Introduction
Graphene is a two-dimensional structured material
with a hexagonal honeycomb lattice composed of carbon atoms, and is
currently the thinnest and most widely used non-metallic
nanomaterial (1). Graphene has
unique electrical and mechanical properties, a large specific
surface area and potential biocompatibility. As a result, it is
widely used in materials, electronics, energy, optics and
biomedical fields, such as cell imaging, drug delivery and
biosensing (2-5).
With the large-scale production and application of graphene,
research into its biological toxicity has been attracting
increasing attention. A large number of studies have confirmed that
the biotoxicity of graphene nanomaterials depends on their
individual physicochemical properties (including size, morphology
and functional groups), concentration, route of biological
ingestion and the organs involved (6-9).
Different forms of graphene and their derivatives display different
physical/chemical properties and biological toxicities. Graphene
may be categorized as single-layer graphene, few-layer graphene,
graphene oxide (GO), reduced graphene oxide (rGO) and graphene
nanoribbons. Among these subtypes, the biological effects of GO
have received the most attention, as it has been more widely used
in biological research due to its higher
hydrophilicity/biocompatibility.
Previous animal studies have demonstrated that
graphene can enter the body through tracheal instillation,
inhalation, intravenous injection, intraperitoneal injection and
oral administration. Graphene penetrates the blood-air, blood-brain
and blood-placenta barriers, and subsequently accumulates in the
lung, liver and spleen, resulting in acute or chronic injury
(10-13).
Different organs exhibit different levels of graphene nanomaterial
accumulation and clearance. The accumulation of GO in the lungs
increases with increasing injection dose and particle size
(14). In the lung, graphene is
engulfed by alveolar macrophages and excreted in the sputum via
mucosal cilia. Furthermore, 28 days after tracheal instillation,
46.2% of graphene layers may be excreted in the feces (15). As graphene can directly act on the
respiratory system, research has mainly focused on graphene-induced
damage to this system. It was previously reported that, in humans,
the majority of inhaled graphene nanoparticles pass through the
upper respiratory tract and are deposited in the lungs. As such,
the deposition rate of graphene nanoparticles in the respiratory
tract is ~4% (16). Therefore, lung
injury is the primary symptom of graphene-induced toxicity, and
mice exposed to GO nanomaterials reportedly experienced acute
injury and chronic fibrosis of the lung (8).
Previous studies on the toxic effects of graphene
have primarily focused on mitochondrial damage, DNA damage, the
inflammatory response, apoptosis and oxidative stress (17-19).
GO-associated lung injury may be relieved with the antioxidant drug
dexamethasone, indicating that GO may cause pulmonary toxicity
through oxidative stress (8).
Autophagy is a process through which cells degrade proteins,
damaged organelles and foreign matter through the lysosomal
degradation pathway. Active autophagy usually results in increased
phosphorylation of mTOR and beclin-1 expression, and a decreased
ratio of LC3B-I/II and p62 expression levels. Therefore, the
expression levels of phosphorylated mTOR, the ratio of LC3B-I/II,
and the expression of p62 and beclin-1, are commonly used
indicators of this process. Autophagy is regulated by multiple
intracellular molecules and plays a key role in the homeostatic
maintenance of cells. Autophagy may be involved in cellular
defense, although excessive uncontrolled autophagy may also result
in tissue damage. A 2012 study demonstrated that GO triggers
Toll-like receptor-mediated autophagy in RAW264.7 mouse macrophages
(20). Chen et al (21) observed that GO also triggered
autologous effects in CT26 cells through Toll-like receptors. They
also determined that co-administration of GO and cisplatin promoted
the nuclear localization of cisplatin and LC3 protein while
triggering autophagy, thereby altering the original LC3 pathway in
the early stages of autophagy and enhancing the antitumor effects
of cisplatin (22). However, the
role of autophagy and its association with oxidative stress and
inflammation in the development of GO-induced lung injury has not
been extensively investigated.
Therefore, the aim of the present study was to
determine the role of GO in lung injury induction, as well as its
involvement in oxidative stress, inflammation and autophagy in a
rat model, in the hope that the findings may help elucidate the
mechanisms underlying GO-induced lung injury.
Materials and methods
Animals and study design
Male Sprague-Dawley rats (age, 10 weeks; weight,
250-300 g) were obtained from the Xinhua Hospital Affiliated to
Shanghai Jiaotong University School of Medicine (Shanghai, China).
The animals were housed in the animal center of Xinhua Hospital at
a constant temperature of 25±2˚C, a relative humidity of 41%, and
on a 12:12 h light/dark cycle. All animals had free access to food
and water. The experiment was conducted according to the principles
of the Bioethics Committee of Shanghai Jiaotong University School
of Medicine for the care and use of laboratory animals (no.
AS-20183265), as well as the Guide for the Care and Use of
Laboratory Animals (NIH publication no. 85-23, revised 1996). The
humane endpoints used to identify the adverse effects of
surgery/treatments were as follows: i) Weight loss of 20-25%; ii)
inability or extreme reluctance to stand, persisting for 24 h; iii)
depression coupled with body temperature <37˚C; iv) infection
involving any organ system failing to respond to antibiotic therapy
within 48 h and accompanied by systemic signs of illness; and v)
signs of severe organ system dysfunction.
To determine the effects of GO on lung injury, rats
were randomly assigned to five groups (n=12 per group) as follows:
i) Control; ii) GO (5 mg/kg); iii) GO (10 mg/kg); iv) GO (50
mg/kg); and v) GO (100 mg/kg). Rats in the control group were fed a
normal diet and received no special treatment. Rats in the GO
groups received 5, 10, 50 and 100 mg/kg GO injections,
respectively. GO was injected into the tail vein once a day for 7
consecutive days. There were no significant differences in age or
weight among the groups. Following treatment, the lung wet-to-dry
(W/D) weight ratio, levels of protein in the bronchoalveolar lavage
fluid (BALF), lung injury scores, oxidative stress, and levels of
autophagy-related proteins and inflammatory factors in the lung
tissue were determined.
Furthermore, to evaluate the involvement of
autophagy in the pathology of GO-induced lung injury, the rats were
randomly assigned to the following six groups (n=12): i) Control;
ii) vehicle; iii) GO50; iv) GO50 + vehicle; v) GO50 +
3-methyladenine (3-MA); and vi) GO50 + chloroquine (CLQ). Rats in
the control group were fed a normal diet and received no special
treatment. Rats in the vehicle group were also fed a normal diet,
and received saline (the same volume as was used for GO
administration) via the tail vein once a day for 7 days. Rats in
the GO50 group received 50 mg/kg GO injections via the tail vein
once a day for 7 days; those in the GO50 + vehicle group received
50 mg/kg GO injected via the tail vein, and the same volume of
saline intraperitoneally for 7 days. The rats in the GO50 + 3-MA
group received 50 mg/kg GO injected via the tail vein, and 15 mg/kg
3-MA (Sigma-Aldrich; Merck KGaA) intraperitoneally once a day for 7
days. Finally, the rats in the GO50 + CLQ group were administered
50 mg/kg GO injected via the tail vein, and 20 mg/kg CLQ
intraperitoneally (Sigma-Aldrich; Merck KGaA), once a day for 7
days. The levels of autophagy-related proteins, lung W/D weight
ratio, protein levels in the BALF, lung injury scores, and levels
of oxidative stress and inflammatory factors in lung tissue were
then determined.
To confirm the role of autophagy in GO-induced lung
cell injury, autophagy was specifically inhibited in BEAS-2B cells
by short hairpin (sh) RNA-mediated autophagy protein 5 (ATG5)
knockdown. The cells were subsequently treated with GO as
previously described (23). BEAS-2B
cells were seeded into 6-well plates (Beyotime Institute of
Biotechnology) at a concentration of 1x105 cells/ml, and
then exposed to 50 µg/ml GO for 24 h. Cell viability was assessed
using the MTT method. The concentration of lactate dehydrogenase
(LDH) in the culture media was determined using an LDH assay kit
(cat. no. C0016; Beyotime Institute of Biotechnology) and the
levels of oxidative stress indictors [malondialdehyde (MDA, cat.
no. S0131S; Beyotime Institute of Biotechnology),
8-hydroxy-2'-deoxyguanosine (8-OHdG; cat. no. ab201734; Abcam) and
protein carbonyl (cat. no. ab235631; Abcam)] were measured using
the corresponding kits as per the manufacturers' protocols.
