1. Introduction
Acute respiratory distress syndrome (ARDS) was first
described in 1967 by Ashbaugh et al (1) in a patient suffering from sudden onset
of dyspnea that was resistant to standard oxygen therapy,
accompanied by a loss of lung compliance and diffuse alveolar
infiltrates. The original definition of ARDS has since been
updated, and the disease is now commonly defined according to the
‘Berlin’ definition proposed by the European Society of Intensive
Care Medicine ARDS Definition Task Force in 2012(2). ARDS is considered a significant health
and economic burden. The incidence of ARDS in the intensive care
units (ICUs) of 50 countries was 10.4%. Furthermore, the incidence
per ICU bed for four weeks in Europe was 0.48; North America, 0.46;
South America, 0.31; Asia, 0.27; Africa, 0.32; and Oceania, 0.57
cases, accounting for 23.4% of all patients who need mechanical
ventilation. Despite advances in supportive treatment, the
mortality of ARDS is still high. As the severity of ARDS increases,
40% of patients with ARDS died in hospitals (3). In China, the prevalence of ARDS among
patients in ICUs can be as high as 4.5%, with a mortality rate of
52% (4,5). Moreover, the long-term prognosis for
ARDS survivors is poor; in fact, a previous study showed that at 5
years, up to 28% of survivors exhibited a decreased capacity for
self-care, as well as various physiological and psychological
sequelae, thereby requiring continuous treatment (4). Clinical ARDS treatment mainly
comprises mechanical ventilation in a prone position and/or
extracorporeal membrane oxygenation (6). Ventilator management varies with the
severity of ARDS:35.1% of patients generally have tidal volumes
above 8 ml/kg predicted body weight, whereas 82.6% received
positive and end-expiratory pressure (PEEP) of <12 cm
H2O. However, from mild to moderate to severe cases, the
decrease in tidal volume and the increase in PEEP could be
beneficial, and the data were statistically significant. A recent
study also showed that, compared with a low PEEP strategy, higher
PEEP did not significantly decrease baro trauma, new organ failure,
or ventilator-free days (medium-level evidence) (7). Compared with control ventilation,
maximal lung recruitment did not reduce the number of days without
ventilation or mortality, which was associated with the increase in
cardiovascular adverse events (8).
Venous extracorporeal membrane oxygenation can be considered as a
strategy for patients with ARDS at the early stages to receive lung
protective ventilation. However, effective drug treatments are not
available for ARDS, and ARDS-associated mortality and disability
have not significantly decreased in recent years. Steroids may be
the most widely studied and discussed drugs for ARDS treatment,
with the results still being considered controversial. Previous
studies (9,10) have confirmed that patients with ARDS
taking methylprednisolone did not result in decreased mortality,
and prolonged steroid use was associated with an increased
mortality compared with that in the placebo group. However, other
studies provide contradicting evidence, demonstrating that patients
receiving steroids (methylprednisolone or hydrocortisone) had a
higher rate of spontaneous breathing on day 28 and lower
in-hospital mortality, and early administration of dexamethasone as
one can decrease the duration of mechanical ventilation and total
mortality in patients with confirmed moderate to severe ARDS
(11). Although it is possible that
subgroups of patients with ARDS will benefit from steroids, it is
unknown who these patients are. In addition, given the underlying
pathological heterogeneity of patients with confirmed ARDS, those
who respond may, in fact, have undiagnosed inflammatory lung
disease, such as tissue pneumonia. The most common adverse effect
of steroid use was hyperglycemia, while other potential side
effects of therapeutic steroids include neuromuscular weakness,
immunosuppression, severe infections, as well as sodium retention,
which is associated with adverse outcomes (11). Thus, presently, steroids are not
recommended for routine treatment of ARDS and further studies
regarding their benefits in patients with ARDS are needed.
Of the various identified infectious and
non-infectious factors, severe sepsis remains a primary cause of
ARDS (12). Acute lung injury (ALI)
is commonly used in experiments to study ARDS.
Histone acetyltransferases (HAT) and deacetylases
(HDACs) are widely expressed in the airways, and they modulate
histone acetylation and deacetylation, respectively (13). HDAC6 is a class-IIb HDAC that,
uniquely, contains two independent catalytic domains (14-18).
While both catalytic domains must remain intact to facilitate
HDAC6-driven deacetylation, each selectively recognizes and
interacts with specific HDAC6 substrates (17), including α-tubulin, heat shock
protein 90 (HSP90), and cortactin (19-22).
Notably, HDAC6 deregulation has been shown to
mediate disease processes including carcinogenesis,
neurodegeneration and autoimmunity (14,15,23).
Accordingly, several HDAC6 and pan-HDAC inhibitors have been
approved for anti-neoplastic clinical trials (24-26).
Recent studies have focused on the potential role of HDAC6 in
inflammation, after Halili et al (27) first demonstrated that HDAC6
inhibition represses macrophage inflammatory responses to
lipopolysaccharide (LPS). Several research groups have since
revealed a role for HDAC6 in sepsis pathogenesis and have shown
that its inhibition protects against both inflammation and
sepsis-induced endothelial barrier dysfunction. These findings
support further investigation of HDAC6 inhibition as a promising
potential therapeutic strategy targeting HDAC6, thereby attenuating
sepsis-induced ARDS.
2. Sepsis-induced ARDS and HDAC6
Sepsis is a systemic disease induced by an
overwhelming infection, resulting in multiple organ system
dysfunction, including ARDS. Inflammation and endothelial barrier
dysfunction are important pathophysiological changes that promote
the incidence and development of sepsis-induced ARDS.
ARDS/ALI is a group of clinically and biologically
heterogeneous diseases. Various pathogenic factors induce increased
pulmonary capillary permeability and cause pneumonia edema, leading
to acute respiratory failure. The characteristic change associated
with ARDS/ALI is refractory hypoxemia, which is difficult to be
corrected through oxygen inhalation and whose common inducements
are sepsis, aspiration pneumonia, shock and trauma, among others.
Treatments include ventilator support, prone position ventilation,
treatment of primary disease, and restriction of liquid intake.
Despite these, ARDS/ALI still show high mortality with a lack of
effective drug treatment (28).
Furthermore, the pathogenesis of ALI is complex, involving a series
of cytokines and inflammatory factors, which can cause vascular
endothelial and alveolar epithelial cell damage, alveolar collapse,
and pulmonary edema. At present, research on the mechanism of ALI
induced by sepsis mainly includes inflammatory pathways, oxidative
stress, apoptosis and endothelial barrier dysfunction. Inflammation
is the protective native immune response of an organism to a
pathogen or tissue injury, which can act as a double-edged sword;
when overactivated or not activated properly, it can lead to some
immune disorders, septic shock, rheumatoid arthritis and
neurodegenerative diseases. Pathogenic factors induce inflammation
by modulating signaling pathways such as the HMGB-RAGE-NF-κB and
MAPK pathways to produce a large number of pro-inflammatory factors
and mediators that can eventually cause sepsis. Oxidative stress is
an important cause of multiple organ damage in sepsis (29), and plays an important role in ALI
caused by various inducements. It can increase lung water content,
damage the integrity of pulmonary endothelial barrier function, and
lead to lung injury. At present, it is generally believed that
decreasing oxidative stress can likewise lower lung injury
(30-32).
