Introduction
The Ras-Raf-MEK pathway is a mitogen-activated
protein kinase (MAPK) pathway. Ras is activated by growth factors
such as epidermal growth factor (EGF), then further activates Raf,
MEK, and ERK, which in turn promote cell proliferation (1,2).
Ras and raf have been reported as the most frequently
mutated genes in cancers. For example, mutations of the ras
gene occur in about 90% of pancreatic cancers and ~50% of colon
cancers. Also, mutations of the raf gene occur in about 70%
of melanomas and about 10% of colon cancers (3). The mutated Ras protein reduces GTPase
function, becomes locked in a permanently activated state, and
continues to send signals downstream (4). The mutated Raf protein also activates
ERK through downstream MEK (5). ERK
activated by mutated Ras or Raf promotes cell proliferation and
enhances EGFR ligand expression, leading to hyperactivation of the
Ras-Raf-MEK pathway. This excessive signal transduction plays role
in carcinogenesis, cancer growth, and drug resistance (6,7). In
the signal transduction of the Ras-Raf-MEK pathway, a scaffold
protein called kinase suppressor of Ras (KSR) is also important.
KSR promotes the complex formation of Raf, MEK, and ERK, thereby
enhancing signal transduction (8-10).
Thus, the inhibition of KSR suppresses the signal transduction of
the Ras-Raf-MEK pathway and is useful as an anti-cancer treatment
(11,12).
In addition, it has been reported that Ras activates
the phosphatidylinositol-3-kinase (PI3K)/Akt pathway (13,14).
Like the Ras-Raf-MEK pathway, the PI3K/Akt pathway is also involved
in promoting cell proliferation and suppressing cell death
(15-17).
Therefore, in anti-cancer treatment, a drug that inhibits only the
Ras-Raf-MEK pathway will not completely eradicate cancer if the
PI3K/Akt pathway remains activated (18). Thus, because of its roles in both
pathways, Ras has been considered the most important target protein
in cancer treatment (19). However,
due to the multi-functionality of Ras, numerous Ras inhibitor
candidates failed to show an anti-cancer effect, and Ras has been
the most difficult target for anti-cancer treatment (20). Now, a number of inhibitors inhibit
downstream of the Ras pathway, but it is desirable to develop drugs
that inhibit the Ras-Raf-MEK and PI3K/Akt pathways simultaneously
(18).
Breast cancer susceptibility gene 1
(BRCA1)-associated protein 2 (BRAP2) was identified as a novel
cytoplasmically localized protein that binds to BRCA1(21). It was later reported that BRAP2 not
only binds BRCA1, but also functions as a cytoplasmic retention
protein for p21 and NF-κB (22,23).
Various studies revealed that BRAP2 plays role in diseases caused
by myocardial infarction, carotid atherosclerosis and inflammation
(23-25).
Taken together, these reports suggest that BRAP2 affects various
types of intracellular signals. Yeast two-hybrid screening revealed
that BRAP2 interact with Ras. BRAP2 is reported to inhibit the
Ras-Raf-MEK pathway by binding to KSR (26). Since the inhibition of complex
formation by the binding of BRAP2 to KSR is an event downstream of
Ras, BRAP2 may suppress the signal transduction of the Ras-Raf-MEK
pathway irrespective of the presence of Ras mutations. Moreover,
BRAP2 has been reported to bind to proteins other than KSR. One of
them is a phosphatase protein known as PH-domain and leucine-rich
repeat protein phosphatase 1 (PHLPP1) (27). This protein is involved in Akt
activation, and the regulation of PHLPP1 leads to inhibition of the
PI3K/Akt pathway (28,29). That is, BRAP2 may inhibit both the
Ras-Raf-MEK pathway and the PI3K/Akt pathway to suppress the
proliferation or induce the death of cancer cells.
However, much remains unclear about the relationship
between BRAP2 and the Ras-Raf-MEK pathway in cancer cells, and the
relation between BRAP2 and the PI3K/Akt pathway is even less clear.
In this study, to investigate the functions of BRAP2 against the
Ras-Raf-MEK and PI3K/Akt pathways, we treated cells of a
BRAP2-deficient cell line with inhibitors of either pathway and
evaluated the changes in signal transduction, apoptosis, and cell
proliferation.
Materials and methods
Cells
Jurkat cells were purchased from DS Pharma
Biomedical. THP-1 was provided by Dr Y. Kobayashi of Toho
University (Chiba, Japan). BALL-1, HL-60 and MOLT-4F were provided
by the Cell Resource Center for Biomedical Research, Tohoku
University (Sendai, Japan).
Reagents
The farnesyl transferase inhibitor tipifarnib and
the PI3K inhibitor LY294002 were purchased from Adooq Bioscience.
The pan-Raf inhibitor LY3009120 was purchased from Selleck
Chemicals. The PKC inhibitor staurosporine was purchased from
Cayman Chemical. All inhibitors were solubilized in dimethyl
sulfoxide (DMSO, Wako). Phorbol 12-myristate 13-acetate (PMA) was
purchased from Wako.
Medium and cell culture
The cells were cultured in RPMI1640 medium
(Sigma-Aldrich; Merck KGaA) containing 3.5 µg/l 2-mercaptoethanol
(Wako), 75 mg/l kanamycin sulfate (Wako), and 2 g/l
NaHCO3 (Wako) supplemented with 10% fetal bovine serum
(Biofill) and maintained at 37˚C in a humidified chamber (ESPEC)
under an atmosphere of 95% air and 5% CO2.
Antibodies
Anti-BRAP2 polyclonal antibody (cat. no. ab77721)
was purchased from Abcam. Anti-KSR polyclonal antibody (cat. no.
AP7202a) was purchased from WuXi AppTec (Shanghai, China).
Anti-β-actin polyclonal antibody (cat. no. 4967S),
anti-phosphorylated (p)-Raf monoclonal antibody (cat. no. 9427S),
anti-Raf polyclonal antibody (cat. no. 9422S), anti-MEK polyclonal
antibody (cat. no. 9122S), p-ERK polyclonal antibody (cat. no.
9101S), anti-ERK polyclonal antibody (cat. no. 9102S), anti-p-Akt
polyclonal antibody (cat. no. 9271S), anti-rabbit IgG-HRP linked
antibody (cat. no. 7074S) and anti-mouse IgG-HRP linked antibody
(cat. no. 7076S) were purchased from Cell Signaling Technology.
Anti-caspase-3 monoclonal antibody (cat. no. sc-7272), anti-p-MEK
monoclonal antibody (cat. no. sc-81503) and anti-Akt polyclonal
antibody (cat. no. sc-8312) were purchased from Santa Cruz
Biotechnology.
Knockout of BRAP2 by CRISPR/Cas9
pSpCas9 (BB)-2A-Puro (PX459) V 2.0 (Plasmid #62988)
was purchased from Addgene (30).
The BbsI site of the plasmid was cut with BbsI (New
England Biolabs) at 37˚C for 1 h. The gRNA of BRAP2 (top:
CACCGGAAAGGCGCTGCGTTCGAAA, bottom: AAACTTTCGAACGCAGCGCCTTTCCC)
designed in CRISPRdirect (https://crispr.dbcls.jp) was ligated to the
BbsI site of the plasmid using a DNA ligation kit (Takara
Bio) at 16˚C for 3 h. The plasmid was transfected into Jurkat cells
by using the Neon transfection system (Thermo Fisher Scientific,
Inc.) under the conditions of pulse voltage 1350 (V), pulse width
10 (ms) and pulse number 3. Before this transfection, the Jurkat
cells were cultured in serum-free RPMI1640 medium containing an
antibiotic agent at 37˚C for 4 h. The cells (2x106) were
washed with Ca²+ and Mg²+-free phosphate
buffered saline (PBS), then supplemented with 30 µl of resuspension
buffer R (Thermo Fisher Scientific, Inc.) and 10 µl of the plasmid
DNA. After transfection, the transfected Jurkat cells were cultured
in serum containing antibiotic-free RPMI-1640 medium containing 10%
FBS. After 72 h, the transfected Jurkat cells were cultured in
RPMI-1640 medium containing 10% FBS and 0.5 µg/ml puromycin
(Sigma-Aldrich; Merck KGaA) for one month. For single cell cloning,
the drug-selected Jurkat cells were diluted and seeded at 1 cell
per well in 96-well plates (Becton, Dickinson and Company). The
wells were confirmed to each have a single cell by examination
under a phase contrast microscope (Olympus) every 2 days. As soon
as they began to grown, single cell clones were transferred to
24-well (Sigma-Aldrich; Merck KGaA), 12-well (Corning) or 6-well
(Thermo Fisher Scientific, Inc.). Finally, stable strains of Jurkat
(Mock) and Jurkat (Δ BRAP2) cell lines were generated.