Lung W/D weight ratio measurement and
BALF collection
Following treatment with GO, the rats were
euthanized with an overdose of pentobarbital (200 mg/kg via i.p.
injection), and the breathing and heartbeat were checked to verify
rat death before opening the thoracic cavity to expose the lungs.
The right middle lobe of the lung was removed and weighed to obtain
the wet weight. The lungs were then dried in an oven at 60˚C for 3
days to obtain the dry weight, and the lung W/D weight ratio was
calculated to evaluate lung edema. To collect BALF, the left lung
was lavaged three times with saline (5 ml, 4˚C). The collected
lavage fluid was centrifuged at 1,200 x g for 10 min at 4˚C, and
the total protein levels were measured using a BCA protein assay
kit (Beyotime Institute of Biotechnology).
Histopathological examination
After the rats were euthanized, the lung tissue was
harvested and stained with hematoxylin and eosin (H&E). Three
specimens were randomly selected from each rat, and five fields for
each section were analyzed under a microscope (magnification, x200)
by two independent pathologists who were blinded to the
experimental groupings. The staining scores were calculated
according to the following variables: Alveolar congestion,
hemorrhage, infiltration or aggregation of neutrophils in the
airspace, and hyaline membrane formation. The lung injury scores
ranged between 0 and 4 as follows: i) 0, no injury; ii) 1, <25%
lung involvement; iii) 2, 25-50% lung involvement; iv) 3, 50-75%
lung involvement; and v) 4, >75% lung involvement.
ShRNA-mediated ATG5 knockdown
Autophagy was inhibited by ATG5 knockdown using
shRNA, as previously described by Domagala et al (24). Briefly, BEAS-2B cells were infected
with lentiviral particles (Sigma-Aldrich; Merck KGaA) encoding
ATG5-specific shRNA (MISSION shRNA TRCN0000151474; shATG5 group) or
a scrambled (non-targeting) shRNA plasmid (SHC002V; shNTC group).
After transduction, 2 µg/ml puromycin was added to the culture
medium.
MTT assay
BEAS-2B cells were harvested with trypsin and
re-suspended in culture medium. The cell suspension was adjusted to
a concentration of 5x104 cell/ml, and 100 µl was added
to each well of a 96-well plate. The cells were maintained in a
CO2 incubator for 12 h at 37˚C, after which time 50
µg/ml GO was added to each well. After a further 24 h, the culture
medium was discarded and the cells were harvested by gentle
centrifugation; 10 µl MTT solution (5 mg/ml, 0.5% MTT; Beyotime
Institute of Biotechnology) was then added to each well and the
cells were incubated for another 4 h at 37˚C. To terminate the
reaction, 150 µl dimethyl sulfoxide (Sigma-Aldrich; Merck KGaA) was
added to each well, and the 96-well plate was placed on a shaking
platform for 10 min to fully dissolve the formazan crystals. The
absorbance of each well was measured at OD490 nm and the cell
survival rate was calculated.
LDH measurement
The release of LDH was measured using the method
described by Zhang et al (25). First, BEAS-2B cells were harvested
as mentioned above, and the cell suspension was adjusted to
5x104/ml, 500 µl of which was added to each well of a
6-well plate. The cells were cultured in a CO2 incubator
for 12 h at 37˚C, after which time 50 µg/ml GO was added to each
well for a further 24-h incubation period (37˚C). The supernatants
were then harvested and LDH level was measured using an LDH assay
kit (Nanjing Jiancheng Bioengineering Institute). The supernatants
were incubated at 37˚C for 15 min, and 2,4-dinitrophenylhydrazine
was added for a further 15 min. Finally, 0.4 M NaOH was added to
each well and the absorbance at 450 nm was measured using a
microplate reader (Bio-Rad 680; Bio-Rad Laboratories, Inc.).
Measurement of oxidative products
The levels of oxidative products (MDA, 8-OHdG and
protein carbonyl) in the lung homogenates were detected using the
respective kits according to the manufacturers' instructions. The
8-OHdG ELISA kit was purchased from Cusabio Technology, Ltd. (cat.
no. CSB-E10526r), and the MDA (cat. no. A003-1-2) and protein
carbonyl (cat. no. A087-1-2) kits were purchased from Nanjing
Jiancheng Bioengineering Institute.
Inflammatory cytokine ELISA
The expression levels of TNF-α (cat. no.
E-EL-R2856c), IL-6 (cat. no. E-EL-R0015c), IL-1β (cat. no.
E-EL-R0012c) and IL-8 (cat. no. SEKR-0071-96T) (all from Beijing
Solarbio Science & Technology Co., Ltd.) in the lung tissue
were measured using commercial ELISA kits (Elabscience, Inc.)
according to the manufacturers' protocols. The cytokine levels were
determined using a spectral scanning plate reader (Varioskan;
Thermo Fisher Scientific, Inc.).
Western blotting
Briefly, the right lung was harvested from each rat
and placed in ice-cold homogenization buffer (100 mmol/l NaCl, 50
mmol/l Tris base, 0.1 mmol/l EDTA, 0.1 mmol/l EGTA and 1% Triton
X-100; pH 7.5), and then homogenized in a 15-ml glass homogenizer.
The homogenate was centrifuged at 1,500 x g for 10 min at 4˚C to
collect the supernatant. The protein concentration was measured
using a BCA protein assay kit (Beyotime Institute of
Biotechnology). The proteins (50 µg/per lane) were separated by 10%
SDS-PAGE and transferred to PVDF membranes, which were then
incubated with a mixture of tris-buffered saline (TBS), Tween-20
(0.1%) and non-fat milk for 1 h at 37˚C. The membranes were
incubated with the following primary antibodies overnight at 4˚C in
Universal Antibody Dilution Buffer (Santa Cruz Biotechnology,
Inc.): Anti-p-mTOR (cat. no. sc-293133), anti-mTOR (cat. no.
sc-517464), anti-LC3B-I (cat. no. sc-398822), anti-LC3B-II (cat.
no. sc-271625), anti-GAPDH (cat. no. sc-365062), anti-p62 (cat. no.
sc-48402), anti-ATG5 (cat. no. sc-133158) and anti-beclin-1 (cat.
no. sc-48341) (all 1:1,000; all from Santa Cruz Biotechnology,
Inc.). The membranes were washed three times with TBST, and then
incubated with secondary antibody (cat. no. sc-516102; 1:5,000;
Santa Cruz Biotechnology, Inc.) for 1 h at room temperature. The
protein bands were visualized using the enhanced chemiluminescence
method with an Electro-Chemi-Luminescence Substrate Kit (Rahn AG)
and quantified using Quantity One software (v4.6.6; Bio-Rad
Laboratories Inc.). The bar graphs show the average values, and the
error bars are quantified from the optical densities of six bands
(experimental repeats) per group.