The important factors involved in oxidative stress include nitrous
oxide, PGE2, iNOS, COX-2, SPD and other factors that cause damage
to alveolar capillaries. Apoptosis is also an important factor in
the progression of sepsis-induced ARDS/ALI. On one hand, it
directly destroys the integrity of the endothelial barrier through
endothelial cell apoptosis, and on the other, it has been reported
that the activity of apoptosis-associated proteins such as
caspase-3 can lead to the rearrangement of cell junction proteins
(such as VE-cadherin and claudin-5). Lastly, in recent years,
research on the function of endothelial cell barrier in ALI has
gradually increased. At present, one of the main signaling pathways
found to be involved in endothelial cell barrier function is the
S1P-SIP1 signaling pathway; with the activation of Gi
protein-combined SIP1 receptor and downstream small GTPase Rac
molecule, S1P induces cortactin transfer and peripheral myosin long
chain (MLC) phosphorylation, which can strengthen the rearrangement
of cytoskeleton structure and form a cortical actin ring, and
strengthen the adhesion connection, tight connection, and local
adhesion complex on the cell surface, thereby decreasing pulmonary
edema induced by LPS in ALI (33).
In addition, the following are some pathways and systems that are
also involved: Ang-2-Tie-2-MLC, renin-angiotensin system (34), Rho-ROCK-LIMK1-MLC, eNOS-Cav1-MLC2
signaling (35), PAR1 moesin
(36), HDAC6-Hsp90 and
α-tubulin-β-catenin (37-39).
HDAC6 is extensively expressed in normal airways,
where it promotes microtubule destabilization and endothelial
barrier hyper-permeability (40).
Accordingly, HDAC6 inhibition has been shown to improve survival in
murine sepsis models, either by blocking endothelial barrier
hyper-permeability or by attenuating airway inflammation (41-46),
as will be further discussed (Fig.
1).
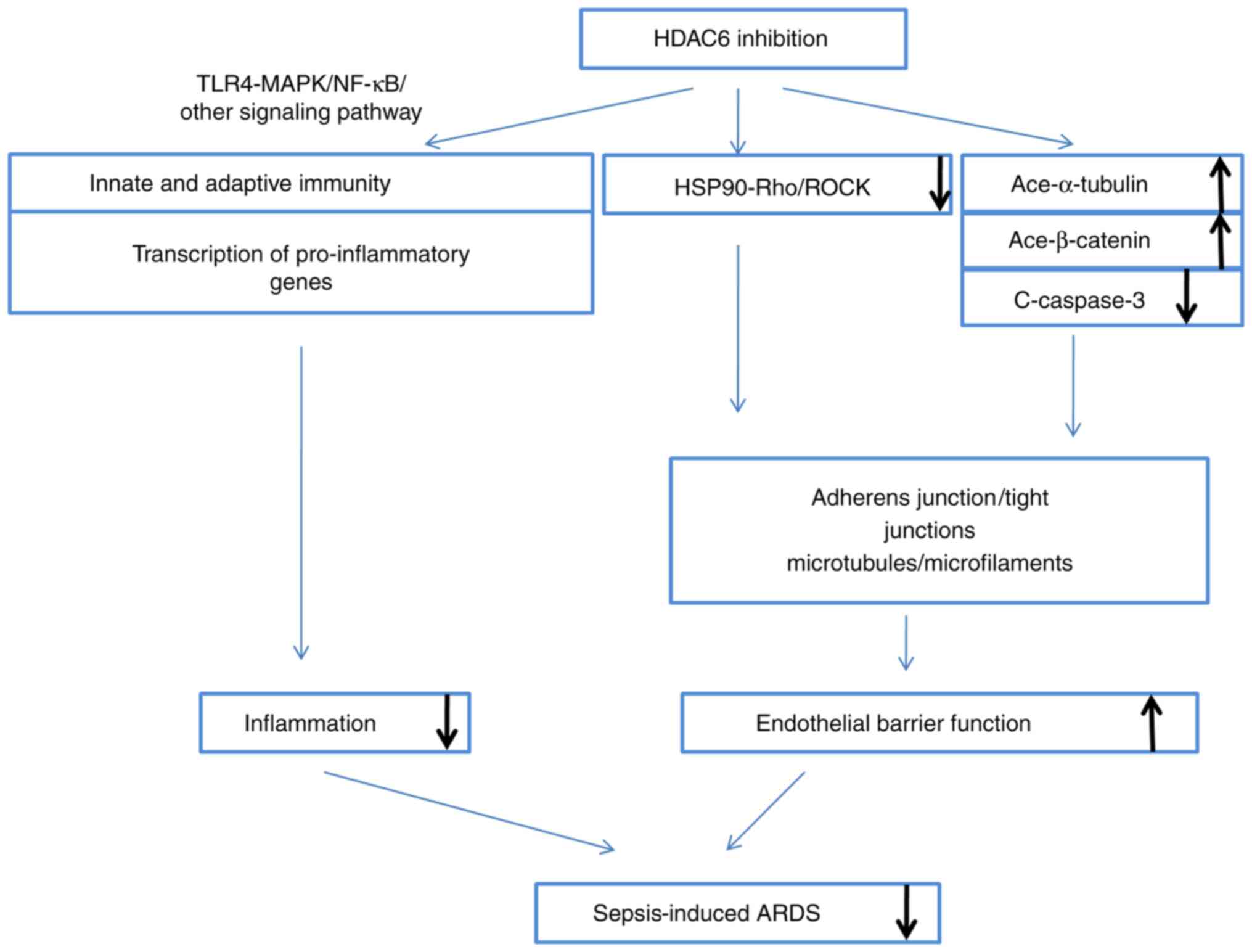 | Figure 1HDAC6 inhibition ameliorates
sepsis-induced ARDS. The inhibition occurs mainly by two ways: i)
Modulation of innate and adaptive immune cells, and the
transcription of pro-inflammatory genes, via the TLR4-MAPK/NF-κB
pathway, which directly attenuates inflammation; and ii) protection
of endothelial barrier function via HSP90, α-tubulin, β-catenin and
caspase-3, thereby enhancing adherents and strengthening the
cytoskeleton. HDAC6, histone deacetylase 6; ARDS, acute respiratory
distress syndrome; TLR, Τoll-like receptor; NF-κB, nuclear
factor-κB; HSP90, heat shock protein 90. |
3. HDAC6 inhibition protects against
inflammation
Halili et al (27) first demonstrated that the HDAC6
inhibitor 17a effectively inhibits LPS-induced pro-inflammatory
endothelin (Edn)-1 and interleukin (IL)-12p40 RNA expression in
macrophages. Various studies have since shown that HDAC6 directly
mediates inflammation and both the innate (i.e., pathogen sensing
and destruction) and adaptive immunity, as well as the modulation
of the transcription of pro-inflammatory genes (47,48).