MTT assay
Cells (2x104) were incubated in 96-well
plates with or without the Ras, pan-Raf or PI3K inhibitors at 37˚C
for 24 h. At 1 h prior to the end of incubation, 10 µl of 5 mg/ml
MTT (Wako) solution was added to each well, and the plates were
further incubated for 1 h. Then, the 96-well plates were
centrifuged at 300 g for 5 min, and the supernatant of each well
was removed. One-hundred microliters of DMSO was added to each
well, and the cell viability was determined by measuring the
absorbance of the formazan at 570 nm using a microplate reader
(Awareness Technology).
SubG1 and cell cycle analysis by
propidium iodide staining
Cells (2x106) were treated with the Ras,
Pan-Raf or PI3K inhibitors at 37˚C for 24 h. The concentration of
cells was adjusted to 1x106 cells and then the cells
were washed with PBS. The cells were added to a solution containing
500 µl of 0.1% Triton X-100 (Wako)-PBS, 5 µl of 5 mg/ml RNase A
(Wako), and 12.5 µl of 1 mg/ml PI (Wako). After the cells were left
in the dark for 20 min at room temperature, they were passed
through a pore-size nylon mesh. PI fluorescence was measured by a
flow cytometer (Becton, Dickinson and Company).
Measurement of cell proliferation
rate
Cells (1x106) were seeded in a BioLite 60
mm tissue culture dish (Thermo Fisher Scientific, Inc.). The cells
were counted at the indicated times with a counting chamber
(Hirschmann). For the measurement of cell growth inhibition by the
PI3K inhibitor, cells (1x106) were seeded in a BioLite
60 mm tissue culture dish, treated with the PI3K inhibitor at 37˚C
for 48 h, and counted with a counting chamber.
SDS-PAGE and western blotting
Cells (2x106) were treated with drugs at
37˚C for the indicated times. The cells were harvested and washed
with PBS. Lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 10%
glycerol, 1% Triton X-100, 1.5 mM MgCl2, 1 mM EGTA, 1 mM
sodium orthovanadate, and 1% protease inhibitor cocktail;
Sigma-Aldrich; Merck KGaA) was added to the cells, and the mixture
was left on ice for 20 min. The cells were centrifuged at 14,500 g
for 15 min, and the supernatants were harvested as lysate samples.
The samples were quantified by a BCA protein assay (Takara Bio) and
adjusted to 2 mg/ml using lysis buffer. After the addition of a
sample application buffer (4% SDS, 125 mM Tris, pH 6.8, 10%
glycerol, 0.02 mg/ml bromophenol blue, 10% 2-mercaptoethanol), the
samples were heated at 100˚C for 3 min. SDS-PAGE was performed on
4% concentrated gel and 12% or 15% running gel. After the protein
was transferred to a polyvinylidene di-fluoride (PVDF) membrane
(Bio-Rad), the PVDF membrane was soaked in 3% skim milk
(Yukijirushi) for 1 h at room temperature. The skim milk dilution
was changed and the membrane was soaked twice more for 30 min each
at room temperature. The PVDF membrane was probed with the
indicated primary antibodies (1:1,000) at 4˚C overnight. The
primary antibodies were collected, and the PVDF membrane was washed
with 0.1% Tween-20 (Wako)-PBS. The PVDF membrane was soaked in
either anti-rabbit IgG-HRP-linked antibody (Cell Signaling
Technology) or anti-mouse IgG-HRP-linked antibody (Cell Signaling
Technology) (1:2,000) for 1 h at room temperature. The secondary
antibodies were washed out and the PVDF membrane was washed three
times with 0.1% Tween-20-PBS for 15 min per wash. The PVDF membrane
was soaked in ECL Western Blotting Substrate (Thermo Fisher
Scientific, Inc.) for 1 min, then imaged with an ImageQuant LAS
4000 (GE Healthcare). For the observation of other proteins, in
case of observing other proteins, the primary and secondary
antibodies were stripped after imaging the PVDF membrane. Stripping
was accomplished by soaking the PVDF membrane in stripping buffer
(100 mM 2-mercaptoethanol, 2% SDS and 62.5 mM Tris, pH 6.7) in 65˚C
water bath for 30 min, then washing with 0.1% Tween-20-PBS for 15
min. The PVDF membrane was soaked in skim milk and reprobed with
another antibody. The relative density of bands was quantified
using ImageJ software (National Institutes of Health).
Statistical analysis
All data are presented as the mean ± SD of three
experiments, and the statistical analyses were performed using
Microsoft Excel for mac ver. 16.0 (Microsoft Corporation) and R
ver. 4.0 software (R Foundation for Statistical Computing). The
Student's t-test was used to compare paired groups. One-way
analysis of variance was used for multi-group analysis, followed by
Bonferroni test as a post hoc test. P<0.05 was considered to
indicate a significant difference.
Results
BRAP2 regulated the signal
transduction of the Ras-Raf-MEK and PI3K/Akt pathways
First, to investigate the role of BRAP2 in the
Ras-Raf-MEK and PI3K/Akt pathways, we knockout BRAP2 expression
using CRISPR/Cas9. After transfecting the mock and designed BRAP2
knockout plasmids, we performed western blotting to determine
whether BRAP2 was knocked out in Jurkat cells. The result showed
that BRAP2 was knocked out in Jurkat (Δ BRAP2) cells (Fig. 1A). Because BRAP2 was reported to be
a cell cycle regulator (31), we
measured the proliferation of Jurkat (Mock) and Jurkat (Δ BRAP2)
cells. We found that Jurkat (Δ BRAP2) cells proliferated more
slowly than the parental cells (Fig.
1B), but the BRAP2 deletion did not induce cell death (data not
shown). In addition, we conducted western blotting to evaluate how
the loss of BRAP2 affected the Ras-Raf-MEK and PI3K/Akt pathways.
The results showed that the levels of p-Raf, p-MEK and p-Akt were
markedly higher in Jurkat (Δ BRAP2) cells compared to Jurkat (Mock)
cells, but the level of p-ERK was markedly lower in the Jurkat (Δ
BRAP2) cells. On the other hand, there was no change in the
expression of KSR (Fig. 1C). These
results suggest that BRAP2 knockout dose not directly downregulate
or upregulate the Ras-Raf-MEK pathway to a significant degree,
which is consistent with a previous study (26). Next, western blotting was performed
to evaluate the effects of the loss of BRAP2 on the Ras, pan-Raf,
and PI3K inhibitors. In Jurkat (Mock) cells, the levels of p-Raf,
p-MEK, and p-ERK were decreased by treatment with the farnesyl
transferase inhibitor tipifarnib, which is used as a Ras inhibitor,
or with the pan-Raf inhibitor LY3009120. Conversely, in the Jurkat
(Δ BRAP2) cells, the levels of these phosphorylations were not
decreased by Ras and pan-Raf inhibitors treatment (Fig. 1D). LY294002, a PI3K inhibitor,
markedly inhibited the phosphorylation of Akt within 24 h, but this
was restored by 48 h treatment in Jurkat (Mock) cells. PI3K
inhibitor did not decrease the level of p-Akt in Jurkat (Δ BRAP2)
cells (Fig. 1E).
