Introduction
Cardiovascular diseases are the most common cause of
disease-associated mortality worldwide (1). Hypertension, hypercholesterolemia,
obesity and smoking pose serious threats to the cardiovascular
system, contributing to vascular inflammation and endothelial cell
dysfunction (2-4).
Atherosclerosis is a lipid-driven progressive inflammatory systemic
disease, which primarily affects the aorta, and the carotid and
coronary arteries (5,6). The accumulation of plasma lipoproteins
is considered as the main factor driving formation of
atherosclerotic lesions. Despite the progress that has been made in
reducing the concentration of lipoproteins, there are residual
risks to these cardiovascular events (7). Anti-inflammatory approaches, including
reducing the levels of pro-inflammatory cytokines, are considered a
promising strategy to supplement the existing lipid-lowering
strategies to decrease the residual risks (8).
Interferon regulatory factor 2-binding protein 2
(IRF2BP2) is a transcriptional repressor that is extensively
involved in the regulation of the expression of a range genes
(9-11).
A recent study reported that IRF2BP2 overexpression improves
cardiomyopathy via suppressing the infiltration of inflammatory
cells and the secretion of inflammatory cytokines (12). Deletion of IRF2BP2 aggravated a high
fat diet-induced increase in systematic and central nervous
inflammation via inhibition of the release of interleukin (IL)-1β
and tumor necrosis factor (TNF)-α (13). IRF2BP2 is a negative regulator of
inflammation via regulation of macrophage polarization. A study
revealed that IRF2BP2-deficient macrophages exacerbated
atherosclerosis in apolipoprotein E null mice by reducing the
expression of the anti-inflammatory transcription factor
Krüppel-like factor 2 (KLF2) in macrophages and disrupting lipid
homeostasis (14). KLFs are members
of the zinc finger family of DNA-binding transcription factors,
which serve a range of roles in several biological processes,
including quiescence, proliferation, differentiation, development,
growth and inflammation (15).
Amongst the members of KLFs, KLF2 is considered as a central
regulator of endothelial biology through regulation of
proinflammatory activation in endothelial cells. Previous studies
have shown that KLF2 signaling served an important role in
mediating endothelial inflammation and atherosclerosis (16,17).
For example, KLF2 was reported to negatively regulate inflammation
and reduce proinflammatory activity of nuclear factor-κB (NF-κB)
(18). Endothelial KLF2 also
exhibited an atheroprotective role as a novel inhibitor of
endothelial inflammasome activation in atherogenesis in vivo
(19). However, the effects of the
regulation of IRF2BP2 gene expression in endothelial cells on
atherosclerosis remains elusive.
In the present study, it was hypothesized that
IRF2BP2 may also serve a role in endothelial cell inflammation and
dysfunction via regulation of KLF2. The aim of the present study
was to investigate whether IRF2BP2 overexpression in endothelial
cells exhibited a beneficial effect on ox-LDL-induced endothelial
cell injury, and further elucidate the underlying regulatory
mechanisms.
Materials and methods
Cell culture
Human umbilical vein endothelial cells (HUVECs) were
provided by The Cell Bank of Type Culture Collection of The Chinese
Academy of Sciences and maintained at 37˚C in a humidified
incubator with 5% CO2. HUVECs were cultured in F12K
medium (Boster Biological Technology Co., Ltd.) supplemented with
10% FBS (Gibco; Thermo Fisher Scientific, Inc.), 100 µg/ml
streptomycin and 100 IU/ml penicillin (Boster Biological Technology
Co., Ltd.). HUVECs were treated with 100 µg/ml oxidized low-density
lipoprotein (ox-LDL; UnionBiol Biotechnologies Co., Ltd.) for 24 h
at 37˚C to simulate atherosclerosis, as previously described
(20).
Cell transfection
To overexpress IRF2BP2, a pcDNA3.1-IRF2BP2 plasmid
was synthesized by Shanghai GenePharma Co., Ltd.; empty pcDNA3.1
vector was used as a negative control. A small interfering RNA
(siRNA) targeting KLF2
(5'-AUUUUGAAAAACAAAACUCGUGAGUUUUGUUUUUCAAAAUGG-3') was synthesized
by Shanghai GenePharma Co., Ltd and transfected into HUVECs to
knockdown KLF2 expression. Scrambled siRNA
(5'-CCUGGCGCCUUCGGUCUUUUU-3') served as a negative control. HUVECs
were grown to 60-70% confluence and 20 µg pcDNA3.1 or siRNA
plasmids were transfected into cells using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) and incubated for 6 h at 37˚C. A total of 48 h
post-transfection, transfected cells were selected for subsequent
experiments.
Cell viability
HUVECs at a density of 5x103 cells/well
were plated into 96-well plates and then incubated with 100 µg/ml
ox-LDL for 24 h at 37˚C. Subsequently, 10 µl Cell Counting Kit-8
reagent (CCK-8; Beijing Solarbio Science & Technology Co.,
Ltd.) was added to each well for 1 h. The absorbance values were
measured at 450 nm using a microplate reader (BioTek Instruments,
Inc.).
ELISA
The levels of pro-inflammatory cytokines in the cell
medium were determined using TNF-α (cat. no. SEKH-0047), IL-1β
(cat. no. SEKH-0002) and IL-6 (cat. no. SEKH-0013) ELISA kits
(Beijing Solarbio Science & Technology Co., Ltd.). Briefly, the
cell culture medium was transferred to a sterile centrifuge tube
and centrifuged (1,000 x g) at 4˚C for 10 min. The supernatants
were then supplemented with the corresponding ELISA kit reagents,
according to manufacturer's protocol. The absorbance values were
measured at a wavelength of 450 nm using a microplate reader
(BioTek Instruments, Inc.).