Role of HDAC6 in innate and adaptive
immunity cells
Inflammation is a protective innate immune response
to invading pathogens and helps to prevent tissue damage. It acts
like a ‘double-edged sword’ and, upon overactivation or improper
activation, causes serious immune disorders, systemic inflammatory
response syndrome, compensatory anti-inflammatory response
syndrome, innate immune and adaptive immune disorders, and
subsequently leads to multiple organ dysfunction such as ARDS. The
process of inflammation begins with the influx of neutrophils,
followed by the recruitment of monocytes, which then differentiate
into inflammatory macrophages or dendritic cells. These cells are
the key effector cells in the inflammatory site, recognizing and
phagocytizing pathogens and necrotic cells to help eliminate
infection. In addition, they activate the adaptive immune response
through communication and coordination with other immune cells.
Macrophages are the main effector cells in the inflammatory
response and play a key role in initiation, regression, tissue
repair, and regeneration of the inflammatory response (49). Monocytes and macrophages, as the
most efficient pathogen scavengers and the main source of
inflammatory cytokines, are the key effector cells that modulate
the innate immune response of the body. However, the progressive
dysfunction of monocytes and macrophages also leads to immune
dysfunction during severe sepsis and septic shock. Thus, HDAC6
inhibition attenuates macrophage-induced inflammation by inhibiting
the overproduction of reactive oxygen species (ROS) and modulating
the expression of pro-inflammatory cytokines induced by LPS
(50). A study has also shown that
HDAC6 deficiency impairs macrophage recruitment to the inflammation
site in a murine model of acute peritonitis, which may suppress the
phagocytic capacity of macrophages, but at the same time serves as
a promising therapeutic strategy for the treatment of
macrophage-associated immune diseases (49); nonetheless, further research is
still needed. The increased preoperative neutrophil lymphocyte
ratio and high and middle ratio of neutrophils can have poor
prognosis in patients with cancer. In addition, the proportion of
neutrophils are found to be significantly higher, with the
lymphocyte count significantly lower, in critical patients with
severe sepsis or septic shock. According to the sequential organ
failure assessment (SOFA) and acute physiological and chronic
health assessment II (APACHE II) scores, the severity of the
clinical course is associated with the difference in the percentage
of neutrophils and lymphocytes in leukocytes. A selective HDAC6
inhibitor, tubastatinA (TubA) has been demonstrated to alter the
composition of blood cells, restore the lymphocyte population, and
decrease the granulocyte-to-lymphocyte ratio by improving the
monocyte and granulocyte count in a murine model of cecal ligation
and puncture (CLP), providing another explanation for the increase
insurvival outcome in septic death model. Furthermore, selective
HDAC6 inhibition is needed to restore innate immune cells in the
bone marrow, lower stress responses, immune organ atrophy and
apoptosis (43,48,51,52).
HDAC6 modulates pro-inflammatory gene
transcription
Cytokines, chemokines and adhesion molecules are
important pro-inflammatory mediators, and HDAC6 overexpression
increases the production of pro-inflammatory factors in
macrophages. Conversely, HDAC6 inhibition suppresses their
expression and circulation, such as tumor necrosis factor (TNF)-α
and IL-6 in peritoneal fluids, decreases myeloperoxidase (MPO)
production, and increases IL-10 production (45,50,53).
HDAC6 inhibition has also been reported to decrease the expression
of adhesion molecules and chemokines, including C-C motif ligand 2
(CCL-2), C-X-C motif (CXC)L-8 and CXCL-10. Additionally, HDAC6
represses extracellular signal-regulated kinase (ERK), c-Jun
N-terminal kinase (JNK), p38 and nuclear factor (NF)-κB. HDAC6 also
represses activator protein (AP)-1 activation in astrocytes
responding to the human immunodeficiency virus (HIV)-1
transactivator of transcription (Tat) protein, by modulating ROS
homeostasis, mitogen-activated protein kinase (MAPK), NF-κB and
AP-1 signaling (54,55). In summary, HDAC6 mediates HIV-1
Tat-induced pro-inflammatory response by regulating the MAPK, NF-κB
and AP-1 signaling pathways in astrocytes. Studies showed that
hindsiipropane B inhibits the expression of HDAC6 induced by HIV-1
Tat, and subsequently inhibiting the expression of CCL2, CXCL8 and
CXCL10 mediated by the former. Another study confirmed that
hindsiipropane B inhibits the expression of CCL2, CXCL8 and CXCL10
by inhibiting the HDAC6-NADPH oxidase-ROS-MAPK-NF-κB-AP-1 axis in
astrocytes (56). Zhang et
al (50,57) similarly found that selective HDAC6
inhibition decreases TNF-α, IL-1β and IL-6 expression in both
LPS-activated RAW264.7 cells and a murine model of acute liver
failure by modulating oxidative stress and toll-like receptor
4(TLR4)-MAPK-NF-κB signaling. Liu et al (58) and Wang et al (59) have since confirmed that HDAC6
inhibition protects against LPS-induced inflammation by suppressing
NF-κB signaling. It also decreases IL-1β expression, as well as
caspase-1 cleavage and activation.
Interferon regulatory factor 3 (IRF-3) is a
transcription factor that critically mediates interferon (IFN)
secretion during inflammation (60). Interestingly, Nusinzon and Horvath
(61) showed that HDAC6 modulates
IRF-3 activity by deacetylating its coactivator, β-catenin.
Moreover, Chattopadhyay et al (62) demonstrated that IRF-3, β-catenin and
CBP form a stable complex in response to viral or bacterial
infection-stimulated TLR-3 signaling to induce IFN production.
Importantly, HDAC6 is required for the formation of this complex,
since its inhibition modulates β-catenin acetylation, thereby
suppressing the interaction of IRF-3 with CBP.
4. HDAC6 inhibition restores endothelial
barrier hyper-permeability
The endothelial barrier is a semi-permeable membrane
that maintains the intra- and extravascular balance of water and
proteins (63). Its composition has
previously been reported in detail; in brief, it comprises the
cytoskeleton and adherents such as adherens junction (AJ) and tight
junction (TJ) proteins, including claudins and occludins, as well
as zonula occludens (ZO) (52) and
local gap junction proteins (64).
Endothelial barrier dysfunction plays an important role in the
pathogenesis of sepsis-induced organ dysfunction; thus, endothelial
barrier protection is a proposed therapeutic modality to treat
sepsis. Nam et al (65)
previously demonstrated that Clostridium difficile toxin A
induces microtubule instability in a murine model of C.
Difficile infection by activating HDAC6, which in turn
modulates α-tubulin deacetylation, thereby inducing acute
inflammation. Moreover, it is now well known that HDAC6 inhibition
promotes endothelial barrier hyper-permeability (37-39,66,67).