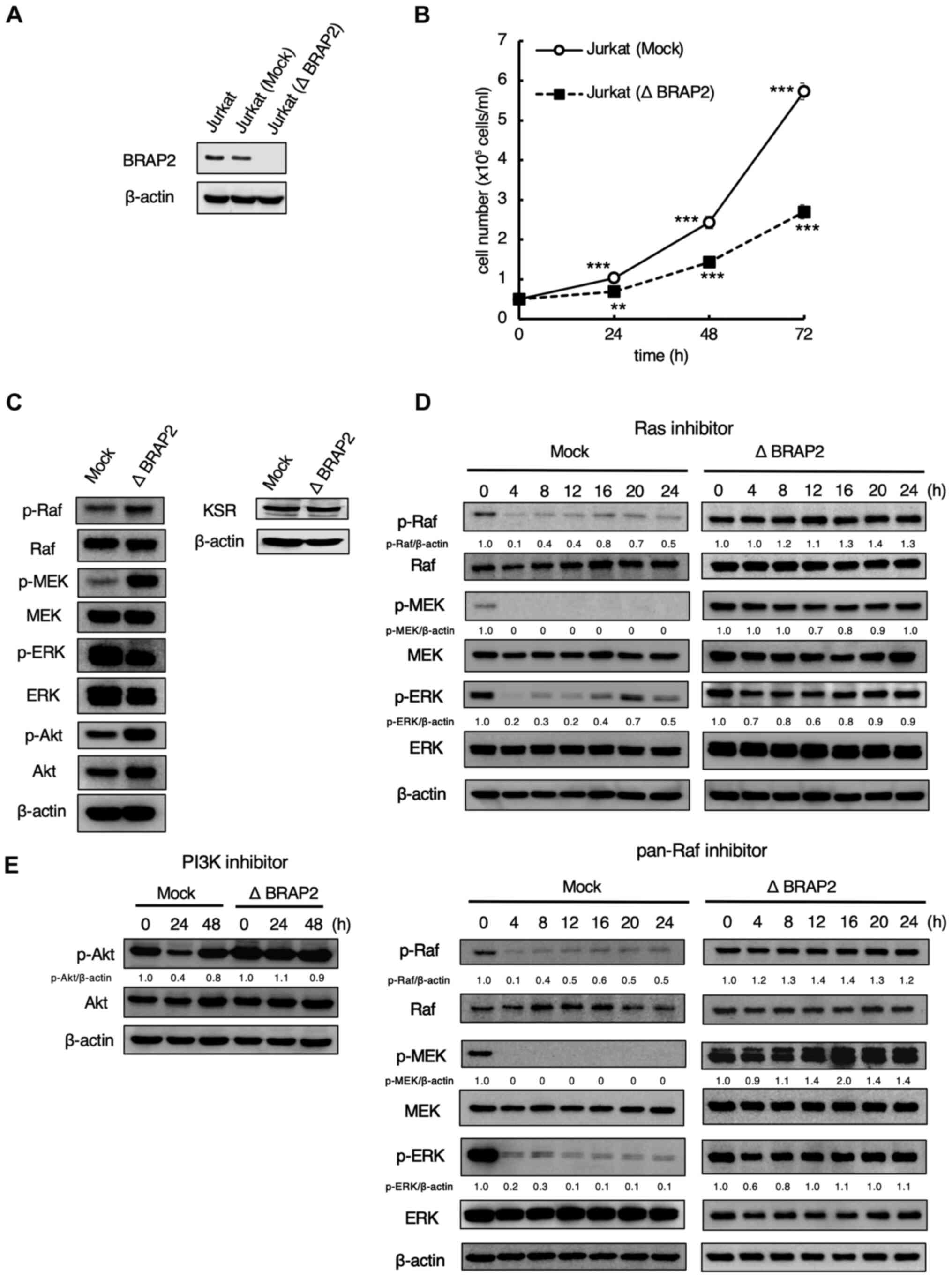 | Figure 1BRAP2 regulates the signal
transduction of the Ras-Raf-MEK and PI3K/Akt pathways. (A) Jurkat
(Mock) and Jurkat (Δ BRAP2) cells were lysed, and BRAP2 and β-actin
were detected via western blotting. (B) Jurkat (Mock) and Jurkat (Δ
BRAP2) cells were counted at the indicated times. Each bar denotes
the standard deviation (n=3). **P<0.01,
***P<0.001 vs. 0 h samples. (C) Jurkat (Mock) and
Jurkat (Δ BRAP2) cells were lysed and p-Raf, Raf, p-MEK, MEK,
p-ERK, ERK, p-Akt, Akt, KSR and β-actin were detected via western
blotting. (D) Jurkat (Mock) and Jurkat (Δ BRAP2) cells were
incubated with 5 µM Ras inhibitor tipifarnib or 10 µM pan-Raf
inhibitor LY3009120 for the indicated times. The cells were lysed
and p-Raf, Raf, p-MEK, MEK, p-ERK, ERK, and β-actin were detected
via western blotting. The relative densities of the p-Raf, p-MEK,
p-ERK bands were estimated and normalized to the β-actin band. (E)
Jurkat (Mock) and Jurkat (Δ BRAP2) cells were incubated with 30 µM
PI3K inhibitor LY294002 for the indicated times. The cells were
lysed and p-Akt, Akt and β-actin were detected via western
blotting. The relative density of the p-Akt band was estimated and
normalized to the β-actin band. BRAP2, breast cancer susceptibility
gene 1-associated protein 2; p-phosphorylated; KSR, kinase
suppressor of Ras. |
Loss of BRAP2 suppressed apoptosis by
Ras and pan-Raf inhibitors
BRAP2 has been shown to bind to KSR, thereby
inhibiting the Ras-Raf-MEK pathway (26), and it also binds to PHLPP, which is
a modulator of Akt (28,29). In our study, because BRAP2 deletion
suppressed the inhibition of signal transduction by the Ras and
Pan-Raf inhibitor (Fig. 1D), we
predicted that BRAP2 deletion also affects the cytotoxicity of
these inhibitors. To investigate this possibility, we performed an
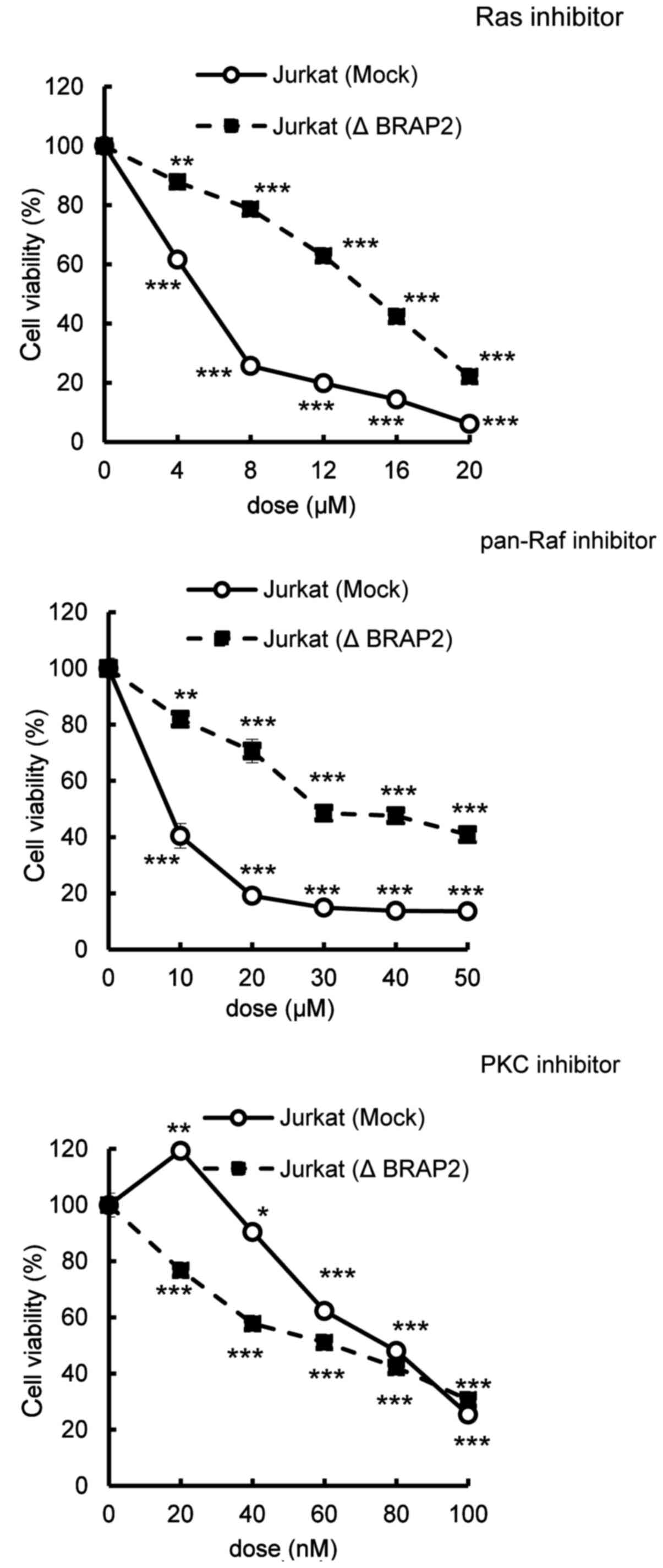
MTT assay of the inhibitor cytotoxicity. We found that the loss of
BRAP2 suppressed the cytotoxicities of the Ras and pan-Raf
inhibitors, but not the cytotoxicity of staurosporine, a PKC
inhibitor, not targeting the Ras-Raf-MEK pathway or the PI3K/Akt
pathway (Fig. 2) (32,33).