Western blot analysis
HUVECs were lysed to extract total protein and the
concentration was quantified using a BCA Protein assay kit (Beijing
Solarbio Science & Technology Co., Ltd.). Protein samples were
loaded on a 12% SDS-gel, resolved using SDS-PAGE and transferred to
a PVDF membrane (EMD Millipore). Following blocking with 5% bovine
serum albumin (BSA; Thermo Fisher Scientific, Inc.) for 2 h at room
temperature, the membranes were then incubated with primary
antibodies against IRF2BP2 (cat. no. 18847-1-AP; 1:1,000), GAPDH
(cat. no. 10494-1-AP; 1:5,000), vimentin (cat. no. 10366-1-AP;
1:1,000; all from ProteinTech Group, Inc.), KLF2 (cat. no.
PA5-40591; 1:1,000), phosphorylated endothelial nitric oxide
synthase (p-eNOS; cat. no. PA5-104858; 1:500), eNOS (cat. no.
PA1-037; 1:1,000; all from Invitrogen; Thermo Fisher Scientific,
Inc.) and VE-cadherin (cat. no. 2500; 1:1,000; Cell Signaling
Technology, Inc.) at 4˚C overnight. The following day, the
membranes were incubated with HRP-conjugated Affinipure goat
anti-mouse IgG (cat. no. SA00001-1; 1:5,000) or HRP-conjugated
Affinipure goat anti-rabbit IgG (cat. no. SA00001-2; 1:5,000; both
from ProteinTech Group, Inc.) for 2 h at room temperature. Signals
were visualized using an ECL Western Blotting Substrate (Thermo
Fisher Scientific, Inc.), and densitometry analysis was performed
using Image Lab system (Bio-Rad Laboratories, Inc.; version 1.52).
GAPDH was used as the internal control.
Immunofluorescence assay
HUVECs were seeded into 24-well plates at a density
of 1x105 cells/well. When cells reached 60-70%
confluence, they were fixed with 4% paraformaldehyde at 4˚C for 15
min and incubated with 0.5% Triton X-100 for 20 min. Subsequently,
5% BSA was added to block non-specific antigen at room temperature
for 30 min, after which cells were incubated with the primary
antibody against KLF2 (cat. no. MA5-24300; Invitrogen; Thermo
Fisher Scientific, Inc.) at 4˚C overnight. Following incubation
with anti-KLF2, cells were incubated with fluorescein
isothiocyanate-conjugated Affinipure goat anti-mouse IgG (cat. no.
SA00003-1; ProteinTech Group, Inc.) for 1 h in the dark at room
temperature. Cell nuclei were stained with DAPI for 5 min at room
temperature, and images were captured under a fluorescence
microscope (Carl Zeiss AG; magnification, x200).
Reverse transcription-quantitative PCR
(RT-qPCR)
HUVECs were harvested, total RNA was extracted using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and cDNA was then synthesized using a reverse transcription
kit, according to the manufacturer's protocol (Takara, Bio, Inc.).
Next, a SYBR Green PCR kit (Takara Bio, Inc.) was used to determine
gene expression on an ABI 7500 Real-Time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The sequences of the
primers used were: KLF2 forward, 5'-GCACGCACACAGGTGAGA-3' (Tm=58˚C)
and reverse, 5'-CACAGATGGCACTGGAATGG-3' (Tm=62˚C), product length,
130 bp; IRF2BP2 forward, 5'-CCCTGACTGCAGTTGCAAGA-3' (Tm=62˚C) and
reverse, 5'-TTGAGCCCCTCTGTGGATGT-3' (Tm=62˚C), product length, 127
bp; vascular cell adhesion molecule 1 (VCAM-1) forward,
5'-GGACCACATCTACGCTGACA-3' (Tm=62˚C) and reverse,
5'-TTGACTGTGATCGGCTTCCC-3' (Tm=62˚C), product length, 184 bp;
intercellular adhesion molecule 1 (ICAM-1) forward,
5'-TCTTCCTCGGCCTTCCCATA-3' (Tm=62˚C) and reverse,
5'-AGGTACCATGGCCCCAAATG-3' (Tm=62˚C), product length, 152 bp; and
GAPDH forward, 5'-ATGGGCAGCCGTTAGGAAAG-3' (Tm=62˚C) and reverse,
5'-ATCACCCGGAGGAGAAATCG-3' (Tm=62˚C), product length, 126 bp.
Co-immunoprecipitation (Co-IP)
Extraction of whole-cell lysates from HUVECs was
performed using RIPA lysis buffer (Beyotime Institute of
Biotechnology). For immunoprecipitation, 500 µg protein was
incubated with 1-2 µg of the specific antibodies against IRF2BP2 or
KLF2 (same antibodies used in western blotting; Invitrogen; Thermo
Fisher Scientific, Inc.) overnight at 4˚C. Subsequently, 40 µl 50%
Protein A/G PLUS-Agarose beads (Invitrogen; Thermo Fisher
Scientific, Inc.) was added, and incubated for a further 2 h at
room temperature. Beads were then washed three times with the lysis
buffer and collected by centrifugation at 12,000 x g for 2 min at
4˚C. Following the final wash, the supernatant was aspirated and
discarded, then the precipitated proteins were eluted from the
beads by resuspending in 2X SDS-PAGE loading buffer and boiling for
5 min. The resulting materials from immunoprecipitation or cell
lysates were analyzed using western blotting.