HDAC6 modulates HSP90-dependent
Rho-associated kinase (Rho-ROCK) activity
Hsp90 protein is a member of the heat shock
molecular chaperone family that regulates protein conformation and
activity, and regulates a variety of cell signaling pathways by
controlling the abundance and activity of several important protein
kinases and cell cycle-associated proteins, such as Rho GTPases and
actin, which is a tripartite framework for effective vesicular
transport. Rho GTPase is a subfamily of small GTP binding proteins
in the Ras super family, composed of Cdc42, Rac1 and RhoA, which
regulates actin dynamics in endothelial cells. For example, RhoA
induces endothelial hyper-permeability by modulating actin stress
fiber formation and cell contraction (68). ROCK increases actin filament
crosslinking activity of myosin II through the
phosphorylation-induced activation of MLC and inactivation of MLC
phosphatase. Inhibition of Rho kinase, the downstream effector of
RhoA, can protect HLMVECS from LPS-mediated high permeability and
eliminate LPS-induced phosphorylation of MLC. Hsp90 plays an
important and special role in regulating Rho activity and
Rho-dependent actin cytoskeleton remodeling. Hsp90 can indirectly
activate Rho GTPase by protecting functional activators from
proteasome degradation (69).
Consistent with these findings, the phosphorylation of MLC induced
by RhoA is also inhibited by 17-AAG, a Hsp90 inhibitor. Studies
have demonstrated that inhibition of Hsp90 can prevent and repair
LPS-induced pulmonary endothelial barrier dysfunction by inhibiting
RhoA activity and signal transduction (70). It is important to note that HSP90 is
a substrate of HDAC6. A previous study by Joshi et al
(37) proposed that HDAC6
inhibition protects against pulmonary endothelial barrier
dysfunction and ALI by modulating HSP90; HDAC6 inhibition
facilitates the acetylation of HSP90 at Lys294, which in turn
suppresses HSP90-dependent Rho-ROCK activity and blocks MLC
phosphorylation to provide protection against LPS-induced
endothelial barrier dysfunction. However, more studies are required
to verify this mechanism.
HDAC6 modulates the acetylation of
α-tubulin and β-catenin
Vascular endothelial (VE)-cadherin and β-catenin are
major AJ components. The majority of β-catenin molecules localize
to the cytoplasmic side of the membrane, where they contact
VE-cadherin (71). In parallel,
β-catenin degradation is controlled by its N-terminal
phosphorylation and ubiquitination; for example, ubiquitination at
Lys49 triggers proteasomal β-catenin degradation (72). VE-cadherin contains five
extracellular cadherin repeats, a transmembrane region, and a
highly conserved cytoplasmic tail that has been shown to form a
complex with β-catenin to maintain endothelial barrier integrity
(73-75).
Microtubules and microfilaments are interacting
cytoskeletal components (76,77).
Microtubules are comprised of two globular proteins, α- and
β-tubulin, which polymerize to regulate endothelial barrier
function. Decreasing α-tubulin acetylation has been shown to affect
microtubule assembly, cytoskeletal stability and cell mobility
(78). Conversely, tubulin
acetylation prolongs the microtubule half-life, increases
cytoskeletal stability, and renders the cytoskeleton more resistant
to drug-induced depolymerization and disassembly (79). Previous studies have suggested that
microtubule disassembly stimulates actin formation and increases
MLC phosphorylation; in contrast, microtubule depolymerization
likely blocks these effects (80,81).
HDAC6 inhibition has been demonstrated to decrease
sepsis-induced endothelial barrier hyper-permeability by restoring
normal α-tubulin and β-catenin acetylation patterns (39,44).
Specifically, HDAC6 inhibition allows β-catenin to be acetylated at
Lys49, preventing its ubiquitination and increasing its
accumulation at the cell membrane, thus promoting the formation of
the VE-cadherin-β-catenin complex (82). Furthermore, HDAC6 inhibition has
been suggested to modulate α-tubulin and β-catenin acetylation by
decreasing caspase-3 cleavage (39,44).
Decreased caspase-3 activation protects endothelial
hyper-permeability, as evidenced by the fact that caspase-3
inhibition restores endothelial barrier permeability in the lung
tissue of septic mice (83).
Increased caspase-3 activity likely contributes to endothelial
barrier dysfunction by inducing endothelial cell apoptosis;
however, it has also been reported to trigger the rearrangement of
connexins, including VE-cadherin, ZO-1and claudin-5 (84-86).
5. A promising and highly selective HDAC6
inhibitor, TubA
In recent years, with an increase in the
understanding of HDAC6 and its roles in different diseases, a large
number of experimental studies on HDAC6 inhibitors have been
reported. There is an increasing number of studies on the role of
HDAC6 inhibition in inflammation, one of which involves the
following: Computer-aided identification of new selective
inhibitors of HDAC6 with anti-sepsis activity, development of new
HDAC inhibitors after virtual screening based on chemical
databases, compound 9A [(E)-n-hydroxy-4-(2-styrylthiazol-4-yl] was
identified as a HDAC6 selective inhibitor (IC50 value of
HDAC6 is 0.199 µm; HDAC, 8 µm). Compound 9A significantly increased
the survival of mice with sepsis induced by LPS and inhibited the
increase of TNF-α and IL-6 mRNA expression induced by LPS (46).
CAY is also a highly selective HDAC6 inhibitor,
which blocks the activation of NF-κB by inhibiting IκB
phosphorylation in LPS-induced ALI, can decrease the activity of
inflammatory corpuscles induced by LPS and decrease the cleavage
and activation of IL-1β and caspase-1(58).
TubA is considered to be a potent and highly
selective HDAC6 inhibitor. Its IC50 value is 15 nm,
1,000 times lower than that of other subtypes, except HDAC8 (57
times lower). TubA can improve the survival of mice with septicemic
induced by lethal CLP (42). In a
hemorrhagic shock model combined with CLP, the survival time of
mice in HDAC6 group was also prolonged (45). The mechanism included changing the
composition of circulating blood cells in lethal septicemia model
(48). Compared with the CLP Group,
the TubA treatment group recovered B lymphocytes and significantly
increased the percentage of innate immune cells and macrophages. In
addition, TubA could significantly decrease the bacterial load in
the spleen and increase the phagocytic capacity of macrophages in
RAW264.7 cells. In this lethal sepsis model, TubA significantly
decreased the stress response and thymus and bone marrow atrophy
(51). The apoptosis of spleen
cells also decreased. WT mice treated with TubA were partially
protected from vascular leakage and inflammation caused by HKSA or
methicillin-resistant Staphylococcus aureus (MRSA) (40). Thus, TubA plays an important
function in endothelial cell barrier protection, and the mechanisms
include an increase in the acetylation of α-tubulin and β-catenin
and a decrease in the activation of caspase-3 as previously
mentioned.
A brief introduction to the role of
TubA in ARDS
As mentioned above, most of the experiments on HDAC6
inhibitors in vitro and in vivo have been conducted with TubA.
Currently, TubA has been reported to prolong the survival period of
septic mice, decrease the ALI from sepsis, and lower the bacterial
load in spleen. It brings about these effects mainly by decreasing
the inflammatory response, restoring endothelial cell barrier
dysfunction, and decreasing the oxidative stress response, as well
as pulmonary fibrosis, by inhibiting the apoptosis of pulmonary
vascular endothelial cells (87).