Statistical analysis
Statistical analysis was conducted with SPSS
software version 19 (IBM Corp.) using one-way ANOVA. The data
fulfilled ANOVA assumptions, such as normality and
homoscedasticity. Dunnett's post hoc test was performed following
ANOVA for comparing experimental groups with the control group
only; Bonferroni's post hoc test was performed following ANOVA for
comparing the data between experimental groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
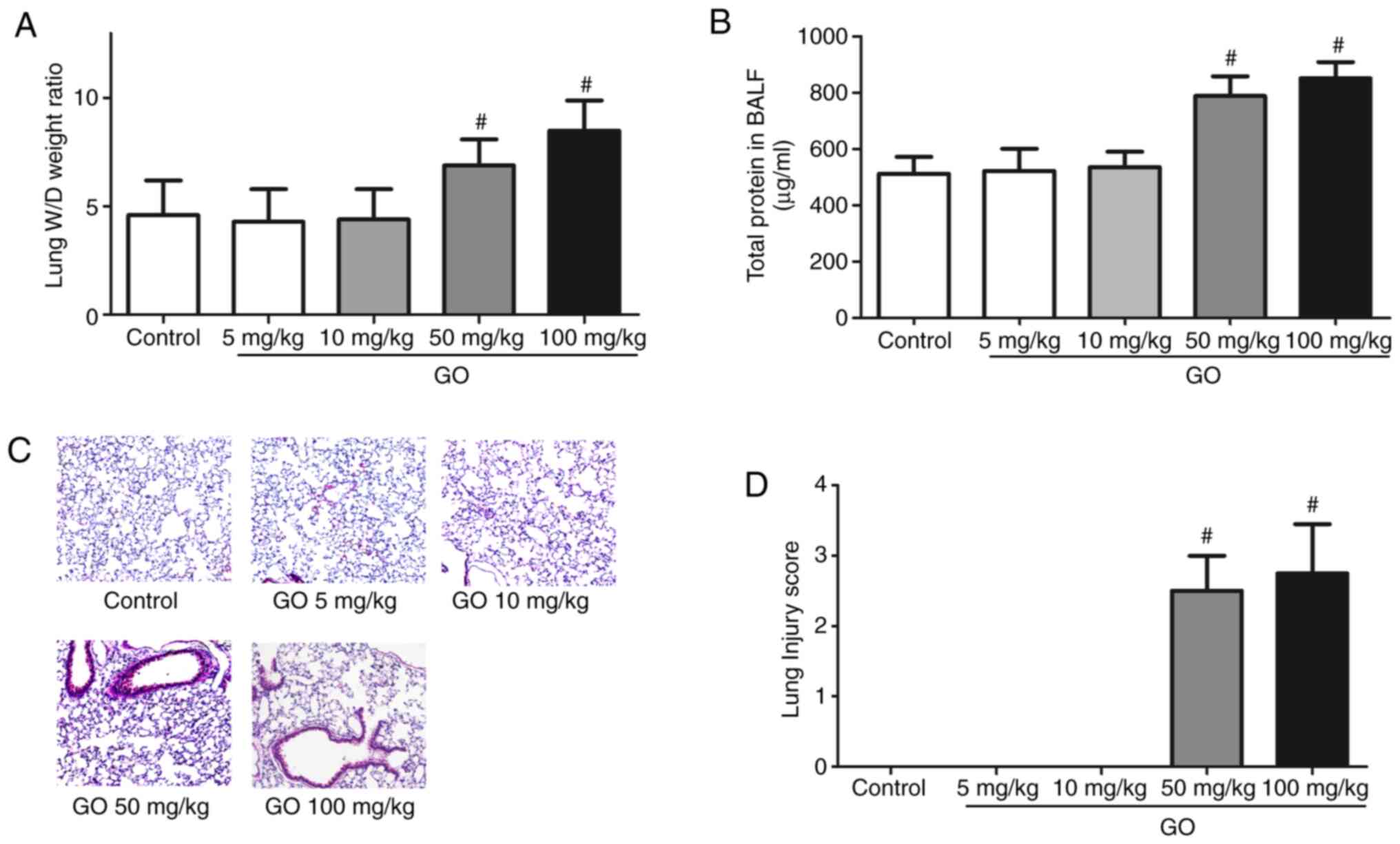
Changes in lung edema,
histopathological appearance and permeability
Changes in the lung W/D weight ratio, the amount of
protein in the BALF, histopathological appearance and lung injury
score are presented in Fig. 1.
Compared with the control group, the lung W/D weight ratio and
total BALF protein levels of the GO 5 and 10 mg/kg groups were not
markedly altered, while those in the GO 50 and 100 mg/kg groups
significantly increased (P<0.05; Fig. 1A and B). As shown by H&E staining, in the GO
50 and 100 mg/kg groups, examination of the lung samples revealed
thickening of the alveolar septa and infiltration by inflammatory
cells (Fig. 1C). The lung injury
scores calculated from the data in Fig.
1C are summarized in Fig. 1D.
The lung injury scores of the GO 50 and 100 mg/kg groups were
significantly higher compared with those of the control group
(P<0.05). These results indicate that the toxic effects of GO
are dose-dependent.
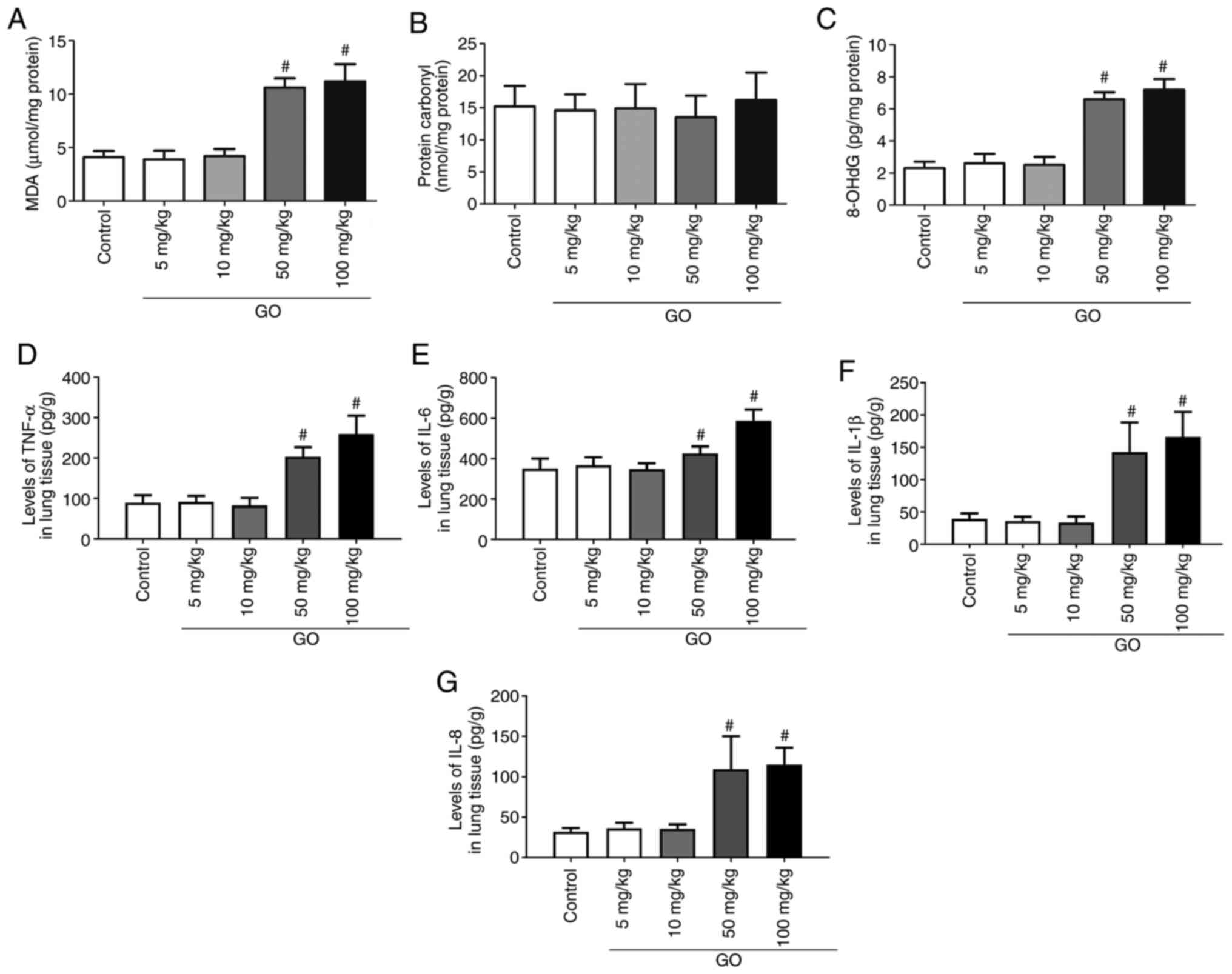
Changes in oxidative products and
inflammatory factors in the lung
Changes in oxidative products (MDA, protein carbonyl
and 8-OHdG) and inflammatory factors (TNF-α, IL-6, IL-1β and IL-8)
in the lung were then assessed. As shown in Fig. 2A and C, in the GO 5 and 10 mg/kg groups, the
levels of MDA and 8-OHdG did not change significantly compared with
those in the control group (P<0.05); however, in the GO 50 and
100 mg/kg groups, the levels of MDA and 8-OHdG were significantly
increased compared with those in the control group (P<0.05). As
shown in Fig. 2B, the levels of
protein carbonyl did not differ significantly among the groups. The
levels of TNF-α, IL-6, IL-1β and IL-8 in the lung tissues of the GO
5 and 10 mg/kg groups were not significantly different from those
in the control group, but their levels in the GO 50 and 100 mg/kg
groups were significantly higher compared with those in the control
group (P<0.05; Fig. 2D-G). These
results indicate that GO induces oxidative injury and inflammatory
reactions in the lung.