Since the MTT assay measures mitochondrial enzyme
activity, it cannot determine whether each inhibitor induces cell
death. However, cell death is usually accompanied by DNA
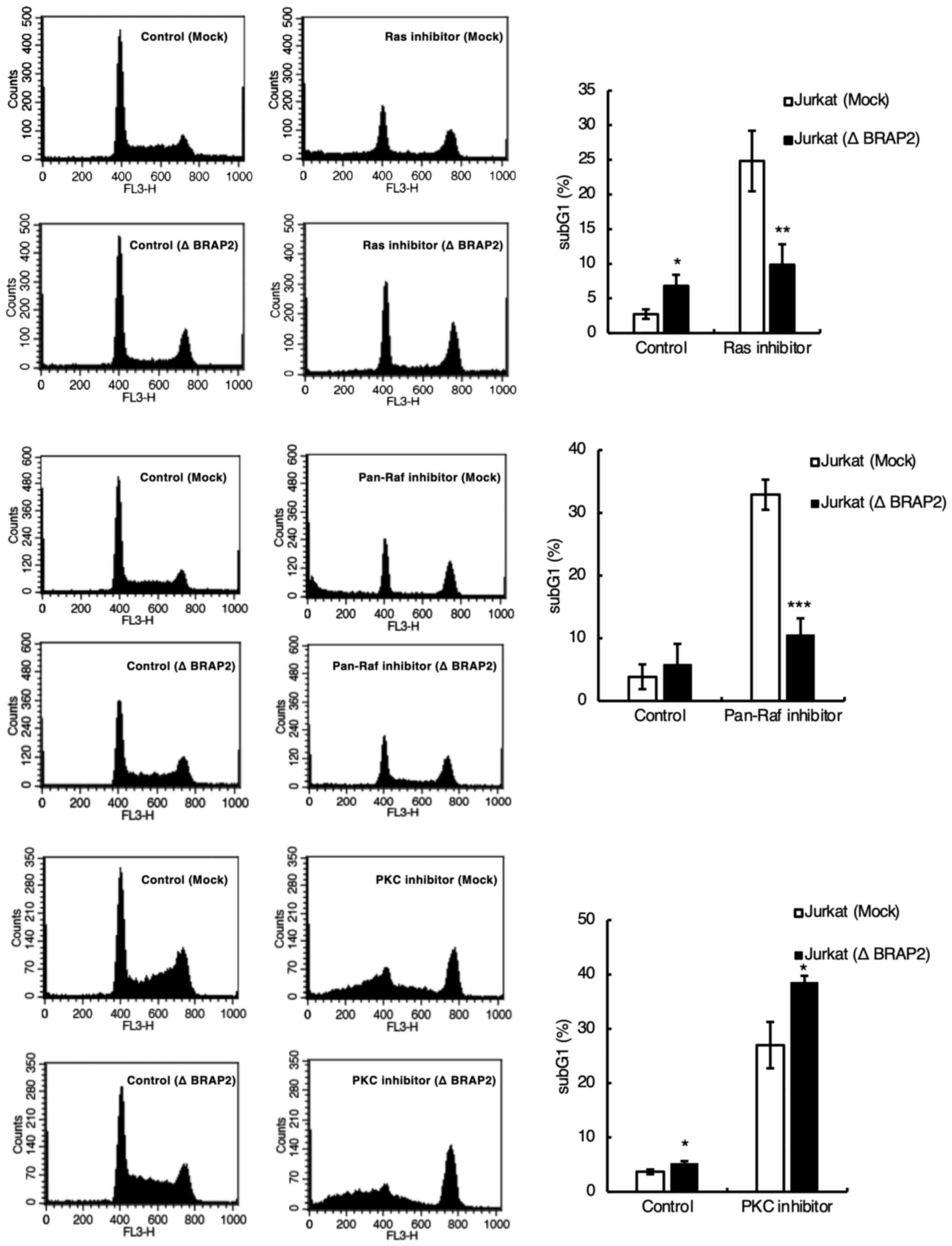
fragmentation. Therefore, we investigated whether each inhibitor
induces cell death by staining DNA with PI and detecting the subG1
phase using flow cytometry (34,35).
We found that the loss of BRAP2 suppressed DNA fragmentation by the
Ras and pan-Raf inhibitors but not by the PKC inhibitor (Fig. 3). We next examined the influence of
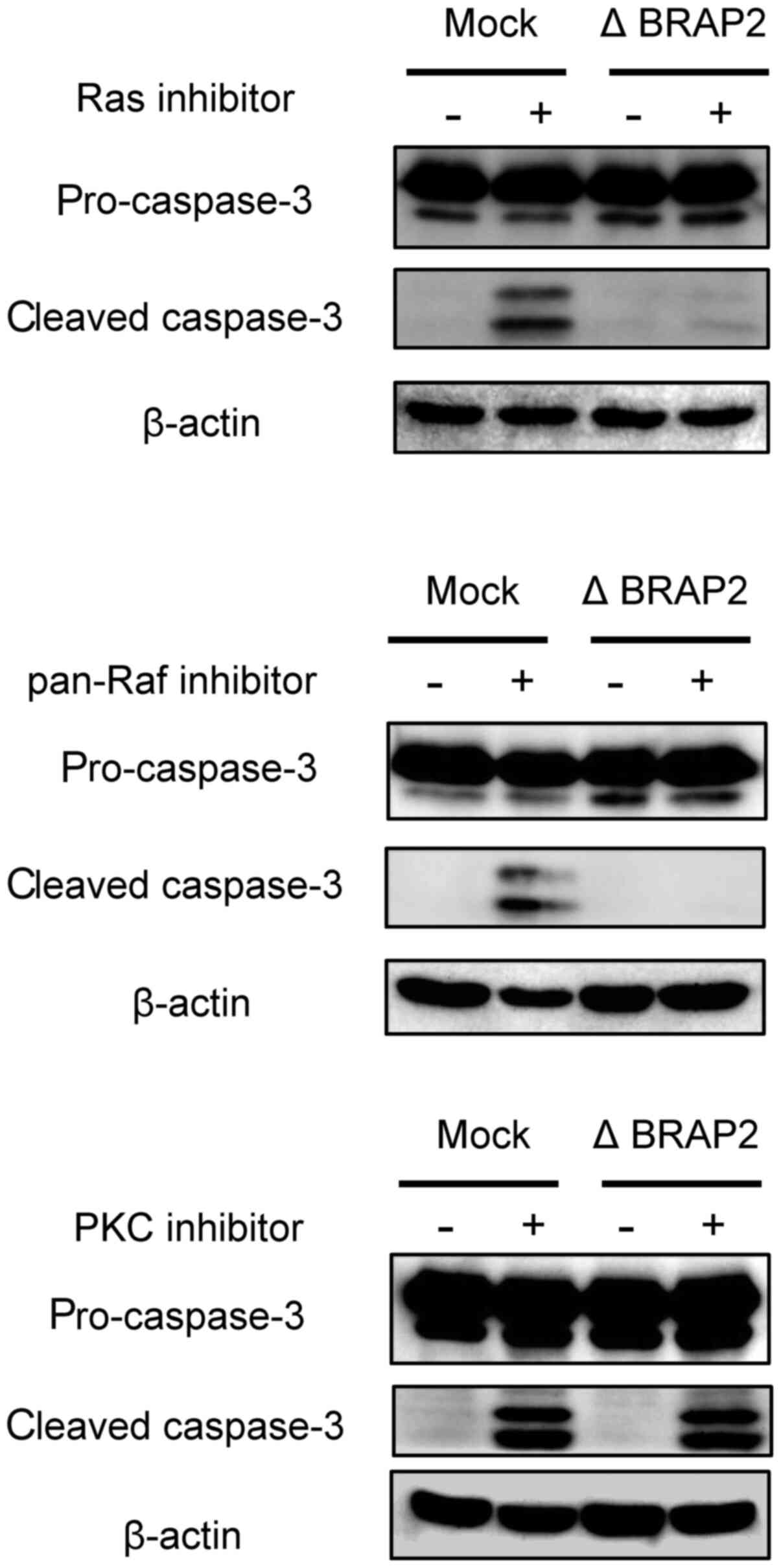
BRAP2 loss on apoptosis. During apoptosis, cleaved caspase-3
appears, so cleaved caspase-3 is often used as a marker of
apoptosis (36). Therefore, to
determine whether each inhibitor induce apoptosis in our present
experiments, we evaluated cleaved caspase-3 by western blotting.