Putative side effects of TubA
As the number of TubA studies increases,
increasingly more attention is being given to its possible side
effects. In one study, a relatively high dose was used (100 mg/kg
body weight of mice) and no significant adverse effects were
observed (88). However, in another
study it was found that TubA exposure significantly decreased the
formation of blastocysts in early embryos of mice, and confocal
microscopy showed that chromosome aggregation failed to occur in
mice embryos treated with HDAC6 inhibitors. In addition, the
inhibition of HDAC6 induced an excessive production of ROS. An
increase in the accumulation of phosphorylated γ-H2AX was also
observed in the embryos after TubA intervention indicating an
increase in DNA damage and blastocyst cell apoptosis (89). TubA interfered with and halted mouse
oocyte meiosis by regulating several key histones (H4K16
acetylation and h3t3 and H3S10 phosphorylation) and messenger RNA
(ccnb1, CDK2, Smad3 and YWHAZ methylation-associated genes DNMT1
and DNMT3b), and by interfering with the organization of spindles
of chromosomes, as well as the attachment of mitotic microtubules.
The first polar body of mouse blastocyst oocyte could not be
expelled after TubA treatment. However, it has been proved that
homozygous knockout of HDAC6 (KO) mice are viable and fertile.
Further studies have confirmed that the HDAC6 protein and mRNA
levels in mouse oocytes treated with TubA were significantly lower.
The SIRT2, HDAC6, SIRT6 and SIRT7 mRNA levels were also
significantly decreased. The mRNA expression levels of Cdk1, Cdk2,
Cdk4, Cdk6, Cdc25B, and other cell cycle-associated kinases
decreased significantly. These abnormalities were associated with
meiosis and cell aging. The abnormal meiotic maturation and cell
senescence induced by TubA may be the result of its interaction
with HDAC and sirtuin (90). It is
presumed that TubA can be used in the treatment of ARDS induced by
sepsis, but effects of HDAC6 on the reproductive function of female
should be of caution.
6. Conclusion
Inflammation and endothelial barrier dysfunction are
important processes that contribute to the pathogenesis of
sepsis-induced ARDS. HDAC6 has been reported to mediate
inflammation, as well as both the innate and adaptive immunity,
including inflammatory cell recruitment and bacterial clearance.
Accordingly, HDAC6 inhibition using an inhibitor such as TubA
increases the survival of septic mice by decreasing the LPS-induced
macrophage and epithelial cell inflammation. It is proposed that
HDAC6 inhibition predominantly ameliorates sepsis-induced ARDS by
modulating the innate and adaptive immunity and the transcription
of pro-inflammatory genes to directly attenuate inflammation. HDAC6
inhibition also protects endothelial barrier function via its
effects on its target substrates by inducing α-tubulin and
β-catenin acetylation and increasing membrane localization of
β-catenin, thereby leading to the stabilization of microtubule and
AJs. It also prevents caspase-3 activation, maintains lung
endothelial cell-cell junctions, and inhibits actin stress fiber
formation and decreases MLC phosphorylation via modulating HSP90.
These findings support that HDAC6 inhibition is a promising
potential therapeutic target to treat sepsis-induced ARDS. However,
the negative effects, if any, of HDAC6 inhibitors on embryo sac and
oocytes need to be taken into account.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science
Foundation of China (grant no. 81800080).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JYu analyzed and interpreted the data and final
approval of the version to be published. JYa analyzed and
interpreted the data and was a major contributor in writing the
manuscript. QZ was a contributor in writing the manuscript. YW and
DQ contributed to literature review. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ashbaugh DG, Bigelow DB, Petty TL and
Levine BE: Acute respiratory distress in adults. Lancet. 2:319–323.
1967.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ferguson ND, Fan E, Camporota L, Antonelli
M, Anzueto A, Beale R, Brochard L, Brower R, Esteban A, Gattinoni
L, et al: The Berlin definition of ARDS: An expanded rationale,
justification, and supplementary material. Intensive Care Med.
38:1573–1582. 2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bellani G, Laffey JG, Pham T, Fan E,
Brochard L, Esteban A, Gattinoni L, van Haren F, Larsson A, McAuley
DF, et al: LUNG SAFE Investigators; ESICM Trials Group:
Epidemiology, patterns of care, and mortality for patients with
acute respiratory distress syndrome in intensive care units in 50
countries. JAMA. 315:788–800. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Herridge MS, Tansey CM, Matté A, Tomlinson
G, Diaz-Granados N, Cooper A, Guest CB, Mazer CD, Mehta S, Stewart
TE, et al: Canadian Critical Care Trials Group: Functional
disability 5 years after acute respiratory distress syndrome. N
Engl J Med. 364:1293–1304. 2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Han S and Mallampalli RK: The acute
respiratory distress syndrome: From mechanism to translation. J
Immunol. 194:855–860. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Griffiths MJD, McAuley DF, Perkins GD,
Barrett N, Blackwood B, Boyle A, Chee N, Connolly B, Dark P, Finney
S, et al: Guidelines on the management of acute respiratory
distress syndrome. BMJ Open Respir Res. 6(e000420)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Walkey AJ, Del Sorbo L, Hodgson CL,
Adhikari NKJ, Wunsch H, Meade MO, Uleryk E, Hess D, Talmor DS,
Thompson BT, et al: Higher PEEP versus Lower PEEP Strategies for
Patients with Acute Respiratory Distress Syndrome. A Systematic
Review and Meta-Analysis. Ann Am Thorac Soc. 14 (Suppl
4):S297–S303. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Hodgson CL, Cooper DJ, Arabi Y, King V,
Bersten A, Bihari S, Brickell K, Davies A, Fahey C, Fraser J, et
al: Maximal Recruitment Open Lung Ventilation in Acute Respiratory
Distress Syndrome (PHARLAP). A Phase II, Multicenter Randomized
Controlled Clinical Trial. Am J Respir Crit Care Med.
200:1363–1372. 2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Steinberg KP, Hudson LD, Goodman RB, Hough
CL, Lanken PN, Hyzy R, Thompson BT and Ancukiewicz M: National
Heart, Lung and Blood Institute Acute Respiratory Distress Syndrome
(ARDS) Clinical Trials Network. Efficacy and safety of
corticosteroids for persistent acute respiratory distress syndrome.
N Engl J Med. 354:1671–1684. 2006.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Peck TJ and Hibbert KA: Recent advances in
the understanding and management of ARDS. F1000 Res.
8(8)2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Villar J, Ferrando C, Martínez D, Ambrós
A, Muñoz T, Soler JA, Aguilar G, Alba F, González-Higueras E,
Conesa LA, et al: dexamethasone in ARDS network: Dexamethasone
treatment for the acute respiratory distress syndrome: A
multicentre, randomised controlled trial. Lancet Respir Med.
8:267–276. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Rubenfeld GD, Caldwell E, Peabody E,
Weaver J, Martin DP, Neff M, Stern EJ and Hudson LD: Incidence and
outcomes of acute lung injury. N Engl J Med. 353:1685–1693.
2005.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ito K, Caramori G, Lim S, Oates T, Chung
KF, Barnes PJ and Adcock IM: Expression and activity of histone
deacetylases in human asthmatic airways. Am J Respir Crit Care Med.