Detection of NO levels
The cell culture medium was collected to estimate
the production of NO. Following mixing of cell culture medium with
Griess Reagent I and Griess Reagent II from the Nitrate/Nitrite
assay kit (Beyotime Institute of Biotechnology), the absorbance
values were determined at a wavelength of 540 nm using a microplate
reader (BioTek Instruments, Inc.).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism version 6.0 (GraphPad Software, Inc.). All experiments were
performed in triplicate. Data are presented as the mean ± standard
deviation. Differences between multiple groups were analyzed using
a one-way ANOVA followed by a post hoc Tukey's test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Protective effects of IRF2BP2 on
ox-LDL-induced inflammation and EMT
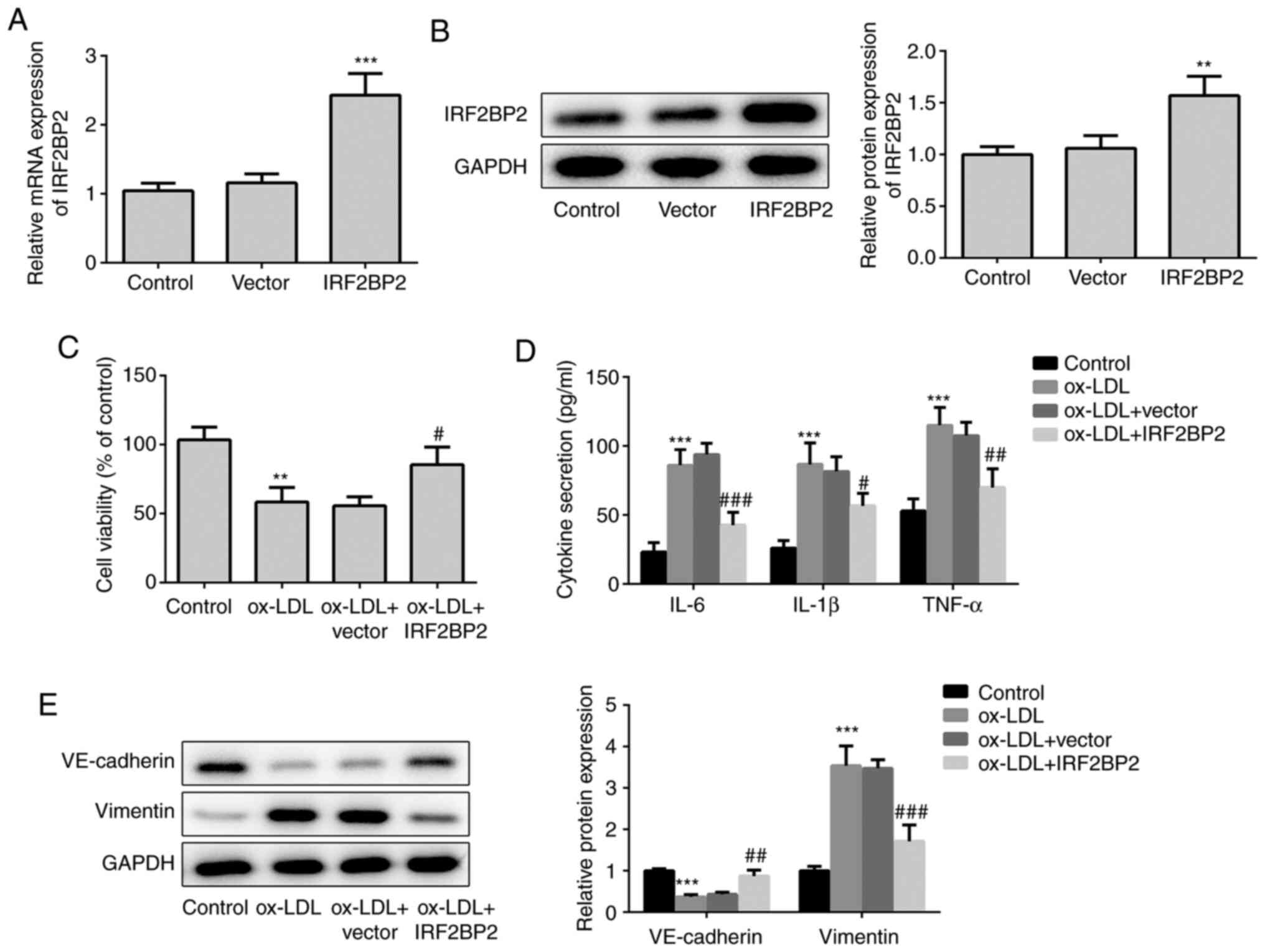
First, IRF2BP2 plasmids were transfected into HUVECs
to overexpress IRF2BP2. As shown in Fig. 1A and B, transfection of HUVECs with IRF2BP2
overexpression plasmids increased the expression of IRF2BP2 at both
the transcriptional (mRNA) and translational (protein) levels.
Subsequently, HUVECs were treated with 100 µg/ml ox-LDL for 24 h,
and the results demonstrated that ox-LDL significantly impaired
cell viability, whereas IRF2BP2 overexpression effectively relieved
ox-LDL-induced cell injury (Fig.
1C). In addition, treatment with ox-LDL induced inflammatory
responses in HUVECs, as determined by the significant increase in
pro-inflammatory cytokines, including TNF-α, IL-1β and IL-6
(Fig. 1D). HUVECs transfected with
IRF2BP2 overexpression plasmid exhibited mild inflammation. As a
crucial risk factor of atherosclerosis, ox-LDL promotes the
secretion of inflammatory cytokines by endothelial cells, thus
inducing EMT (21,22). Therefore, the decreased expression
of the endothelial marker VE-cadherin, along with the upregulation
of the mesenchymal marker vimentin, indicated that HUVECs underwent
EMT following stimulation with ox-LDL (Fig. 1E). By contrast, IRF2BP2
overexpression inhibited EMT.