The Ras and Pan-Raf inhibitors increased the level of cleaved
caspase-3 in Jurkat (Mock) cells, but cleaved caspase-3 was not
observed in the Jurkat (Δ BRAP2) cells following treatment with
these inhibitors. BRAP2. On the other hand, cleaved caspase-3 was
present in both Jurkat (Mock) and Jurkat (Δ BRAP2) cells treated
with the PKC inhibitor (Fig.
4).
Inhibition of the Ras-Raf-MEK pathway
by a Ras inhibitor was involved in apoptosis
In order to investigate whether the Ras inhibitor
used in this study induces apoptosis through the Ras-Raf-MEK
pathway, the cells were subjected to co-treatment with the Ras
inhibitor and the activator PMA. As shown in Fig. 5A, the phosphorylations of Raf, MEK,
and ERK were inhibited by Ras inhibitor treatment alone but were
restored by co-treatment with PMA and the Ras inhibitor (Fig. 5A). In addition, we examined whether
PMA would suppress the Ras inhibitor-induced apoptosis. We found
that Ras inhibitor treatment increased the levels of subG1-phase
cells and cleaved caspase-3, while co-treatment with the Ras
inhibitor and PMA suppressed the increase in subG1 and cleaved
caspase-3 (Fig. 5B and C). This demonstrated that the Ras
inhibitor induced apoptosis through the Ras-Raf-MEK pathway.
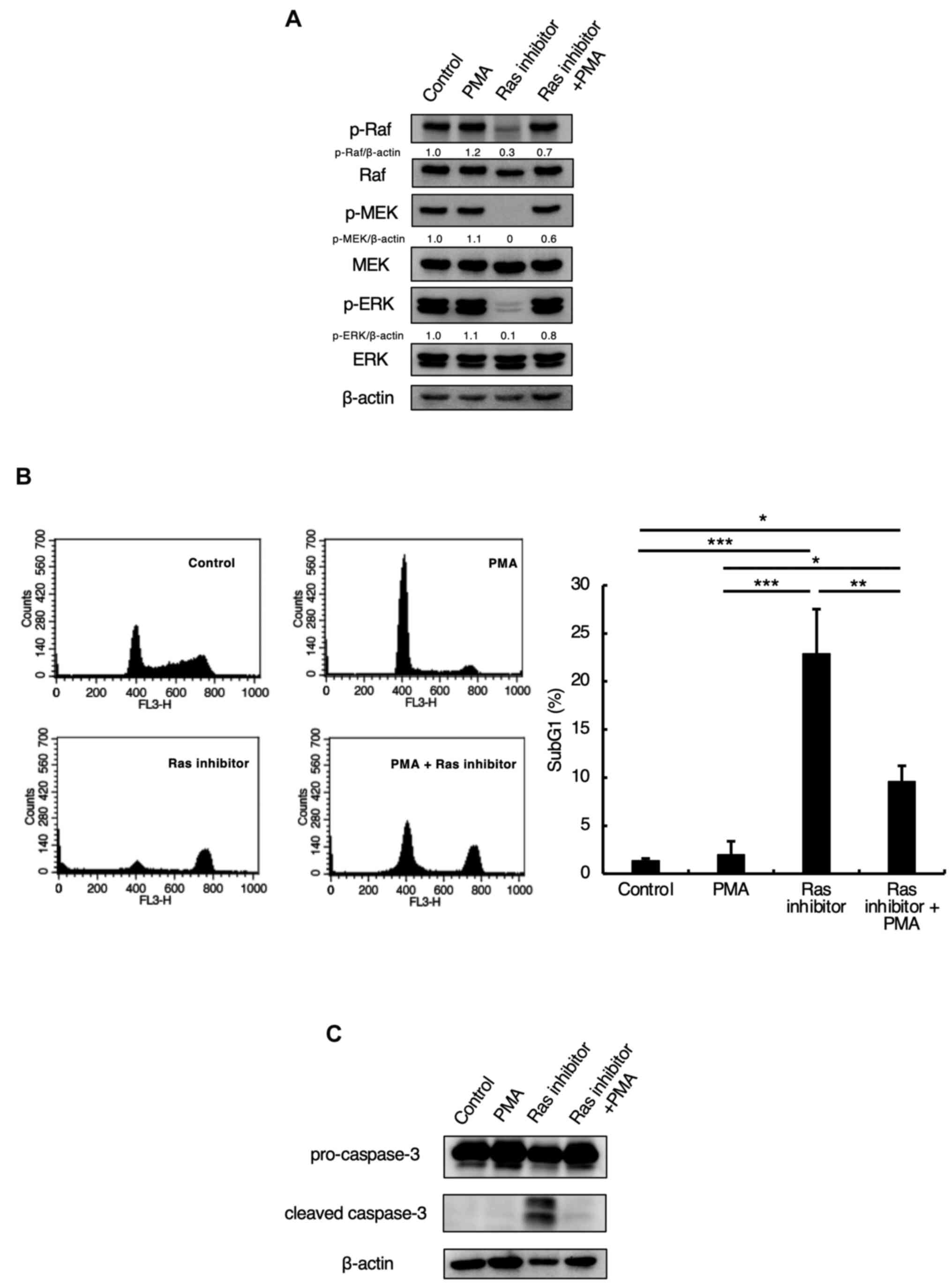 | Figure 5Inhibition of the Ras-Raf-MEK pathway
by a Ras inhibitor influences apoptosis. (A) Jurkat cells were
preincubated with 100 nM PMA for 1 h and incubated with 5 µM Ras
inhibitor for 24 h. Cells were lysed and p-Raf, Raf, p-MEK, MEK,
p-ERK, ERK, and β-actin were detected by western blotting. The
relative density of the p-Raf, p-MEK, p-ERK bands were estimated
and normalized by β-actin band. (B) Jurkat cells were preincubated
with 100 nM PMA for 1 h and incubated with 5 µM Ras inhibitor for
24 h. SubG1 phase was detected by flow cytometric analysis with PI
staining, as described in the Materials and methods section.
Representative histograms of one of three independent measurements
are shown. The bar graph shows the percentages of cells in the
sub-G1 phase. Each bar denotes the standard deviation (n=3).
*P<0.05, **P<0.01,
***P<0.001. (C) Jurkat cells were preincubated with
100 nM PMA for 1 h and incubated with 5 µM Ras inhibitor for 24 h.