166:392–396. 2002.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Sadoul K, Boyault C, Pabion M and Khochbin
S: Regulation of protein turnover by acetyltransferases and
deacetylases. Biochimie. 90:306–312. 2008.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Dekker FJ and Haisma HJ: Histone acetyl
transferases as emerging drug targets. Drug Discov Today.
14:942–948. 2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zou H, Wu Y, Navre M and Sang BC:
Characterization of the two catalytic domains in histone
deacetylase 6. Biochem Biophys Res Commun. 341:45–50.
2006.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhang Y, Gilquin B, Khochbin S and
Matthias P: Two catalytic domains are required for protein
deacetylation. J Biol Chem. 281:2401–2404. 2006.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Schäfer S, Saunders L, Eliseeva E, Velena
A, Jung M, Schwienhorst A, Strasser A, Dickmanns A, Ficner R and
Schlimme S: Phenylalanine-containing hydroxamic acids as selective
inhibitors of class IIb histone deacetylases (HDACs). Bioorg Med
Chem. 16:2011–2033. 2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhang X, Yuan Z, Zhang Y, Yong S,
Salas-Burgos A, Koomen J, Olashaw N, Parsons JT, Yang XJ, Dent SR,
et al: HDAC6 modulates cell motility by altering the acetylation
level of cortactin. Mol Cell. 27:197–213. 2007.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Bali P, Pranpat M, Bradner J, Balasis M,
Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, et
al: Inhibition of histone deacetylase 6 acetylates and disrupts the
chaperone function of heat shock protein 90: A novel basis for
antileukemia activity of histone deacetylase inhibitors. J Biol
Chem. 280:26729–26734. 2005.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Parab S, Shetty O, Gaonkar R, Balasinor N,
Khole V and Parte P: Correction to: HDAC6 deacetylates alpha
tubulin in sperm and modulates sperm motility in Holtzman rat. Cell
Tissue Res. 371(375)2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Wang XX, Wan RZ and Liu ZP: Recent
advances in the discovery of potent and selective HDAC6 inhibitors.
Eur J Med Chem. 143:1406–1418. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Jia YJ, Liu ZB, Wang WG, Sun CB, Wei P,
Yang YL, You MJ, Yu BH, Li XQ and Zhou XY: HDAC6 regulates
microRNA-27b that suppresses proliferation, promotes apoptosis and
target MET in diffuse large B-cell lymphoma. Leukemia. 32:703–711.
2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Vogl DT, Raje N, Jagannath S, Richardson
P, Hari P, Orlowski R, Supko JG, Tamang D, Yang M, Jones SS, et al:
Ricolinostat, the first selective histone deacetylase 6 inhibitor,
in combination with bortezomib and dexamethasone for relapsed or
refractory multiple myeloma. Clin Cancer Res. 23:3307–3315.
2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Yee AJ, Bensinger WI, Supko JG, Voorhees
PM, Berdeja JG, Richardson PG, Libby EN, Wallace EE, Birrer NE,
Burke JN, et al: Ricolinostat plus lenalidomide, and dexamethasone
in relapsed or refractory multiple myeloma: A multicentre phase 1b
trial. Lancet Oncol. 17:1569–1578. 2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Tu Y, Hershman DL, Bhalla K, Fiskus W,
Pellegrino CM, Andreopoulou E, Makower D, Kalinsky K, Fehn K,
Fineberg S, et al: A phase I-II study of the histone deacetylase
inhibitor vorinostat plus sequential weekly paclitaxel and
doxorubicin-cyclophosphamide in locally advanced breast cancer.
Breast Cancer Res Treat. 146:145–152. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Halili MA, Andrews MR, Labzin LI, Schroder
K, Matthias G, Cao C, Lovelace E, Reid RC, Le GT, Hume DA, et al:
Differential effects of selective HDAC inhibitors on macrophage
inflammatory responses to the Toll-like receptor 4 agonist LPS. J
Leukoc Biol. 87:1103–1114. 2010.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Estenssoro E and Dubin A: Acute
respiratory distress syndrome. Medicina (B Aires). 76:235–241.
2016.PubMed/NCBI(In Spanish).
|
|
29
|
Kasznica J, Helmann M, Collins JP and
Akhtar R: Bilateral Ebstein-like anomaly with atrial septal defect.
Jpn Heart J. 36:119–125. 1995.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Fu P, Murley JS, Grdina DJ, Birukova AA
and Birukov KG: Induction of cellular antioxidant defense by
amifostine improves ventilator-induced lung injury. Crit Care Med.
39:2711–2721. 2011.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Kratzer E, Tian Y, Sarich N, Wu T, Meliton
A, Leff A and Birukova AA: Oxidative stress contributes to lung
injury and barrier dysfunction via microtubule destabilization. Am
J Respir Cell Mol Biol. 47:688–697. 2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Zhao B, Gao W, Gao X, Leng Y, Liu M, Hou J
and Wu Y: Sulforaphane attenuates acute lung injury by inhibiting
oxidative stress via Nrf2/HO-1 pathway in a rat sepsis model. Int J
Clin Exp Pathol. 10:9021–9028. 2017.PubMed/NCBI
|
|
33
|
Peng X, Hassoun PM, Sammani S, McVerry BJ,
Burne MJ, Rabb H, Pearse D, Tuder RM and Garcia JG: Protective
effects of sphingosine 1-phosphate in murine endotoxin-induced
inflammatory lung injury. Am J Respir Crit Care Med. 169:1245–1251.
2004.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Kong J, Zhu X, Shi Y, Liu T, Chen Y, Bhan
I, Zhao Q, Thadhani R and Li YC: VDR attenuates acute lung injury
by blocking Ang-2-Tie-2 pathway and renin-angiotensin system. Mol
Endocrinol. 27:2116–2125. 2013.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Thangavel J, Malik AB, Elias HK, Rajasingh
S, Simpson AD, Sundivakkam PK, Vogel SM, Xuan YT, Dawn B and
Rajasingh J: Combinatorial therapy with acetylation and methylation
modifiers attenuates lung vascular hyperpermeability in
endotoxemia-induced mouse inflammatory lung injury. Am J Pathol.
184:2237–2249. 2014.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Xu Q, Liu J, Wang Z, Guo X, Zhou G, Liu Y,
Huang Q and Su L: Heat stress-induced disruption of endothelial
barrier function is via PAR1 signaling and suppressed by Xuebijing
injection. PLoS One. 10(e0118057)2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Joshi AD, Barabutis N, Birmpas C,
Dimitropoulou C, Thangjam G, Cherian-Shaw M, Dennison J and
Catravas JD: Histone deacetylase inhibitors prevent pulmonary
endothelial hyperpermeability and acute lung injury by regulating
heat shock protein 90 function. Am J Physiol Lung Cell Mol Physiol.
309:L1410–L1419. 2015.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Borgas D, Chambers E, Newton J, Ko J,
Rivera S, Rounds S and Lu Q: Cigarette Smoke Disrupted Lung
Endothelial Barrier Integrity and Increased Susceptibility to Acute
Lung Injury via Histone Deacetylase 6. Am J Respir Cell Mol Biol.