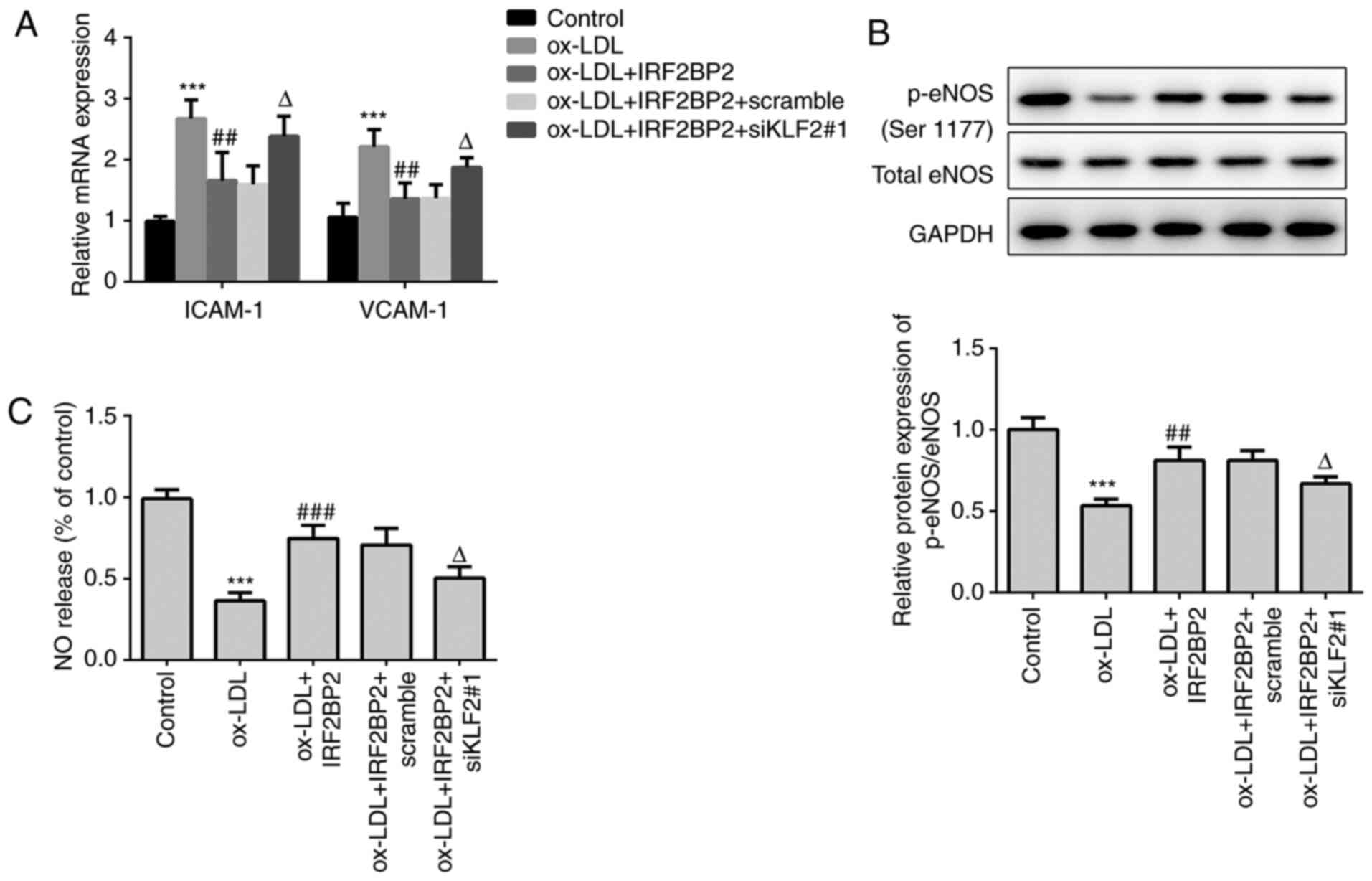
IRF2BP2 upregulates KLF2 and improves
endothelial NO production
VCAM-1 and ICAM-1 are members of the endothelial
adhesion molecules, which are involved in atherogenesis and may be
key mediators of atherosclerosis (23,24).
The cellular mRNA expression levels of VCAM-1 and ICAM-1 were
upregulated by ox-LDL (Fig. 2A),
whereas IRF2BP2 overexpression significantly reduced the
ox-LDL-induced upregulation of both VCAM-1 and ICAM-1.
Additionally, the expression of the anti-inflammatory transcription
factor KLF2 was reduced in HUVECs exposed to ox-LDL. This effect
was reversed following IRF2BP2 overexpression (Fig. 2B). It has been reported that the
activity of eNOS maintains endothelial function (25). NO, produced by eNOS, has a profound
effect on angiogenesis, vascular remodeling and endothelial
permeability. Therefore, a decrease in NO release is a hallmark of
endothelial cell dysfunction (24,26).
In the present study, the phosphorylation of eNOS at Ser1177 was
reduced after stimulation of HUVECs with ox-LDL.
IRF2BP2-overexpressing cells exhibited higher eNOS phosphorylation
levels compared with the empty vector group in the presence of
ox-LDL (Fig. 2B). Furthermore, NO
release in the ox-LDL treated group was significantly reduced,
whereas IRF2BP2 overexpression partly restored its production
(Fig. 2C). The expression of KLF2
was determined using immunofluorescence analysis (Fig. 2D), and the results were consistent
with those of the western blot analysis.
 | Figure 2IRF2BP2 increases the expression of
KLF2 and improves endothelial function. (A) The expression levels
of ICAM-1 and VCAM-1 were determined using reverse
transcription-quantitative PCR analysis. (B) The protein expression
levels of KLF2, p-eNOS, total eNOS and GAPDH were measured using
western blot analysis. (C) A Nitrate/Nitrite assay kit was utilized
to estimate the release of NO. (D) The expression of KLF2 was
evaluated by immunofluorescence analysis (magnification, x200).