The cells were lysed, and caspase-3 and β-actin expression levels
were detected via western blotting. BRAP2, breast cancer
susceptibility gene 1-associated protein 2; p-, phosphorylated-;
PMA, phorbol 12-myristate 13-acetate. |
Loss of BRAP2 suppressed cell cycle
arrest by a PI3K inhibitor
Next, we evaluated the involvement of BRAP2 in the
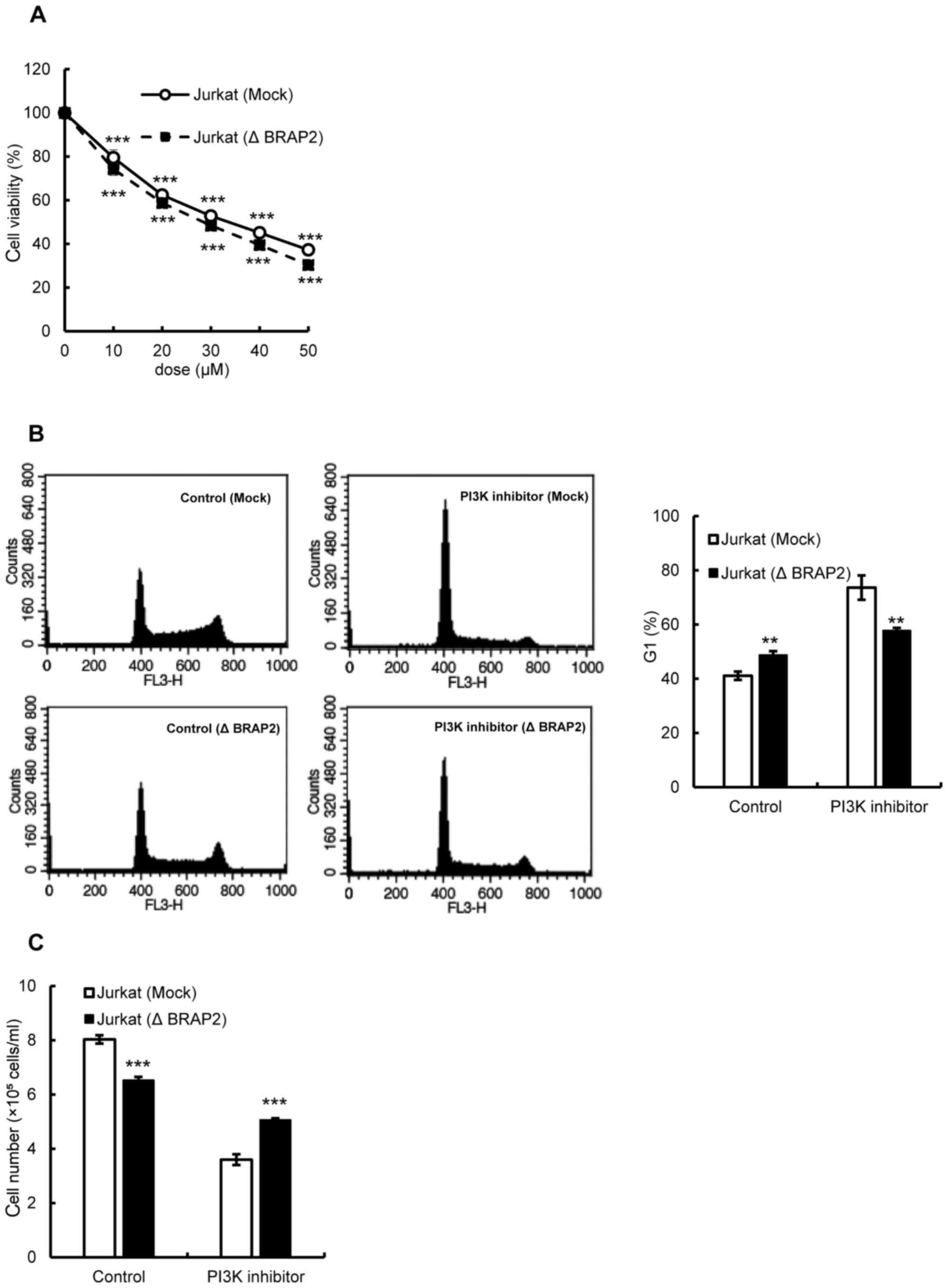
PI3K/Akt pathway. An MTT assay was used to examine whether the loss
of BRAP2 changed the susceptibility of cells to PI3K
inhibitor-induced cell death. However, the results showed that the
presence or absence of BRAP2 expression had no effect on the
susceptibility of cells to PI3K inhibitor-induced cell death
(Fig. 6A). Therefore, we further
investigated the effect of the PI3K inhibitor on the cell cycle.
Unlike the Ras and Pan-Raf inhibitors, the PI3K inhibitor increased
the G1 phase cells in Jurkat (Mock) cells, and the loss of BRAP2
suppressed the increase in the G1 phase (Fig. 6B). Finally, we assessed the
potential inhibition of cell proliferation by the PI3K inhibitor
and found that the PI3K inhibitor did indeed inhibit the
proliferation of Jurkat (Mock) cells, and the BRAP2 deletion
suppressed this inhibition (Fig.
6C).
BRAP2 expression was similar among
various leukemia cells
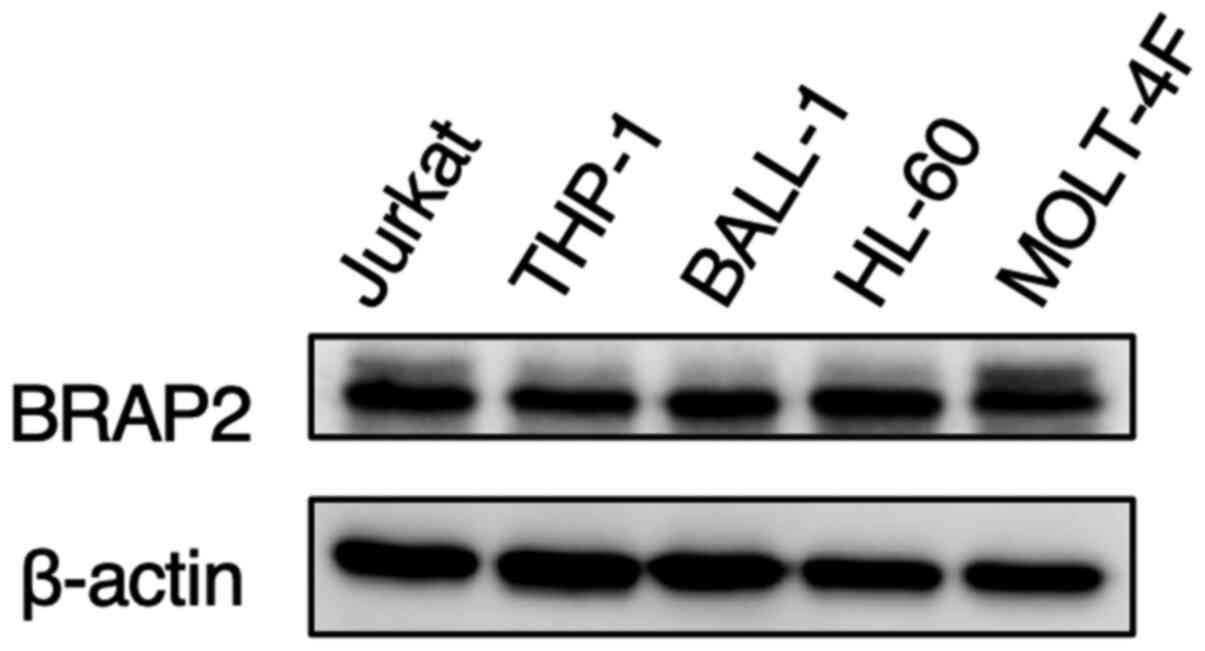
Finally, we investigated the BRAP2 expression levels
in other leukemia cell lines by western blotting. The levels of
BRAP2 protein expression in human monocytic THP-1 cells, human
B-cell lymphoma BALL-1 cells, human acute promyelocytic HL-60
cells, and human acute T-lymphoblastic MOLT-4F cells were almost
the same as those in human lymphoid helper T-cell line Jurkat cells
(Fig. 7).
Discussion
Because BRAP2 is required for homeostasis, the
mutation or deletion of BRAP2 may cause carcinogenesis (37). In addition, BRAP2 has been shown to
interact with regulators of the Ras-Raf-MEK and PI3K/Akt pathways,
thereby contributing to carcinogenesis (26,27).
These reports suggest that BRAP2 plays a role in carcinogenesis by
regulating the Ras-Raf-MEK and PI3K/Akt pathways; however, the
relationships between BRAP2 and these pathways in cancer are still
unclear. Therefore, in this study, to clarify the role of BRAP2 on
the Ras-Raf-MEK and PI3K/Akt pathways, we deleted BRAP2. We found
that the deletion suppressed apoptosis by a Ras inhibitor and a
pan-Raf inhibitor and inhibited the cell cycle arrest by a PI3K
inhibitor.