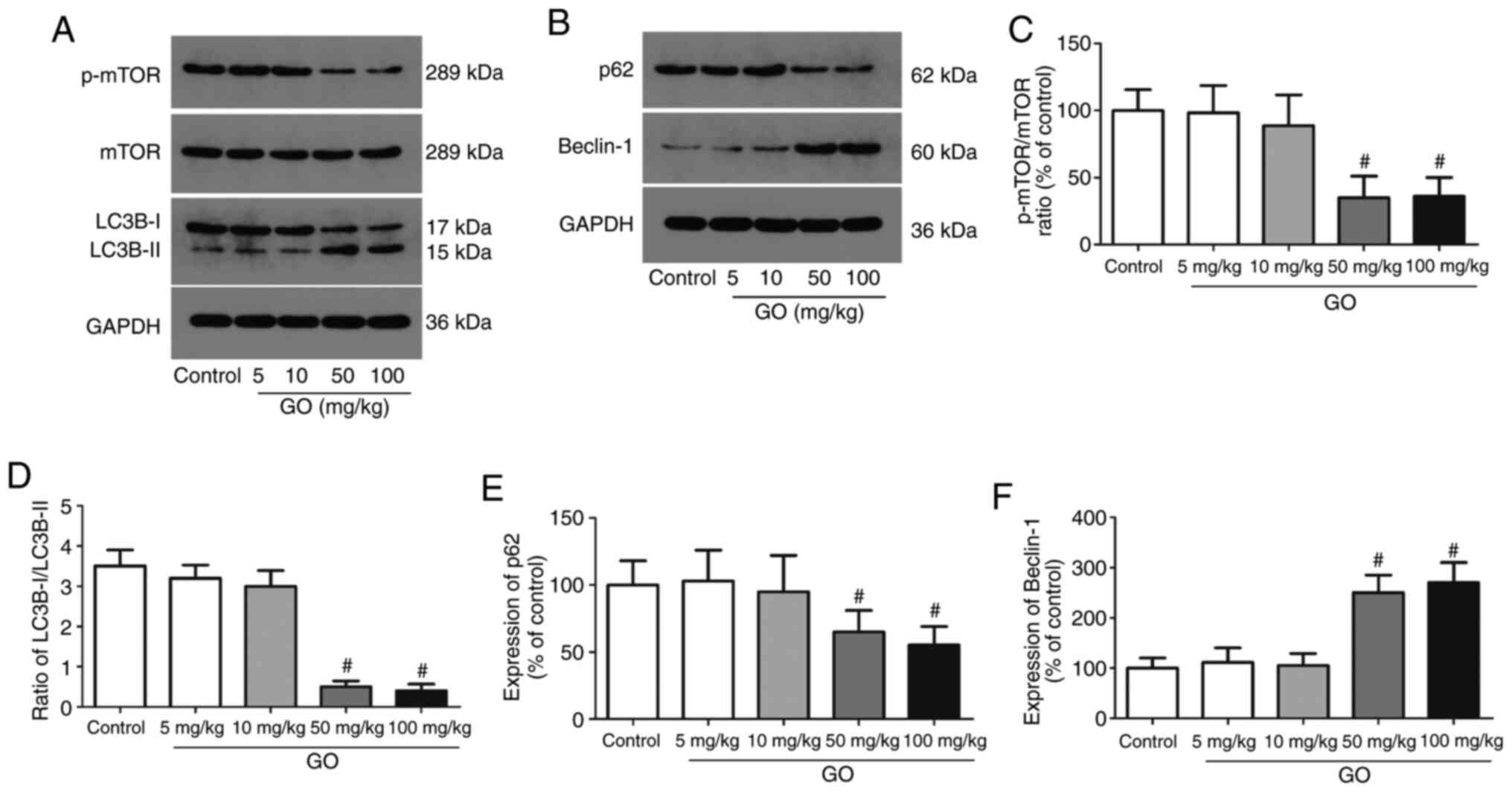
Changes in autophagic protein levels
in the lung tissue
The expression levels of autophagic proteins (mTOR,
LC3B-I/II, p62 and beclin-1) were then detected in lung tissues;
representative western blot images are displayed in Fig. 3A and B. The levels of phosphorylated mTOR were
significantly decreased in the GO 50 and 100 mg/kg groups, and the
ratio of LC3B-I/II was also significantly decreased in these groups
(Fig. 3C and D). Compared with the control group, the
expression of p62, an autophagy inhibitor, was significantly
decreased in the GO 50 and 100 mg/kg groups, while the expression
of beclin-1, an autophagy promoter, was significantly enhanced
(Fig. 3E and F; P<0.05). The expression levels of
these proteins in the GO 5 and 10 mg/kg groups were not
significantly altered. These data directly indicate that a higher
concentration of GO induces autophagy in the lung in an
mTOR-dependent manner.
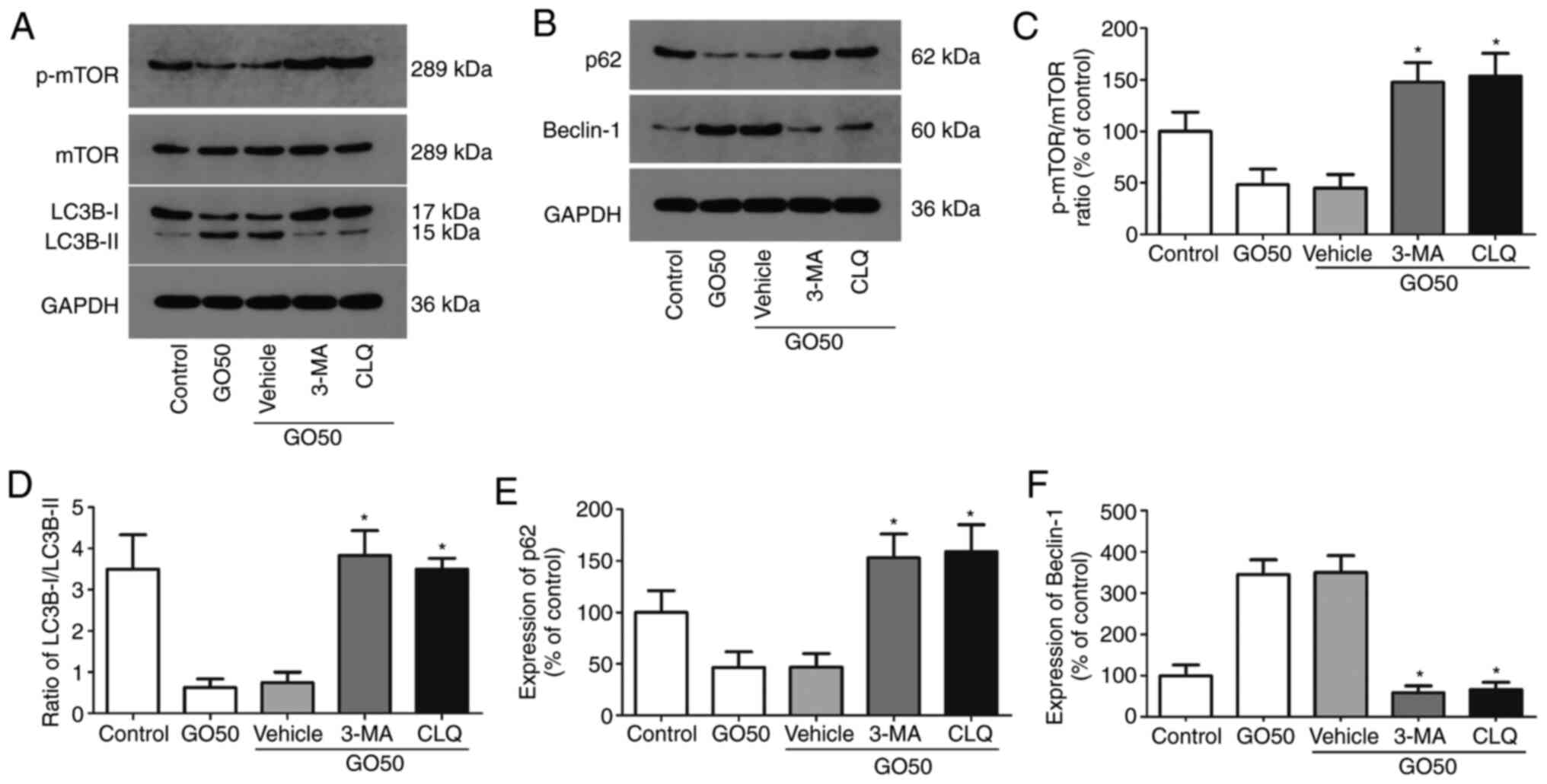
Effects of autophagy inhibitors on
autophagic protein expression in the lung
The expression levels of autophagy-related proteins
(mTOR, LC3B-I/II, p62 and beclin-1) in rat lung tissues after
treatment with the autophagy inhibitors 3-MA and CLQ are shown in
Fig. 4; representative western blot
images are shown in Fig. 4A and
B. 3-MA and CLQ significantly
increased the level of phosphorylated mTOR, the LC3B-I/II ratio and
the expression of beclin-1 (Fig.