Scale bar, 50 µm. **P<0.01, ***P<0.001
vs. the control group; ##P<0.05,
###P<0.001 vs. the ox-LDL + vector group. IRF2BP2,
interferon regulatory factor binding protein 2; KLF2, Krüppel-like
factor 2; ICAM-1, intercellular adhesion molecule 1; VCAM-1,
vascular cell adhesion molecule 1; p-eNOS, phosphorylated
endothelial nitric oxide synthase; NO, nitric oxide; ox-LDL,
oxidized low-density lipoprotein. |
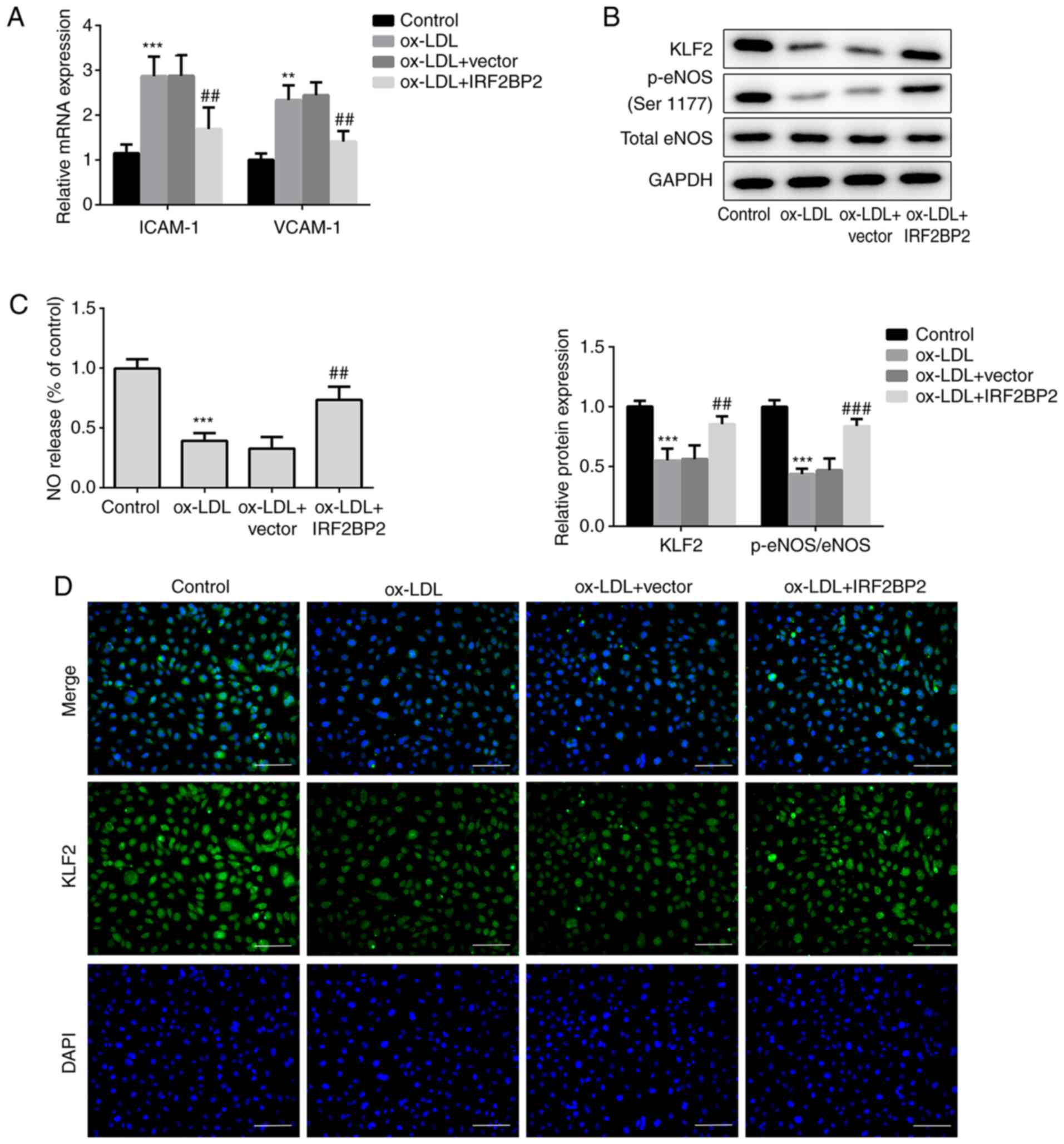
IRF2BP2 attenuates inflammation and
EMT via KLF2 expression
The interaction between IRF2BP2 and KLF2 was
verified using Co-IP (Fig. 3A).
Subsequently, two pairs of siRNAs targeting KLF2 were transfected
into HUVECs, and the transfection efficiency is shown in Fig. 3B and C. The siKLF2#1 clone exhibited better
knockdown of KLF2 expression compared with siKLF2#2. Cell viability
assays revealed that siKLF2#1 could reduce the viability of
IRF2BP2-overexpressing HUVECs following treatment with ox-LDL
(Fig. 3D). Furthermore, the IRF2BP2
overexpression-mediated reduction in secretion of TNF-α, IL-1β and
IL-6 were reversed following transfection with siKLF2#1 in the
presence of ox-LDL (Fig. 3E).
Additionally, siKLF2#1 abrogated the inhibitory effect of IRF2BP2
overexpression on EMT (Fig.
