies.
Acknowledgements
In partial fulfillment of the requirements for an MD
thesis (Christian Reiter).
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
AR analyzed and interpreted the data and wrote the
manuscript. KF helped to analyze the data and wrote the manuscript.
NH performed the in vivo experiments. CR performed the
western blot analysis. TB helped to analyze and interpret the data.
AH helped to design the study. MWH helped to analyze and interpret
the data. RH helped to design the study, analyzed and interpreted
the data and wrote the manuscript. CT analyzed and interpreted the
data and wrote the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The current study was performed after obtaining
approval from the State Agency for Nature, Environment and Consumer
Protection (LANUV), North-Rhine Westphalia, Germany
(8.87-50.10.37.09.148). The current study complies with the ARRIVE
guidelines.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Global Health Estimates 2016: Deaths by
Cause, Age, Sex, by Country and by Region, 2000-2016. Geneva, World
Health Organization, 2018.
|
|
2
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ferdinandy P, Hausenloy DJ, Heusch G,
Baxter GF and Schulz R: Interaction of risk factors, comorbidities,
and comedications with ischemia/reperfusion injury and
cardioprotection by preconditioning, postconditioning, and remote
conditioning. Pharmacol Rev. 66:1142–1174. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Murry CE, Jennings RB and Reimer KA:
Preconditioning with ischemia: A delay of lethal cell injury in
ischemic myocardium. Circulation. 74:1124–1136. 1986.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Heusch G: Molecular basis of
cardioprotection: signal transduction in ischemic pre-, post-, and
remote conditioning. Circ Res. 116:674–699. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Cho YJ and Kim WH: Perioperative
cardioprotection by remote ischemic conditioning. Int J Mol Sci.
20(4839)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Rossello X and Yellon DM: The RISK pathway
and beyond. Basic Res Cardiol. 113(2)2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Li J, Xuan W, Yan R, Tropak MB,
Jean-St-Michel E, Liang W, Gladstone R, Backx PH, Kharbanda RK and
Redington AN: Remote preconditioning provides potent
cardioprotection via PI3K/Akt activation and is associated with
nuclear accumulation of β-catenin. Clin Sci (Lond). 120:451–462.
2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hadebe N, Cour M and Lecour S: The SAFE
pathway for cardioprotection: Is this a promising target? Basic Res
Cardiol. 113(9)2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wu Q, Wang T, Chen S, Zhou Q, Li H, Hu N,
Feng Y, Dong N, Yao S and Xia Z: Cardiac protective effects of
remote ischaemic preconditioning in children undergoing tetralogy
of fallot repair surgery: A randomized controlled trial. Eur Heart
J. 39:1028–1037. 2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Heusch G, Musiolik J, Kottenberg E, Peters
J, Jakob H and Thielmann M: STAT5 activation and cardioprotection
by remote ischemic preconditioning in humans: Short communication.
Circ Res. 110:111–115. 2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Chen H, Jing XY, Shen YJ, Wang TL, Ou C,
Lu SF, Cai Y, Li Q, Chen X, Ding YJ, et al: Stat5-dependent
cardioprotection in late remote ischaemia preconditioning.
Cardiovasc Res. 114:679–689. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Weber NC, Riedemann I, Smit KF, Zitta K,
van de Vondervoort D, Zuurbier CJ, Hollmann MW, Preckel B and
Albrecht M: Plasma from human volunteers subjected to remote
ischemic preconditioning protects human endothelial cells from
hypoxia-induced cell damage. Basic Res Cardiol.
110(17)2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Hildebrandt HA, Kreienkamp V, Gent S,
Kahlert P, Heusch G and Kleinbongard P: Kinetics and signal
activation properties of circulating factor(s) from healthy
volunteers undergoing remote ischemic pre-conditioning. JACC Basic
Transl Sci. 1:3–13. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Kilkenny C, Browne WJ, Cuthill IC, Emerson
M and Altman DG: Improving bioscience research reporting: The
ARRIVE guidelines for reporting animal research. PLoS Biol.
8(e1000412)2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Brandenburger T, Huhn R, Galas A, Pannen
BH, Keitel V, Barthel F, Bauer I and Heinen A: Remote ischemic
preconditioning preserves connexin 43 phosphorylation in the rat
heart in vivo. J Transl Med. 12(228)2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Chow SC, Shao J and Wang H: Sample Size
Calculations in Clinical Research. 2nd edition. Chapman &
Hall/CRC Biostatistics Series, pp58. 2008.
|
|
18
|
Donato M, Goyeneche MA, Garces M, Marchini
T, Pérez V, Del Mauro J, Höcht C, Rodriguez M, Evelson P and Gelpi
RJ: Myocardial triggers involved in activation of remote ischaemic
preconditioning. Exp Physiol. 101:708–716. 2016.PubMed/NCBI View
Article : Google Scholar
|
|
19
|
Skyschally A, Gent S, Amanakis G, Schulte
C, Kleinbongard P and Heusch G: Across-species transfer of
protection by remote ischemic preconditioning with species-specific
myocardial signal transduction by reperfusion injury salvage kinase
and survival activating factor enhancement pathways. Circ Res.
117:279–288. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Ma LL, Kong FJ, Guo JJ, Zhu JB, Shi HT, Li
Y, Sun RH and Ge JB: Hypercholesterolemia abrogates remote ischemic
preconditioning-induced cardioprotection: Role of reperfusion
injury salvage kinase signals. Shock. 47:363–369. 2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Hong J, Ge HW, Liu JQ, Sun RH and Kong FJ:
Pharmacological inhibition of PTEN restores remote ischemic
postconditioning cardioprotection in hypercholesterolemic mice:
Potential role of PTEN/AKT/GSK3β SIGNALS. Shock. 52:522–531.
2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Gedik N, Thielmann M, Kottenberg E, Peters
J, Jakob H, Heusch G and Kleinbongard P: No evidence for activated
autophagy in left ventricular myocardium at early reperfusion with
protection by remote ischemic preconditioning in patients
undergoing coronary artery bypass grafting. PLoS One.
9(e96567)2014.PubMed/NCBI View Article : Google Scholar
|