4C-F). On the other hand, p62 expression was significantly
increased by 3-MA and CLQ compared with the saline vehicle
(P<0.05). These results indicate that both 3-MA and CLQ
successfully inhibited autophagy.
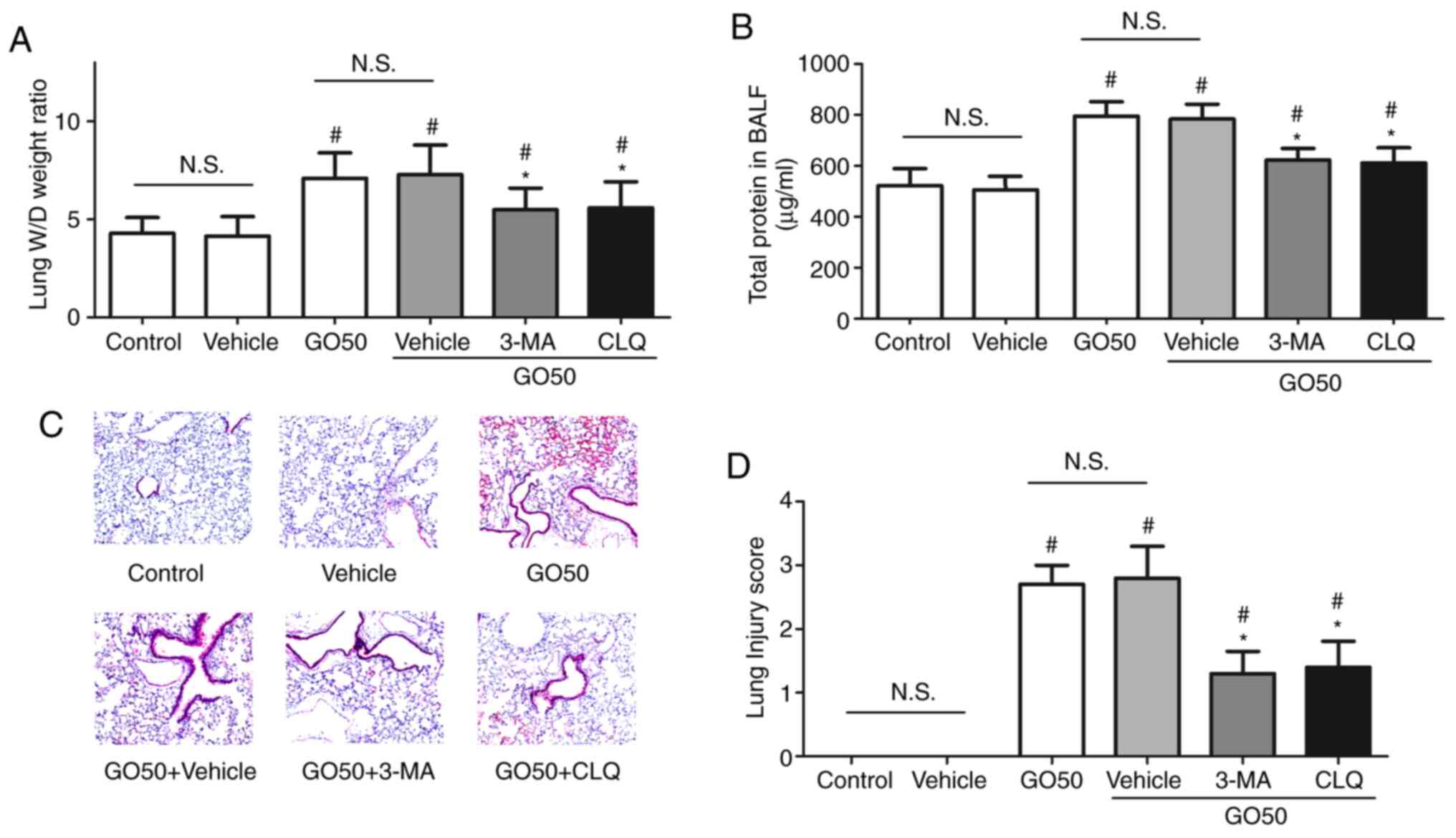
Effects of autophagy inhibitors on the
severity of GO-induced lung injury
To confirm the role of autophagy in the development
of GO-induced lung injury, the effects of 3-MA and CLQ on the
severity of GO-induced lung injury were assessed. First, treatment
with the vehicle control did not induce lung injury. The lung W/D
weight ratio and amount of protein in the BALF were significantly
decreased, but not completely restored by 3-MA or CLQ (Fig. 5A and B). Following treatment with 3-MA and CLQ,
thickening of the alveolar septa and inflammatory cell infiltration
were significantly attenuated compared with the GO50 + vehicle
group (the same dose of saline; P<0.05; Fig. 5C). The lung injury score was found
to be significantly decreased, but not completely restored by 3-MA
and CLQ, compared with GO50 + vehicle treatment (the same dose of
saline; P<0.05; Fig. 5D). These
results indicate that 3-MA and CLQ inhibited autophagy in the lung
and attenuated GO-induced lung injury.
Effects of autophagy inhibitors on
oxidative stress and inflammation
To confirm the role of autophagy in the development
of GO-induced oxidative stress and inflammation in lung tissue, the
effects of 3-MA and CLQ on the production of MDA, 8-OHdG and
inflammatory factors were investigated. First, treatment with
vehicle did not induce oxidative stress or inflammation. The levels
of MDA and 8-OHdG were significantly decreased, but not completely
restored by 3-MA and CLQ (Fig. 6A
and B). The levels of TNF-α, IL-1β
and IL-8 in the lung tissue were significantly decreased, but not
completely restored by 3-MA and CLQ, compared with the GO + vehicle
group (the same dose of saline; P<0.05; Fig. 6C). Notably, the levels of IL-6 in
lung tissue were completely restored by 3-MA and CLQ, compared with
the vehicle (P<0.05). These results indicate that 3-MA and CLQ
significantly reduced the production of MDA, 8-OHdG and
inflammatory factors in the lung, suggesting that autophagy also
mediates the development of oxidative injury and inflammation.