54:683–696. 2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Yu J, Ma Z, Shetty S, Ma M and Fu J:
Selective HDAC6 inhibition prevents TNF-α-induced lung endothelial
cell barrier disruption and endotoxin-induced pulmonary edema. Am J
Physiol Lung Cell Mol Physiol. 311:L39–L47. 2016.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Karki P, Ke Y, Tian Y, Ohmura T, Sitikov
A, Sarich N, Montgomery CP and Birukova AA: Staphylococcus
aureus-induced endothelial permeability and inflammation are
mediated by microtubule destabilization. J Biol Chem.
294:3369–3384. 2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Rosenjack J, Hodges CA, Darrah RJ and
Kelley TJ: HDAC6 depletion improves cystic fibrosis mouse airway
responses to bacterial challenge. Sci Rep. 9(10282)2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Deng Q, Zhao T, Pan B, Dennahy IS, Duan X,
Williams AM, Liu B, Lin N, Bhatti UF, Chen E, et al: Protective
Effect of Tubastatin A in CLP-Induced Lethal Sepsis. Inflammation.
41:2101–2109. 2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Li Y, Zhao T, Liu B, Halaweish I,
Mazitschek R, Duan X and Alam HB: Inhibition of histone deacetylase
6 improves long-term survival in a lethal septic model. J Trauma
Acute Care Surg. 78:378–385. 2015.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Yu J, Ma M, Ma Z and Fu J: HDAC6
inhibition prevents TNF-α-induced caspase 3 activation in lung
endothelial cell and maintains cell-cell junctions. Oncotarget.
7:54714–54722. 2016.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Cheng X, Liu Z, Liu B, Zhao T, Li Y and
Alam HB: Selective histone deacetylase 6 inhibition prolongs
survival in a lethal two-hit model. J Surg Res. 197:39–44.
2015.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Yoo J, Kim SJ, Son D, Seo H, Baek SY,
Maeng CY, Lee C, Kim IS, Jung YH, Lee SM, et al: Computer-aided
identification of new histone deacetylase 6 selective inhibitor
with anti-sepsis activity. Eur J Med Chem. 116:126–135.
2016.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Moreno-Gonzalo O, Mayor F Jr and
Sánchez-Madrid F: HDAC6 at Crossroads of Infection and Innate
Immunity. Trends Immunol. 39:591–595. 2018.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Zhao T, Li Y, Liu B, Pan B, Cheng X,
Georgoff P and Alam HB: Inhibition of histone deacetylase 6
restores innate immune cells in the bone marrow in a lethal septic
model. J Trauma Acute Care Surg. 80:34–40; discussion 40-41.
2016.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Yan B, Xie S, Liu Y, Liu W, Li D, Liu M,
Luo HR and Zhou J: Histone deacetylase 6 modulates macrophage
infiltration during inflammation. Theranostics. 8:2927–2938.
2018.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Zhang WB, Yang F, Wang Y, Jiao FZ, Zhang
HY, Wang LW and Gong ZJ: Inhibition of HDAC6 attenuates LPS-induced
inflammation in macrophages by regulating oxidative stress and
suppressing the TLR4-MAPK/NF-κB pathways. Biomed Pharmacother.
117(109166)2019.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Zhao T, Li Y, Bronson RT, Liu B, Velmahos
GC and Alam HB: Selective histone deacetylase-6 inhibition
attenuates stress responses and prevents immune organ atrophy in a
lethal septic model. Surgery. 156:235–242. 2014.PubMed/NCBI View Article : Google Scholar
|
|
52
|
González-Mariscal L, Tapia R and Chamorro
D: Crosstalk of tight junction components with signaling pathways.
Biochim Biophys Acta. 1778:729–756. 2008.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Di Liddo R, Valente S, Taurone S, Zwergel
C, Marrocco B, Turchetta R, Conconi MT, Scarpa C, Bertalot T,
Schrenk S, et al: Histone deacetylase inhibitors restore IL-10
expression in lipopolysaccharide-induced cell inflammation and
reduce IL-1β and IL-6 production in breast silicone implant in
C57BL/6J wild-type murine model. Autoimmunity: Jan 20, 2016 (Epub
ahead of print).
|
|
54
|
Youn GS, Lee KW, Choi SY and Park J:
Overexpression of HDAC6 induces pro-inflammatory responses by
regulating ROS-MAPK-NF-κB/AP-1 signaling pathways in macrophages.
Free Radic Biol Med. 97:14–23. 2016.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Youn GS, Ju SM, Choi SY and Park J: HDAC6
mediates HIV-1 tat-induced proinflammatory responses by regulating
MAPK-NF-kappaB/AP-1 pathways in astrocytes. Glia. 63:1953–1965.
2015.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Jo H, Jang HY, Youn GS, Kim D, Lee CY,
Jang JH, Choi SY, Jun JG and Park J: Hindsiipropane B alleviates
HIV-1 Tat-induced inflammatory responses by suppressing HDAC6-NADPH
oxidase-ROS axis in astrocytes. BMB Rep. 51:394–399.
2018.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Zhang WB, Zhang HY, Jiao FZ, Wang LW,
Zhang H and Gong ZJ: Histone deacetylase 6 inhibitor ACY-1215
protects against experimental acute liver failure by regulating the
TLR4-MAPK/NF-κB pathway. Biomed Pharmacother. 97:818–824.
2018.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Liu L, Zhou X, Shetty S, Hou G, Wang Q and
Fu J: HDAC6 inhibition blocks inflammatory signaling and caspase-1
activation in LPS-induced acute lung injury. Toxicol Appl
Pharmacol. 370:178–183. 2019.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Wang J, Zhao L, Wei Z, Zhang X, Wang Y, Li
F, Fu Y and Liu B: Inhibition of histone deacetylase reduces
lipopolysaccharide-induced-inflammation in primary mammary
epithelial cells by regulating ROS-NF-кB signaling pathways. Int
Immunopharmacol. 56:230–234. 2018.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Hiscott J: Convergence of the NF-kappaB
and IRF pathways in the regulation of the innate antiviral
response. Cytokine Growth Factor Rev. 18:483–490. 2007.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Nusinzon I and Horvath CM: Positive and
negative regulation of the innate antiviral response and beta
interferon gene expression by deacetylation. Mol Cell Biol.
26:3106–3113. 2006.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Chattopadhyay S, Fensterl V, Zhang Y,
Veleeparambil M, Wetzel JL and Sen GC: Inhibition of viral
pathogenesis and promotion of the septic shock response to
bacterial infection by IRF-3 are regulated by the acetylation and
phosphorylation of its coactivators. MBio. 4(4)2013.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Trani M and Dejana E: New insights in the
control of vascular permeability: Vascular endothelial-cadherin and
other players. Curr Opin Hematol. 22:267–272. 2015.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Rodrigues SF and Granger DN: Blood cells
and endothelial barrier function. Tissue Barriers.