After generating BRAP2-deficient cells, we conducted
a western blot analysis to evaluate changes in the Ras-Raf-MEK and
the PI3K/Akt pathways in the normal state, since BRAP2 has been
reported to be involved in both pathways (26,27).
Interestingly, BRAP2 deletion increased the phosphorylation levels
of Raf and MEK (Fig. 1C). KSR is a
scaffold protein and enhances the Ras-Raf-MEK pathway (8-10).
Because BRAP2 inhibits KSR and thereby impedes the signal
transduction from Raf to MEK (26),
we considered that BRAP2 deletion may increase the phosphorylation
levels of Raf and MEK. In our present experiments, it was also of
interest that BRAP2 deletion caused an increase in the
phosphorylation level of Akt (Fig.
1C). A recent report showed that BRAP2 knockdown led to an
increase in the phosphorylation levels of Akt and mTOR in glioma
cells (38), and our result was
consistent with that report. BRAP2 binds PHLPP1 (27,39)
which is involved in Akt activation (28,29),
and it has been suggested that BRAP2 suppresses Akt through PHLPP1
to promote apoptosis induction (39). Therefore, we thought that the
phosphorylation level of Akt may have been increased because BRAP2
deletion could not suppress Akt through PHLPP1. Taken together,
these results suggest that BRAP2 negatively regulates the
Ras-Raf-MEK and PI3K/Akt pathways through binding partners in a
normal state. BRAP2 deletion also attenuated the Ras
inhibitor-mediated and pan-Raf inhibitor-mediated apoptosis
(Figs. 3 and 4). KSR acts as a scaffold to bind Raf,
MEK, and ERK, but KSR also possesses an intrinsic Raf activating
mechanism independent of Ras (40).
That is, KSR and Raf heterodimerization directly activate the
Raf-MEK-ERK pathway. The Ras inhibitor used in this study, inhibits
farnesyl transferase and target the region upstream of KSR, while
the pan-Raf inhibitor LY3009120 inhibits Raf dimerization by the
Ras signal, which is also upstream of KSR (41). Therefore, we considered that BRAP2
deletion increases the scaffolding and Raf-activating ability of
KSR and further activates MEK and ERK independently of Ras
(7,26,40),
which is in agreement with our present findings.
If the inhibitors used in this study targeted
further downstream than KSR, the influence of BRAP2 deletion might
be unaffected. For instance, the MEK inhibitor U-0126 is an
inhibitor further downstream than KSR. However, U-0126
nonspecifically inhibited Akt as well as ERK phosphorylation at
concentrations that induced cell death (data not shown), and thus
U-0126 treatment could not be used to confirm our hypothesis. In
light of all the above, we conclude that BRAP2 deletion attenuated
Ras and pan-Raf inhibitor activity in this study.
In the PI3K/Akt pathway, the PI3K inhibitor blocked
the level of p-Akt within 24 h; however, it was found to restore
p-Akt within 48 h in the presence of BRAP2. Deletion of BRAP2
suppressed the inhibition of Akt phosphorylation by the PI3K
inhibitor for 48 h (Fig. 1E). BRAP2
was found to bind to not only KSR, but other proteins as well. One
of them is PHLPP, which controls Akt phosphorylation (28,29).
Also, the PI3K/Akt pathway is frequently overactive in T-ALL
(42). Thus, we predicted that
BRAP2 can regulate the level of p-Akt within 24 h, but, due to the
abnormal activation of the PI3K/Akt pathway, the decreased levels
of p-Akt were restored within 48 h. Similarly, it is considered
that the PI3K inhibitor induced cell growth inhibition rather than
apoptosis, unlike the Ras-Raf-MEK pathway inhibitors, because of
the abnormal activation of the PI3K/Akt pathway in Jurkat cells
(Fig. 6C). It was reported that the
Jurkat cell line used in this study is deficient in phosphatase and
tensin homolog deleted on chromosome 10 (PTEN) (43). PTEN is a tumor suppressor and
regulates the survival of T-cells through the PI3K/Akt pathway.
Because Jurkat cells are deficient in PTEN, apoptosis is suppressed
(43,44). Therefore, we predicted that the PI3K
inhibitor inhibited cell proliferation, not apoptosis. However, the
interaction between BRAP2 and the mutated protein or pathway is
still unclear and needs further study.
In summary, the present study focused on signal
transduction, apoptosis, and cell proliferation to clarify the role
of BRAP2 on the Ras-Raf-MEK and PI3K/Akt pathways. The results
showed that BRAP2 induces apoptosis and cell growth inhibition
against Jurkat cells by negatively regulating the Ras-Raf-MEK and
PI3K/Akt pathways. These pathways are important targets in cancer
treatment, and BRAP2 negatively regulates them. Moreover, we found
that BRAP2 expression was mostly similar among various leukemia
cells, suggesting that BRAP2 regulates the Ras-Raf-MEK and PI3K/Akt
pathways in different leukemia cell lines (Fig. 7). Moreover, in a recent report, a
reduction in BRAP2 expression was shown to promote cancer cell
proliferation both in vitro and in vivo (38). Further studies are warranted to
investigate the BRAP2 expression and cytotoxicities compared with
normal lymphocytes for cancer cell progression, and we will examine
in the future study. Therefore, the development of a drug that
enhances the function of BRAP2 would be a new approach in cancer
treatment.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YN and TS designed the research. HS performed the
experiments. YN and HS drafted the manuscript and analyzed data.
IS, TS and HS interpreted data and revised the manuscript. YN and
HS confirm the authenticity of all the raw data. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Raman M, Chen W and Cobb MH: Differential
regulation and properties of MAPKs. Oncogene. 26:3100–3112.
2007.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Brunet A, Roux D, Lenormand P, Dowd S,
Keyse S and Pouysségur J: Nuclear translocation of p42/p44
mitogen-activated protein kinase is required for growth
factor-induced gene expression and cell cycle entry. EMBO J.
18:664–674. 1999.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Downward J: Targeting RAS signalling
pathways in cancer therapy. Nat Rev Cancer. 3:11–22.
2003.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Corcoran RB, Ebi H, Turke AB, Coffee EM,
Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D,
Hung KE, et al: EGFR-mediated re-activation of MAPK signaling
contributes to insensitivity of BRAF mutant colorectal cancers to
RAF inhibition with vemurafenib. Cancer Discov. 2:227–235.
2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Misale S, Yaeger R, Hobor S, Scala E,
Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M,
Siravegna G, et al: Emergence of KRAS mutations and acquired
resistance to anti-EGFR therapy in colorectal cancer. Nature.
28:532–536. 2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Vakana E, Pratt S, Blosser W, Dowless M,
Simpson N, Yuan XJ, Jaken S, Manro J, Stephens J, Zhang Y, et al:
LY3009120, a pan RAF inhibitor, has significant anti-tumor activity
in BRAF and KRAS mutant preclinical models of colorectal cancer.
Oncotarget. 8:9251–9266. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Therrien M, Michaud NR, Rubin GM and
Morrison DK: KSR modulates signal propagation within the MAPK
cascade. Gene Dev. 10:2684–2695. 1996.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Kortum RL and Lewis RE: The molecular
scaffold KSR1 regulates the proliferative and oncogenic potential
of cells. Mol Cell Biol. 24:4407–4416. 2004.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Razidlo GL, Kortum RL, Haferbier JL and
Lewis RE: Phosphorylation regulates KSR1 stability, ERK activation,
and cell proliferation. J Biol Chem. 279:47808–47814.
2004.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wang L, Jiang CF, Li DM, Ge X, Shi ZM, Li
CY, Liu X, Yin Y, Zhen L, Liu LZ and Jiang BH: MicroRNA-497
inhibits tumor growth and increases chemosensitivity to
5-fluorouracil treatment by targeting KSR1. Oncotarget.
7:2660–2671. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhou L, Lyons-Rimmer J, Ammoun S, Muller
J, Lasonder E, Sharma V, Ercolano E, Hilton D, Taiwo I, Barczyk M
and Hanemann CO: The scaffold protein KSR1, a novel therapeutic
target for the treatment of merlin-deficient tumors. Oncogene.