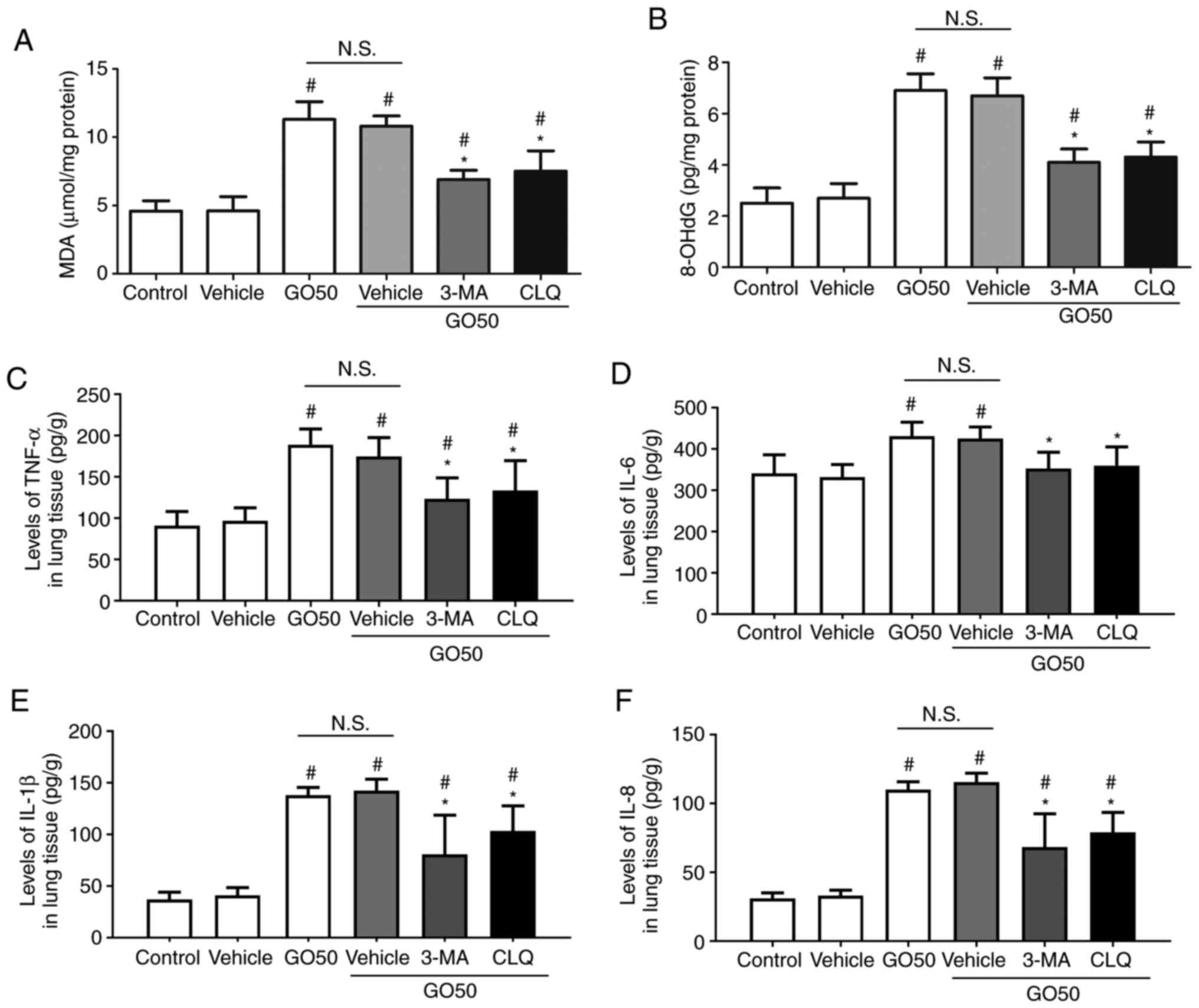 | Figure 6Effects of autophagy inhibitors on
oxidative products and inflammatory factors in the lung. Expression
levels of (A) MDA, (B) 8-OHdG, (C) TNF-α, (D) IL-6, (E) IL-1β and
(F) IL-8. Data are expressed as the mean ± SEM; n=12.
#P<0.05 vs. vehicle (the same dose of saline);
*P<0.05 vs. GO50 + vehicle (same dose of saline).
N.S., not significant. Bonferroni's post hoc test was performed
following ANOVA to compare the data between experimental groups.
MDA, malondialdehyde; 8-OHdG, 8-hydroxy-2'-deoxyguanosine; 3-MA,
3-methyladenine; CLQ, chloroquine; GO, graphene oxide. |
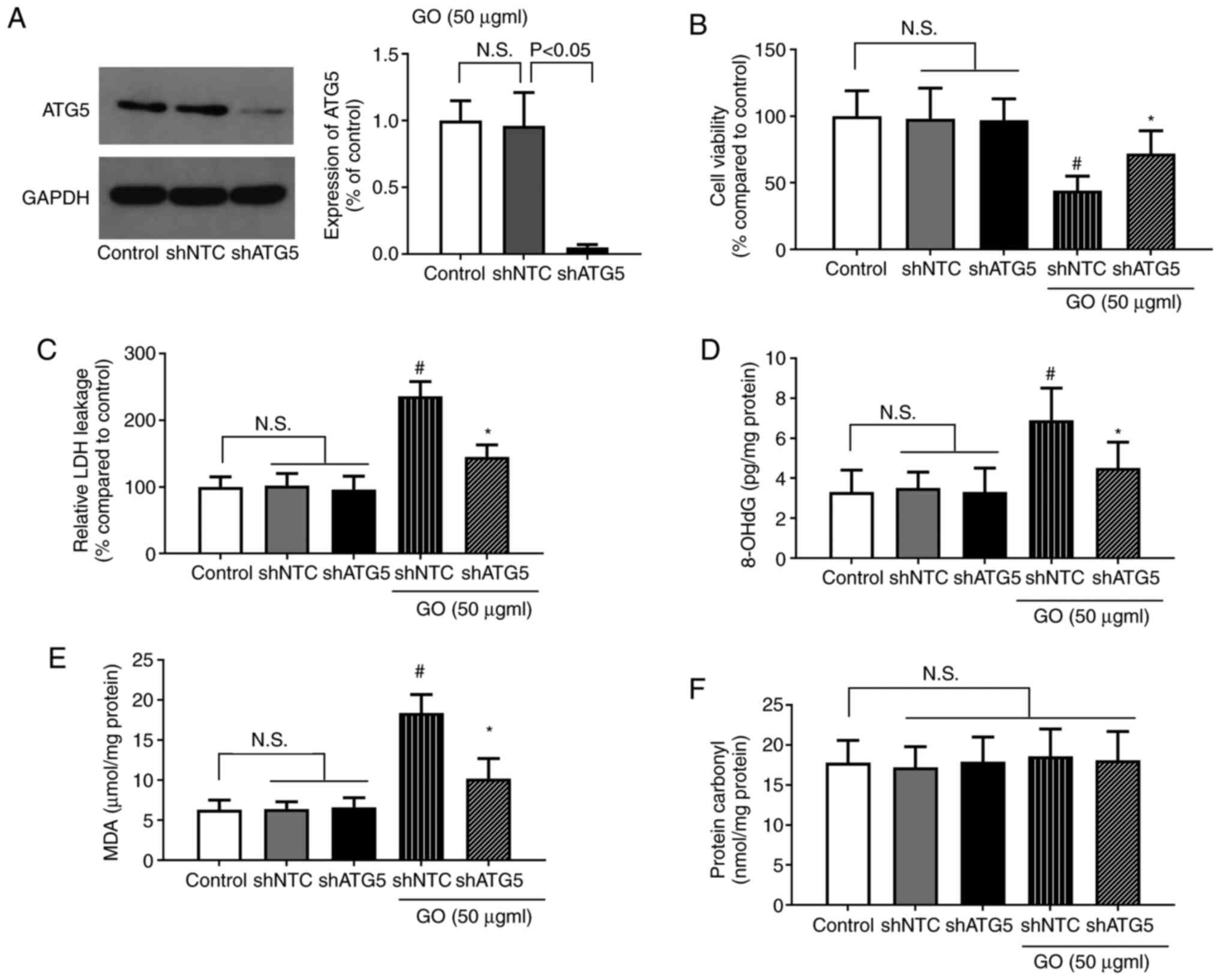
Effects of ATG5 knockdown on the
GO-induced extent of injury and levels of oxidative stress in
BEAS-2B cells
To confirm the role of autophagy in GO-induced lung
cell injury, autophagy was specifically inhibited in BEAS-2B cells
via shRNA-mediated ATG5 knockdown, and the changes in cell
viability, LDH levels and oxidative stress were determined.
Treatment with shNTC or shATG5 alone did not significantly affect
cell viability, or the levels of LDH and oxidative stress. The
protein expression of ATG5 following shRNA transfection is shown in
Fig. 7A. The reduction of ATG5
expression in the shATG5 group indicated that the shRNA
transfection targeting ATG5 was successful. As shown in Fig. 7B, cell viability was significantly
increased in the GO + shATG5 group compared with that in the GO +
shNTC group (P<0.05). By contrast, LDH release was significantly
lower in the GO + shATG5 group compared with that in the GO + shNTC
group (P<0.05). Furthermore, both the MDA and 8-OHdG levels were
significantly decreased by GO + shATG5 treatment compared with GO +
shNTC treatment (P<0.05). The level of protein carbonyl,
however, was not significantly affected by these treatments. As
inhibiting autophagy significantly decreased the levels of cellular
injury and oxidative stress, autophagy induction was suggested to
be the key event for lung injury during exposure to GO.