3F).
 | Figure 3IRF2BP2 attenuates inflammation and
EMT via KLF2. (A) The interaction between IRF2BP2 and KLF2 was
confirmed using a Co-IP assay. (B) HUVECs were transfected with
scrambled siRNA, siKLF2#1 or siKLF2#2. The mRNA expression levels
of KLF2 were measured using reverse transcription-quantitative PCR
analysis. ***P<0.001 vs. the scramble group. (C) The
protein expression levels of KLF2 were evaluated using western blot
analysis. **P<0.01, ***P<0.001 vs. the
scrambled group. (D) The cell viability in different groups was
determined using a CCK-8 assay. (E) The levels of TNF-α, IL-1β and
IL-6 in the culture medium were assessed using ELISA kits. (F) The
protein expression levels of VE-cadherin and vimentin were
evaluated using western blot analysis. ***P<0.001 vs.
the control group; ##P<0.05, ###P<0.001
vs. the ox-LDL group; ΔP<0.05,
ΔΔP<0.01, ΔΔΔP<0.001 vs. the ox-LDL +
IRF2BP2 + scramble group. IRF2BP2, interferon regulatory factor
binding protein 2; KLF2, Krüppel-like factor 2; EMT,
endothelial-to-mesenchymal transition; HUVECs, human umbilical vein
endothelial cells; siRNA, small interfering RNA; TNF-α, tumor
necrosis factor α; IL-1β, interleukin 1β; ox-LDL, oxidized
low-density lipoprotein; Co-IP, co-immunoprecipitation. |
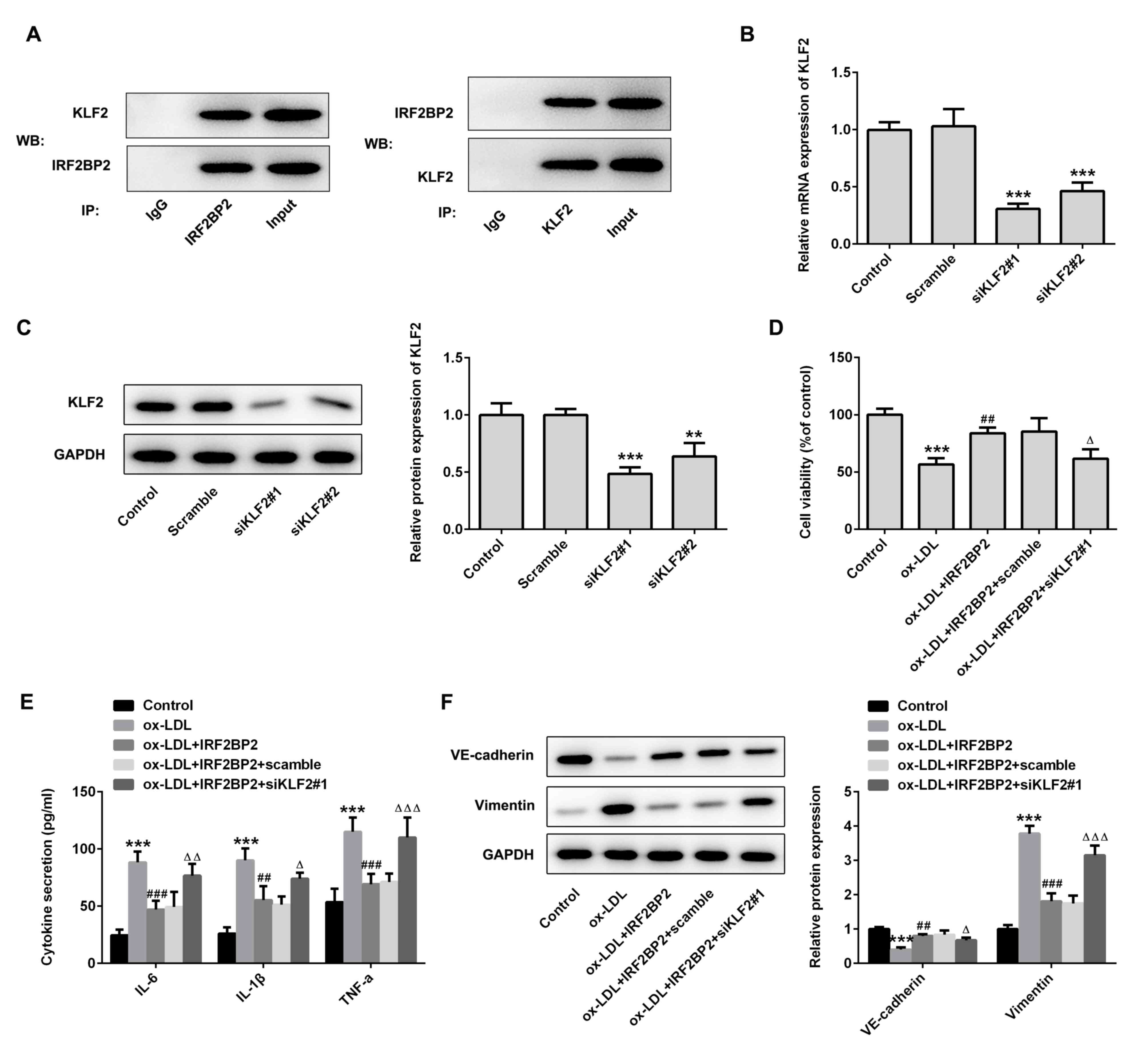
KLF2 is required for the effects of
IRF2BP2 on endothelial cells
Subsequently, the role of KLF2 in ox-LDL-regulated
endothelial dysfunction were determined. The mRNA expression levels
of ICAM-1 and VCAM-1 were increased in ox-LDL-induced HUVECs.
IRF2BP2 overexpression repressed the expression of both ICAM-1 and
VCAM-1, which was restored following KLF2 knockdown (Fig. 4A). Additionally, the phosphorylation
levels of eNOS and the production of NO were also evaluated. As
shown in Fig. 4B and C, ox-LDL reduced the levels of p-eNOS and
NO. IRF2BP2 overexpression significantly increased the levels of
p-eNOS and NO, whereas transfection with siKLF2#1 counteracted the
effects of IRF2BP2.
Discussion
Atherosclerosis is the most common disease of the
cardiovascular system (27).
Coronary heart disease, stroke and other cardiovascular diseases
caused by atherosclerosis are significant causes of death worldwide
(28). During the pathogenesis of
atherosclerosis, endothelial cells undergo a series of pathological
changes, from early dysfunction to advanced plaque transition
(29). Ox-LDL triggers inflammation
and EMT of endothelial cells, and facilitates the formation of
atherosclerotic plaques (30,31).