35:3443–3453. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Posch C, Moslehi H, Feeney L, Green GA,
Ebaee A, Feichtenschlager V, Chong K, Peng L, Dimon MT, Phillips T,
et al: Combined targeting of MEK and PI3K/mTOR effector pathways is
necessary to effectively inhibit NRAS mutant melanoma in vitro and
in vivo. Proc Natl Acad Sci USA. 110:4015–4020. 2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Mendoza MC, Er EE and Blenis J: The
Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends
Biochem Sci. 36:320–328. 2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Schult C, Dahlhaus M, Ruck S, Sawitzky M,
Amoroso F, Lange S, Etro D, Glass A, Fuellen G, Boldt S, et al: The
multikinase inhibitor Sorafenib displays significant
antiproliferative effects and induces apoptosis via caspase 3, 7
and PARP in B- and T-lymphoblastic cells. BMC Cancer.
10(560)2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Coloff JL, Mason EF, Altman BJ, Gerriets
VA, Liu T, Nichols AN, Zhao Y, Wofford JA, Jacobs SR, Ilkayeva O,
et al: Akt requires glucose metabolism to suppress puma expression
and prevent apoptosis of leukemic T cells. J Biol Chem.
286:5921–5933. 2011.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Huang Y, Wu S, Zhang Y, Wang L and Guo Y:
Antitumor effect of triptolide in T-cell lymphoblastic lymphoma by
inhibiting cell viability, invasion, and epithelial-mesenchymal
transition via regulating the PI3K/AKT/mTOR pathway. Onco Targets
Ther. 11:769–779. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kiessling MK, Curioni-Fontecedro A,
Samaras P, Atrott K, Cosin-Roger J, Lang S, Scharl M and Rogler G:
Mutant HRAS as novel target for MEK and mTOR inhibitors.
Oncotarget. 6:42183–42196. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Rodriguez-Viciana P, Warne PH, Dhand R,
Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD and Downward J:
Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature.
370:527–532. 1994.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Diaz-Flores E and Shannon K: Targeting
oncogenic ras. Gene Dev. 21:1989–1992. 2007.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Li S, Ku CY, Farmer AA, Cong YS, Chen CF
and Lee WH: Identification of a novel cytoplasmic protein that
specifically binds to nuclear localization signal motifs. J Biol
Chem. 273:6183–6189. 1998.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Asada M, Ohmi K, Delia D, Enosawa S,
Suzuki S, You A, Suzuki H and Mizutani S: Brap2 functions as a
cytoplasmic retention protein for p21 during monocyte
differentiation. Mol Cell Biol. 24:8236–8243. 2004.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Takashima O, Tsuruta F, Kigoshi Y,
Nakamura S, Kim J, Katoh MC, Fukuda T, Irie K and Chiba T: Brap2
regulates temporal control of NF-κB localization mediated by
inflammatory response. PLoS One. 8(e58911)2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ozaki K, Sato H, Inoue K, Tsunoda T,
Sakata Y, Mizuno H, Lin TH, Miyamoto Y, Aoki A, Onouchi Y, et al:
SNPs in BRAP associated with risk of myocardial infarction in Asian
populations. Nat Genet. 41:329–333. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
25
|
Liao YC, Wang YS, Guo YC, Ozaki K, Tanaka
T, Lin HF, Chang MH, Chen KC, Yu ML, Sheu SH and Juo SH: BRAP
activates inflammatory cascades and increases the risk for carotid
atherosclerosis. Mol Med. 17:1065–1074. 2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Matheny SA, Chen C, Kortum RL, Razidlo GL,
Lewis RE and White MA: Ras regulates assembly of mitogenic
signalling complexes through the effector protein IMP. Nature.
427:256–260. 2004.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Fatima S, Wagstaff KM, Loveland KL and
Jans DA: Interactome of the negative regulator of nuclear import
BRCA1-binding protein 2. Sci Rep. 5(9459)2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Gao T, Furnari F and Newton AC: PHLPP: A
phosphatase that directly dephosphorylates Akt, promotes apoptosis,
and suppresses tumor growth. Mol Cell. 18:13–24. 2005.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Brognard J, Sierecki E, Gao T and Newton
AC: PHLPP and a second isoform, PHLPP2, differentially attenuate
the amplitude of Akt signaling by regulating distinct Akt isoforms.
Mol Cell. 25:917–931. 2007.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Ran FA, Hsu PD, Wright J, Agarwala V,
Scott DA and Zhang F: Genome engineering using the CRISPR-Cas9
system. Nat Protoc. 11:2281–2308. 2013.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Lanctot AA, Guo Y, Le Y, Edens BM,
Nowakowski RS and Feng Y: Loss of brap results in premature G1/S
phase transition and impeded neural progenitor differentiation.
Cell Rep. 20:1148–1160. 2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Shi L, Weng XQ, Sheng Y, Wu J, Ding M and
Cai X: Staurosporine enhances ATRA-induced granulocytic
differentiation in human leukemia U937 cells via the MEK/ERK
signaling pathway. Oncol Rep. 36:3072–3080. 2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Liu AH, Cao YN, Liu HT, Zhang WW, Liu Y,
Shi TW, Jia GL and Wang XM: DIDS attenuates staurosporine-induced
cardiomyocyte apoptosis by PI3K/Akt signaling pathway: Activation
of eNOS/NO and inhibition of Bax translocation. Cell Physiol
Biochem. 22:177–186. 2008.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Darzynkiewicz Z, Bruno S, Del Bino G,
Gorczyca W, Hotz MA, Lassota P and Traganos F: Features of
apoptotic cells measured by flow cytometry. Cytometry. 13:795–808.
1992.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Bedner E, Li X, Gorczyca W, Melamed MR and
Darzynkiewicz Z: Analysis of apoptosis by laser scanning cytometry.
Cytometry. 35:181–195. 1999.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Cohen GM: Caspase: The executioners of
apoptosis. Biochem J. 326:1–16. 1997.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Koon JC and Kubiseski TJ: Developmental
arrest of caenorhabditis elegans BRAP-2 mutant oxidative stress is
dependent on BRC-1. J Biol Chem. 285:13437–13443. 2010.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Wang B, Cao C, Liu X, He X, Zhuang H, Wang
D and Chen B: BRCA1-associated protein inhibits glioma cell
proliferation and migration and glioma stem cell self-renewal via
the TGF-β/PI3K/AKT/mTOR signalling pathway. Cell Oncol (Dordr).
43:223–235. 2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
D'Amora DR, Hu Q, Pizzardi M and Kubiseski
TJ: BRAP-2 promotes DNA damage induced germline apoptosis in C.
elegans through the regulation of SKN-1 and AKT-1. Cell Death
Differ. 25:1276–1288. 2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Rajakulendran T, Sahmi M, Lefraancois M,
Sicheri F and Therrien M: A dimerization-dependent mechanism drives
RAF catalytic activation. Nature. 461:542–545. 2009.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Peng SB, Henry JR, Kaufman MD, Lu WP,
Smith BD, Vogeti S, Rutkoski TJ, Wise S, Chun L, Zhang Y, et al:
Inhibition of RAF isoforms and active dimers by LY3009120 leads to
anti-tumor activities in RAS or BRAF mutant cancers. Cancer Cell.
28:384–398. 2015.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Zhao WL: Targeted therapy in T-cell
malignancies: Dysregulation of the cellular signaling pathways.
Leukemia. 24:13–21. 2010.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Xu Z, Stokoe D, Kane LP and Weiss A: The
inducible expression of the tumor suppressor gene PTEN promotes
apoptosis and decreases cell size by inhibiting the PI3K/Akt
pathway in Jurkat T cells. Cell Growth Differ. 13:285–296.
2002.PubMed/NCBI
|
|
44
|
Wang Z, Gjörloff-Wingren A, Saxena M,
Pathan N, Reed JC and Mustelin T: The tumor suppressor PTEN
regulates T cell survival and antigen receptor signaling by acting
as a phosphatidylinositol 3-phosphatase. J Immunol. 164:1934–1939.
2000.PubMed/NCBI View Article : Google Scholar
|