Discussion
Due to their unique physical and chemical
properties, graphene nanomaterials have been widely used in various
fields, such as those surrounding energy and the environment. As a
result, the environmental behavior and biological toxicity of
graphene have been attracting increasing attention. GO may promote
acute inflammatory reactions and chronic injury by interfering with
the normal physiological functions of important organ systems. The
results of various in vitro studies have demonstrated that
graphene nanomaterials may alter cell viability and morphology,
destroy the integrity of cell membranes (26), and enter lysosomes, mitochondria,
nuclei and endoplasmic reticulum to induce DNA damage (27) and cell apoptosis (28). Whether graphene maintains cell
viability or promotes cell death depends on the cell type in
question, as well as the type and amount of graphene material. For
example, the same dose of GO decreased the viability of A549 cells,
but did not decrease the viability of colorectal cancer cells
(29). Exposure to GO
concentrations >50 µg/ml resulted in A549 cytotoxicity, while
concentrations >20 µg/ml were found to be cytotoxic to human
fibroblasts and lung epithelial cells (30). The results of the present study
revealed that the injection of lower doses of GO (5 and 10 mg/kg)
via the tail vein did not induce significant lung edema, vascular
permeability or histopathological changes. In addition, low doses
exerted no significant effect on oxidative injury and inflammatory
reactions in the lung. However, higher concentrations of GO (50 and
100 mg/kg) induced lung edema and increased lung vascular
permeability and histopathological changes, as well as triggered
oxidative injury and inflammation These results indicate that the
toxic effects of GO are dose-dependent. This phenomenon has also
been reported in previous studies. In an in vivo experiment
on mice, low- (0.1 mg) and medium- (0.25 mg) level exposure to GO
was not associated with significant toxicity, but high-level
exposure (0.4 mg) was chronically toxic (31). Furthermore, inflammatory cell
infiltration, granulomatosis and pulmonary edema were observed in
the lungs of mice injected with 10 mg/kg GO, whereas those injected
with 1 mg/kg excreted the GO after 1 week, and no significant
pathological response was observed (32).
The GO-induced production of excess reactive oxygen
species (ROS) is associated with acute lung injury, inflammatory
response, cell apoptosis and DNA damage (33). The activity of superoxide dismutase
and glutathione has been reported to decrease with increasing time
and dose of GO exposure (30). As
shown in Fig. 2, higher
concentrations of GO induced membrane lipid breakdown and DNA
fragmentation, but not protein denaturation. Previous studies have
also revealed that graphene activates ROS-mediated MAPKs (JNK, ERK
and p38), TGF-β signaling pathways, apoptosis-related signaling
proteins (Bcl-2 and caspase-3) and downstream genes of poly(ADP
ribose) polymerase (34). Due to
its small size, large surface area and high surface charge, GO can
cause genotoxicity in the form of chromosomal breaks, DNA strand
damage, point mutations and the formation of DNA adducts (24), which may explain the accumulation of
8-OHdG in the present study.
Intratracheal infusion and intravenous injection of
high doses of graphene nanomaterials cause significant inflammatory
reactions in animals, such as inflammatory cell infiltration,
pulmonary edema and granuloma formation (9). In the GO 50 and 100 mg/kg groups, the
lung tissues exhibited thickening of the alveolar septa and
infiltration by inflammatory cells. Furthermore, the levels of
TNF-α, IL-6, IL-1β and IL-8 in the lung tissue were significantly
increased, suggesting the activation of an inflammatory reaction.
As regards the mechanism of GO-induced inflammation, previous
publications have reported that intravenous administration of GO
may directly stimulate platelet accumulation, induce blood clot
formation and occlude pulmonary blood vessels (35). It was also suggested that graphene
and rGO bind to intracellular Toll-like receptors, activating the
NF-κB signaling pathway and triggering inflammation (36).
The number of reports on GO-induced autophagy has
increased in recent years. In the present study, the expression
levels of autophagy-related proteins (mTOR, LC3B-I/II, p62 and
beclin-1) were assessed to confirm the role of autophagy in rat
GO-induced lung injury, following treatment with different
concentrations of GO. The results revealed that GO at 50 and 100
mg/kg significantly increased the level of mTOR phosphorylation and
beclin-1 expression, but decreased the LC3B-I/II ratio and the
expression of p62. These data directly confirm that higher
concentrations of GO can induce autophagy in the lung in an
mTOR-dependent manner. To elucidate the role of autophagy in
GO-induced lung injury, rats were then treated with 50 mg/kg GO and
the autophagy inhibitors 3-MA and CLQ, and the levels of
autophagy-related proteins and the severity of GO-induced lung
injury were assessed. Autophagy was also specifically inhibited in
BEAS-2B cells via shRNA-mediated ATG5 knockdown, and changes in
cell viability, LDH release and oxidative stress were determined.
The results revealed that 3-MA and CLQ inhibited autophagy in the
lung and attenuated GO-induced lung injury. 3-MA and CLQ also
significantly reduced the production of MDA, 8-OHdG and
inflammatory factors in the lung tissue, suggesting that autophagy
may mediate the development of oxidative injury and inflammation in
the lung. Furthermore, cell viability was significantly increased
by shATG5 compared with the control shRNA. By contrast, LDH release
in the shATG5 group was significantly lower compared with that in
the shNTC group. Furthermore, the levels of both MDA and 8-OHdG
were significantly decreased by shATG5 treatment.
There is a close association between oxidative
stress and autophagy. ROS can induce autophagy via various
associated signaling pathway proteins (37), such as mTOR (38), a key negative regulator of
autophagy. mTOR activity is affected by multiple signaling
pathways, such as AMPK and P13K/Akt. It was previously demonstrated
that excess ROS can activate autophagy by inhibiting the
P13K/Akt/mTOR pathway (39).
Conversely, autophagy activation promotes the production of
catalase via the degradation of selective autophagic inhibitors,
leading to ROS accumulation, ultimately resulting in a positive
feedback regulatory loop between autophagy and ROS (40). In the present study, the fact that
3-MA and CLQ significantly reduced the production of MDA and 8-OHdG
also reflects the existence of this regulatory feedback loop. As
inhibiting autophagy via ATG5 knockdown significantly decreased
cellular injury and oxidative stress, it may be inferred that the
induction of autophagy is the key event leading to GO-associated
lung cell injury.
In conclusion, the results of the present study
confirmed that the intravenous administration of GO induces lung
injury in a dose-dependent manner, as demonstrated by lung edema
and increased lung vascular permeability, histopathological
changes, oxidative injury and inflammation. GO also significantly
induced autophagy in lung cells, while autophagy inhibitors
attenuated GO-induced lung injury. These results suggest that GO
promotes autophagy-induced lung injury.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
LZ conducted lung W/D weight ratio measurement and
BALF collection, histopathological examination, MTT assays and
wrote the manuscript; SO conducted shRNA-mediated ATG5 knockdown
and LDH measurements; HZ conducted the measurement of oxidative
products; MQ conducted western blotting; YD conducted inflammatory
cytokine ELISAs; SW collected and analyzed the data; YW interpreted
the results; JO designed the study, financially supported the study
and revised the manuscript. All authors read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
The experiment was conducted according to the
principles of the Bioethics Committee of Shanghai Jiaotong
University School of Medicine for the care and use of laboratory
animals (no. AS-20183265), as well as the Guide for the Care and
Use of Laboratory Animals (NIH publication no. 85-23, revised
1996).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bianco A: Graphene: Safe or toxic? The two
faces of the medal. Angew Chem Int Ed Engl. 52:4986–4997.
2013.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Novoselov KS, Fal'ko VI, Colombo L,
Gellert PR, Schwab MG and Kim K: A roadmap for graphene. Nature.
490:192–200. 2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Jastrzębska AM, Kurtycz P and Olszyna AR:
Recent advances in graphene family materials toxicity
investigations. J Nanopart Res. 14(1320)2012.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Loh KP, Bao Q, Eda G and Chhowalla M:
Graphene oxide as a chemically tunable platform for optical
applications. Nat Chem. 2:1015–1024. 2010.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Rong P, Yang K, Srivastan A, Kiesewetter
DO, Yue X, Wang F, Nie L, Bhirde A, Wang Z, Liu Z, et al:
Photosensitizer loaded nano-graphene for multimodality imaging
guided tumor photodynamic therapy. Theranostics. 4:229–239.
2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lv M, Zhang Y, Liang L, Wei M, Hu W, Li X
and Huang Q: Effect of graphene oxide on undifferentiated and
retinoic acid-differentiated SH-SY5Y cells line. Nanoscale.
4:3861–3866. 2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yue H, Wei W, Yue Z, Wang B, Luo N, Gao Y,
Ma D, Ma G and Su Z: The role of the lateral dimension of graphene
oxide in the regulation of cellular responses. Biomaterials.