In the present study, treatment with ox-LDL reduced the viability
of HUVECs, and IRF2BP2 overexpression ameliorated ox-LDL-induced
cell injury. Furthermore, ox-LDL-induced inflammation and EMT were
also impeded following IRF2BP2 overexpression.
EMT of endothelial cells promotes the expression of
VCAM-1 and ICAM-1, which are key mediators of atherosclerosis,
thereby recruiting monocytes to the arterial intima (32). Endothelial dysfunction usually
refers to endothelium-dependent vasodilation and constriction
impairment. Emerging evidence has suggested that NO, produced by
eNOS, regulates vasomotor tone (29). In the present study, treatment of
HUVECs with ox-LDL inhibited the activity of eNOS and reduced the
synthesis of NO, and these effects were reversed following IRF2BP2
overexpression. Of note, IRF2BP2 overexpression also increased the
protein expression levels of KLF2. The direct interaction between
IRF2BP2 and KLF2 was also verified by Co-IP. Chen et al
(14) demonstrated that IRF2BP2
could bind to the promoter of KLF2, resulting in the upregulation
of KLF2 expression. Kim et al (33) also reported that IRF2BP2
overexpression increased KLF2 expression, suggesting that IRF2BP2
acts upstream of KLF2 in bone cells. As an anti-atherogenic factor
in the vascular endothelium, KLF2 improves the endothelial
functions and suppresses the development of atherosclerosis via
regulating multiple pathways, including the NF-κB, MAPK/ERK, JNK
and p38 signaling pathways (34-36).
Therefore, it is hypothesized that IRF2BP2 may exert its effect
through binding to KLF2 and positively regulating KLF2 expression,
thereby affecting the downstream signaling transduction of KLF2;
however, this requires further study. In accordance with this
hypothesis, KLF2 knockdown exacerbated cell injury, inflammation
and EMT. A recent report demonstrated that KLF2 orchestrates its
effects on endothelial function via the eNOS/NO signaling pathway
(17). In the present study, KLF2
knockdown attenuated the phosphorylation of eNOS and the generation
of NO.
In summary, the present study showed that IRF2BP2
overexpression could protect HUVECs from ox-LDL-induced
inflammation and EMT, whilst also increasing NO production in
endothelial cells by enhancing the expression of KLF2. These
findings reflect the role of IRF2BP2/KLF2 in endothelial cells and
may improve our understanding of the pathogenesis of
atherosclerosis, and may also highlight potentially novel targets
for management of this disease. However, further in-depth studies
are required to elucidate the mechanisms underlying
endothelium-dependent vasodilation and vasoconstriction, and the
vulnerability of arterial plaques.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QS conceived and designed the current study. QS and
YJ contributed to the acquisition of data. YJ analyzed and
interpreted the data. QS drafted the initial manuscript and revised
it critically for important intellectual content. QS and YJ confirm
the authenticity of all the raw data. Both authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Piepoli MF, Hoes AW, Agewall S, Albus C,
Brotons C, Catapano AL, Cooney MT, Corrà U, Cosyns B, Deaton C, et
al: 2016 European Guidelines on cardiovascular disease prevention
in clinical practice: The Sixth Joint Task Force of the European
Society of Cardiology and Other Societies on Cardiovascular Disease
Prevention in Clinical Practice (constituted by representatives of
10 societies and by invited experts)Developed with the special
contribution of the European Association for Cardiovascular
Prevention & Rehabilitation (EACPR). Eur Heart J. 37:2315–2381.
2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Mauricio MD, Aldasoro M, Ortega J and Vila
JM: Endothelial dysfunction in morbid obesity. Curr Pharm Des.
19:5718–5729. 2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Vanhoutte PM, Shimokawa H, Tang EH and
Feletou M: Endothelial dysfunction and vascular disease. Acta
Physiol (Oxf). 196:193–222. 2009.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Molinaro R, Boada C, Del Rosal GM, Hartman
KA, Corbo C, Andrews ED, Toledano-Furman NE, Cooke JP and Tasciotti
E: Vascular inflammation: A novel access route for nanomedicine.
Methodist Debakey Cardiovasc J. 12:169–174. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Seneviratne AN and Monaco C: Role of
inflammatory cells and toll-like receptors in atherosclerosis. Curr
Vasc Pharmacol. 13:146–160. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Hansson GK: Inflammation, atherosclerosis,
and coronary artery disease. N Engl J Med. 352:1685–1695.
2005.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Packard CJ: LDL cholesterol: How low to
go? Trends Cardiovasc Med. 28:348–354. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bäck M and Hansson GK: Anti-inflammatory
therapies for atherosclerosis. Nat Rev Cardiol. 12:199–211.
2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Childs KS and Goodbourn S: Identification
of novel co-repressor molecules for interferon regulatory factor-2.
Nucleic Acids Res. 31:3016–3026. 2003.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Carneiro FR, Ramalho-Oliveira R, Mognol GP
and Viola JP: Interferon regulatory factor 2 binding protein 2 is a
new NFAT1 partner and represses its transcriptional activity. Mol
Cell Biol. 31:2889–2901. 2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Stadhouders R, Cico A, Stephen T,
Thongjuea S, Kolovos P, Baymaz HI, Yu X, Demmers J, Bezstarosti K,
Maas A, et al: Control of developmentally primed erythroid genes by
combinatorial co-repressor actions. Nat Commun.
6(8893)2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Li T, Luo Q, He L, Li D, Li Q, Wang C, Xie
J and Yi C: Interferon regulatory factor-2 binding protein 2
ameliorates sepsis-induced cardiomyopathy via AMPK-mediated
anti-inflammation and anti-apoptosis. Inflammation. 43:1464–1475.