3(e978720)2015.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Nam HJ, Kang JK, Kim SK, Ahn KJ, Seok H,
Park SJ, Chang JS, Pothoulakis C, Lamont JT and Kim H:
Clostridium difficile toxin A decreases acetylation of
tubulin, leading to microtubule depolymerization through activation
of histone deacetylase 6, and this mediates acute inflammation. J
Biol Chem. 285:32888–32896. 2010.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Saito S, Lasky JA, Guo W, Nguyen H, Mai A,
Danchuk S, Sullivan DE and Shan B: Pharmacological inhibition of
HDAC6 attenuates endothelial barrier dysfunction induced by
thrombin. Biochem Biophys Res Commun. 408:630–634. 2011.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Wang F, Zheng L, Yi Y, Yang Z, Qiu Q, Wang
X, Yan W, Bai P, Yang J, Li D, et al: SKLB-23bb, A HDAC6-Selective
Inhibitor, Exhibits Superior and Broad-Spectrum Antitumor Activity
via Additionally Targeting Microtubules. Mol Cancer Ther.
17:763–775. 2018.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Majolée J, Pronk MCA, Jim KK, van Bezu
JSM, van der Sar AM, Hordijk PL and Kovačević I: CSN5 inhibition
triggers inflammatory signaling and Rho/ROCK-dependent loss of
endothelial integrity. Sci Rep. 9(8131)2019.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Croisé P, Estay-Ahumada C, Gasman S and
Ory S: Rho GTPases, phosphoinositides, and actin: A tripartite
framework for efficient vesicular trafficking. Small GTPases.
5(e29469)2014.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Joshi AD, Dimitropoulou C, Thangjam G,
Snead C, Feldman S, Barabutis N, Fulton D, Hou Y, Kumar S, Patel V,
et al: Heat shock protein 90 inhibitors prevent LPS-induced
endothelial barrier dysfunction by disrupting RhoA signaling. Am J
Respir Cell Mol Biol. 50:170–179. 2014.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Valenta T, Hausmann G and Basler K: The
many faces and functions of β-catenin. EMBO J. 31:2714–2736.
2012.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Winer IS, Bommer GT, Gonik N and Fearon
ER: Lysine residues Lys-19 and Lys-49 of beta-catenin regulate its
levels and function in T cell factor transcriptional activation and
neoplastic transformation. J Biol Chem. 281:26181–26187.
2006.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Sauteur L, Krudewig A, Herwig L,
Ehrenfeuchter N, Lenard A, Affolter M and Belting HG:
Cdh5/VE-cadherin promotes endothelial cell interface elongation via
cortical actin polymerization during angiogenic sprouting. Cell
Rep. 9:504–513. 2014.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Dejana E and Vestweber D: The role of
VE-cadherin in vascular morphogenesis and permeability control.
Prog Mol Biol Transl Sci. 116:119–144. 2013.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Huber AH and Weis WI: The structure of the
beta-catenin/E-cadherin complex and the molecular basis of diverse
ligand recognition by beta-catenin. Cell. 105:391–402.
2001.PubMed/NCBI View Article : Google Scholar
|
|
76
|
D'Alessandro M, Hnia K, Gache V, Koch C,
Gavriilidis C, Rodriguez D, Nicot AS, Romero NB, Schwab Y, Gomes E,
et al: Amphiphysin 2 orchestrates nucleus positioning and shape by
linking the nuclear envelope to the actin and microtubule
cytoskeleton. Dev Cell. 35:186–198. 2015.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Coles CH and Bradke F: Coordinating
neuronal actin-microtubule dynamics. Curr Biol. 25:R677–R691.
2015.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Hubbert C, Guardiola A, Shao R, Kawaguchi
Y, Ito A, Nixon A, Yoshida M, Wang XF and Yao TP: HDAC6 is a
microtubule-associated deacetylase. Nature. 417:455–458.
2002.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Matsuyama A, Shimazu T, Sumida Y, Saito A,
Yoshimatsu Y, Seigneurin-Berny D, Osada H, Komatsu Y, Nishino N,
Khochbin S, et al: In vivo destabilization of dynamic microtubules
by HDAC6-mediated deacetylation. EMBO J. 21:6820–6831.
2002.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Shivanna M and Srinivas SP: Microtubule
stabilization opposes the (TNF-alpha)-induced loss in the barrier
integrity of corneal endothelium. Exp Eye Res. 89:950–959.
2009.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Bogatcheva NV and Verin AD: The role of
cytoskeleton in the regulation of vascular endothelial barrier
function. Microvasc Res. 76:202–207. 2008.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Iaconelli J, Huang JH, Berkovitch SS,
Chattopadhyay S, Mazitschek R, Schreiber SL, Haggarty SJ and
Karmacharya R: HDAC6 inhibitors modulate Lys49 acetylation and
membrane localization of β-catenin in human iPSC-derived neuronal
cells. ACS Chem Biol. 10:883–890. 2015.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Matsuda N, Takano Y, Kageyama S,
Hatakeyama N, Shakunaga K, Kitajima I, Yamazaki M and Hattori Y:
Silencing of caspase-8 and caspase-3 by RNA interference prevents
vascular endothelial cell injury in mice with endotoxic shock.
Cardiovasc Res. 76:132–140. 2007.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Sawant DA, Tharakan B, Tobin RP, Reilly J,
Hunter FA, Newell MK, Smythe WR and Childs EW: Microvascular
endothelial cell hyperpermeability induced by endogenous caspase 3
activator staurosporine. J Trauma Acute Care Surg. 74:516–523.
2013.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Lopez-Ramirez MA, Fischer R,
Torres-Badillo CC, Davies HA, Logan K, Pfizenmaier K, Male DK,
Sharrack B and Romero IA: Role of caspases in cytokine-induced
barrier breakdown in human brain endothelial cells. J Immunol.
189:3130–3139. 2012.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Zehendner CM, Librizzi L, de Curtis M,
Kuhlmann CR and Luhmann HJ: Caspase-3 contributes to ZO-1 and Cl-5
tight-junction disruption in rapid anoxic neurovascular unit
damage. PLoS One. 6(e16760)2011.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Leyk J, Daly C, Janssen-Bienhold U,
Kennedy BN and Richter-Landsberg C: HDAC6 inhibition by tubastatin
A is protective against oxidative stress in a photoreceptor cell
line and restores visual function in a zebrafish model of inherited
blindness. Cell Death Dis. 8(e3028)2017.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Wang X, Tang X, Zhou Z and Huang Q:
Histone deacetylase 6 inhibitor enhances resistance to
Mycobacterium tuberculosis infection through innate and adaptive
immunity in mice. Pathog Dis. 76(76)2018.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Wang H, Ling L, Ai L and Bai L: HDAC6
inhibition induces the failure of mouse early embryonic
development. J Cell Physiol. 234:8752–8759. 2019.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Choi YJ, Kang MH, Hong K and Kim JH:
Tubastatin A inhibits HDAC and Sirtuin activity rather than being a
HDAC6-specific inhibitor in mouse oocytes. Aging (Albany NY).
11:1759–1777. 2019.PubMed/NCBI View Article : Google Scholar
|