33:4013–4021. 2012.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Li B, Yang J, Huang Q, Zhang Y, Peng C,
Zhang Y, He Y, Shi J, Li W, Hu J and Fan C: Biodistribution and
pulmonary toxicity of intratracheally instilled graphene oxide in
mice. NPG Asia Materials. 5(e44)2013.
|
|
9
|
Mao L, Hu M, Pan B, Xie Y and Petersen EJ:
Biodistribution and toxicity of radio-labeled few layer graphene in
mice after intratracheal instillation. Part Fibre Toxicol.
13(7)2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Han SG, Kim JK, Shin JH, Hwang JH, Lee JS,
Kim TG, Lee JH, Lee GH, Kim KS, Lee HS, et al: Pulmonary responses
of sprague-dawley rats in single inhalation exposure to graphene
oxide nanomaterials. Biomed Res Int. 2015(376756)2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zhang D, Zhang Z, Liu Y, Chu M, Yang C, Li
W, Shao Y, Yue Y and Xu R: The short- and long-term effects of
orally administered high-dose reduced graphene oxide nanosheets on
mouse behaviors. Biomaterials. 68:100–113. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Ema M, Gamo M and Honda K: A review of
toxicity studies on graphene-based nanomaterials in laboratory
animals. Regul Toxicol Pharmacol. 85:7–24. 2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ou L, Song B, Liang H, Liu J, Feng X, Deng
B, Sun T and Shao L: Toxicity of graphene-family nanoparticles: A
general review of the origins and mechanisms. Part Fibre Toxicol.
13(57)2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Liu JH, Yang ST, Wang H, Chang Y, Cao A
and Liu Y: Effect of size and dose on the biodistribution of
graphene oxide in mice. Nanomedicine (Lond). 7:1801–1812.
2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Park EJ, Lee GH, Han BS, Lee BS, Lee S,
Cho MH, Kim JH and Kim DW: Toxic response of graphene nanoplatelets
in vivo and in vitro. Arch Toxicol. 89:1557–1568. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Su WC, Ku BK, Kulkarni P and Cheng YS:
Deposition of graphene nanomaterial aerosols in human upper
airways. J Occup Environ Hyg. 13:48–59. 2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Jarosz A, Skoda M, Dudek I and Szukiewicz
D: Oxidative stress and mitochondrial activation as the main
mechanisms underlying graphene toxicity against human cancer cells.
Oxid Med Cell Longev. 2016(5851035)2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kumari R, Mondal T, Bhowmick AK and Das P:
Impeded repair of abasic site damaged lesions in DNA adsorbed over
functionalized multiwalled carbon nanotube and graphene oxide.
Mutat Res Genet Toxicol Environ Mutagen. 804:39–46. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhang W, Sun Y, Lou Z, Song L, Wu Y, Gu N
and Zhang Y: In vitro cytotoxicity evaluation of graphene oxide
from the peroxidase-like activity perspective. Colloids Surf B
Biointerfaces. 151:215–223. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Hirsch LR, Stafford RJ, Bankson JA,
Sershen SR, Rivera B, Price RE, Hazle JD, Halas NJ and West JL:
Nanoshell-mediated near-infrared thermal therapy of tumors under
magnetic resonance guidance. Proc Natl Acad Sci USA.
100:13549–13554. 2003.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Chen GY, Chen CL, Tuan HY, Yuan PX, Li KC,
Yang HJ and Hu YC: Graphene oxide triggers toll-like
receptors/autophagy responses in vitro and inhibits tumor growth in
vivo. Adv Healthc Mater. 3:1486–1495. 2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Chen GY, Meng CL, Lin KC, Tuan HY, Yang
HJ, Chen CL, Li KC, Chiang CS and Hu YC: Graphene oxide as a
chemosensitizer: Diverted autophagic flux, enhanced nuclear import,
elevated necrosis and improved antitumor effects. Biomaterials.
40:12–22. 2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Kang Y, Liu J, Wu J, Yin Q, Liang H, Chen
A and Shao L: Graphene oxide and reduced graphene oxide induced
neural pheochromocytoma-derived PC12 cell lines apoptosis and cell
cycle alterations via the ERK signaling pathways. Int J
Nanomedicine. 12:5501–5510. 2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Domagala A, Stachura J, Gabrysiak M,
Muchowicz A, Zagozdzon R, Golab J and Firczuk M: Inhibition of
autophagy sensitizes cancer cells to photofrin-based photodynamic
therapy. BMC Cancer. 18(210)2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhang X, Li M, Wang YB, Cheng Y, Zheng YF,
Xi TF and Wei SC: Cell response of nanographene platelets to human
osteoblast-like MG63 cells. J Biomed Mater Res A. 102:732–742.
2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Chatterjee N, Eom HJ and Choi J: A systems
toxicology approach to the surface functionality control of
graphene-cell interactions. Biomaterials. 35:1109–1127.
2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Liu Y, Luo Y, Wu J, Wang Y, Yang X, Yang
R, Wang B, Yang J and Zhang N: Graphene oxide can induce in vitro
and in vivo mutagenesis. Sci Rep. 3(3469)2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Vallabani NV, Mittal S, Shukla RK, Pandey
AK, Dhakate SR, Pasricha R and Dhawan A: Toxicity of graphene in
normal human lung cells (BEAS-2B). J Biomed Nanotechnol. 7:106–107.
2011.PubMed/NCBI View Article : Google Scholar
|
|
29
|
De Marzi L, Ottaviano L, Perrozzi F,
Nardone M, Santucci S, De Lapuente J, Borras M, Treossi E, Palermo
V and Poma A: Flake size-dependent cyto and genotoxic evaluation of
graphene oxide on in vitro A549, CaCo2 and vero cell lines. J Biol
Regul Homeost Agents. 28:281–289. 2014.PubMed/NCBI
|
|
30
|
Chang Y, Yang ST, Liu JH, Dong E, Wang Y,
Cao A, Liu Y and Wang H: In vitro toxicity evaluation of graphene
oxide on A549 cells. Toxicol Lett. 200:201–210. 2011.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Wang K, Ruan J, Song H, Zhang J, Wo Y, Guo
S and Cui D: Biocompatibility of graphene oxide. Nanoscale Res
Lett. 6(8)2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Jasim DA, Ménard-Moyon C, Begin D, Bianco
A and Kostarelos K: Tissue distribution and urinary excretion of
intravenously administered chemically functionalized graphene oxide
sheets. Chem Sci. 6:3952–3964. 2015.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Singh Z: Applications and toxicity of
graphene family nanomaterials and their composites. Nanotechnol Sci
Appl. 9:15–28. 2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Li Y, Liu Y, Fu Y, Wei T, Le Guyader L,
Gao G, Liu RS, Chang YZ and Chen C: The triggering of apoptosis in
macrophages by pristine graphene through the MAPK and TGF-beta
signaling pathways. Biomaterials. 33:402–411. 2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Fujimi S, MacConmara MP, Maung AA, Zang Y,
Mannick JA, Lederer JA and Lapchak PH: Platelet depletion in mice
increases mortality after thermal injury. Blood. 107:4399–4406.
2006.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zhou H, Zhao K, Li W, Yang N, Liu Y, Chen
C and Wei T: The interactions between pristine graphene and
macrophages and the production of cytokines/chemokines via TLR- and
NF-κB-related signaling pathways. Biomaterials. 33:6933–6942.
2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Finkel T: Signal transduction by reactive
oxygen species. J Cell Biol. 194:7–15. 2011.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Yan J, Feng Z and Liu J, Shen W, Wang Y,
Wertz K, Weber P, Long J and Liu J: Enhanced autophagy plays a
cardinal role in mitochondrial dysfunction in type 2 diabetic
Goto-Kakizaki (GK) rats: Ameliorating effects of
(-)-epigallocatechin-3-gallate. J Nutr Biochem. 23:716–724.
2012.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Yu L, McPhee CK, Zheng L, Mardones GA,
Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al: Termination of
autophagy and reformation of lysosomes regulated by mTOR. Nature.
465:942–946. 2010.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Matsuda N, Sato S, Shiba K, Okatsu K,
Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al:
PINK1 stabilized by mitochondrial depolarization recruits Parkin to
damaged mitochondria and activates latent Parkin for mitophagy. J
Cell Biol. 189:211–221. 2010.PubMed/NCBI View Article : Google Scholar
|