2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ma YL, Xia JL and Gao X: Suppressing
Irf2bp2 expressions accelerates metabolic syndrome-associated brain
injury and hepatic dyslipidemia. Biochem Biophys Res Commun.
503:1651–1658. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Chen HH, Keyhanian K, Zhou X, Vilmundarson
RO, Almontashiri NA, Cruz SA, Pandey NR, Lerma Yap N, Ho T, Stewart
CA, et al: IRF2BP2 reduces macrophage inflammation and
susceptibility to atherosclerosis. Circ Res. 117:671–683.
2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Jha P and Das H: KLF2 in regulation of
NF-κB-mediated immune cell function and inflammation. Int J Mol
Sci. 18(2383)2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhou Z, Tang AT, Wong WY, Bamezai S,
Goddard LM, Shenkar R, Zhou S, Yang J, Wright AC, Foley M, et al:
Cerebral cavernous malformations arise from endothelial gain of
MEKK3-KLF2/4 signalling. Nature. 532:122–126. 2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Wu W, Geng P, Zhu J, Li JW, Zhang L, Chen
WL, Zhang DF, Lu Y and Xu XH: KLF2 regulates eNOS uncoupling via
Nrf2/HO-1 in endothelial cells under hypoxia and reoxygenation.
Chem Biol Interact. 305:105–111. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Das H, Kumar A, Lin Z, Patino WD, Hwang
PM, Feinberg MW, Majumder PK and Jain MK: Kruppel-like factor 2
(KLF2) regulates proinflammatory activation of monocytes. Proc Natl
Acad Sci USA. 103:6653–6658. 2006.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhuang T, Liu J, Chen X, Zhang L, Pi J,
Sun H, Li L, Bauer R, Wang H, Yu Z, et al: Endothelial Foxp1
suppresses atherosclerosis via modulation of Nlrp3 inflammasome
activation. Circ Res. 125:590–605. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhao D, Sun X, Lv S, Sun M, Guo H, Zhai Y,
Wang Z, Dai P, Zheng L, Ye M and Wang X: Salidroside attenuates
oxidized low-density lipoprotein-induced endothelial cell injury
via promotion of the AMPK/SIRT1 pathway. Int J Mol Med.
43:2279–2290. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Lee WJ, Ou HC, Hsu WC, Chou MM, Tseng JJ,
Hsu SL, Tsai KL and Sheu WHH: Ellagic acid inhibits oxidized
LDL-mediated LOX-1 expression, ROS generation, and inflammation in
human endothelial cells. J Vasc Surg. 52:1290–1300. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Li H, Zhao Q, Chang L, Wei C, Bei H, Yin
Y, Chen M, Wang H, Liang J and Wu Y: LncRNA MALAT1 modulates ox-LDL
induced EndMT through the Wnt/β-catenin signaling pathway. Lipids
Health Dis. 18(62)2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Cybulsky MI, Iiyama K, Li H, Zhu S, Chen
M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW and Milstone
DS: A major role for VCAM-1, but not ICAM-1, in early
atherosclerosis. J Clin Invest. 107:1255–1262. 2001.PubMed/NCBI View
Article : Google Scholar
|
|
24
|
Galkina E and Ley K: Vascular adhesion
molecules in atherosclerosis. Arterioscler Thromb Vasc Biol.
27:2292–2301. 2007.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Huang PL: Endothelial nitric oxide
synthase and endothelial dysfunction. Curr Hypertens Rep.
5:473–480. 2003.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Lyons D: Impairment and restoration of
nitric oxide-dependent vasodilation in cardiovascular disease. Int
J Cardiol. 62 (Suppl 2):S101–S109. 1997.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Skuratovskaia D, Vulf M, Komar A,
Kirienkova E and Litvinova L: Promising directions in
atherosclerosis treatment based on epigenetic regulation using
MicroRNAs and long noncoding RNAs. Biomolecules.
9(226)2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zhao Y, Evans MA, Allison MA, Bertoni AG,
Budoff MJ, Criqui MH, Malik S, Ouyang P, Polak JF and Wong ND:
Multisite atherosclerosis in subjects with metabolic syndrome and
diabetes and relation to cardiovascular events: The multi-ethnic
study of atherosclerosis. Atherosclerosis. 282:202–209.
2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Gimbrone MA Jr and García-Cardeña G:
Endothelial cell dysfunction and the pathobiology of
atherosclerosis. Circ Res. 118:620–636. 2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhang Y, Mu Q, Zhou Z, Song H, Zhang Y, Wu
F, Jiang M, Wang F, Zhang W, Li L, et al: Protective effect of
irisin on atherosclerosis via suppressing oxidized low density
lipoprotein induced vascular inflammation and endothelial
dysfunction. PLoS One. 11(e0158038)2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Yang H, Mohamed AS and Zhou SH: Oxidized
low density lipoprotein, stem cells, and atherosclerosis. Lipids
Health Dis. 11(85)2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Chen PY, Qin L, Baeyens N, Li G, Afolabi
T, Budatha M, Tellides G, Schwartz MA and Simons M:
Endothelial-to-mesenchymal transition drives atherosclerosis
progression. J Clin Invest. 125:4514–4528. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
33
|
Kim I, Kim JH, Kim K, Seong S and Kim N:
The IRF2BP2-KLF2 axis regulates osteoclast and osteoblast
differentiation. BMB Rep. 52:469–474. 2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Atkins GB, Wang Y, Mahabeleshwar GH, Shi
H, Gao H, Kawanami D, Natesan V, Lin Z, Simon DI and Jain MK:
Hemizygous deficiency of Krüppel-like factor 2 augments
experimental atherosclerosis. Circ Res. 103:690–693.
2008.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Xu Y, Liu P, Xu S, Koroleva M, Zhang S, Si
S and Jin ZG: Tannic acid as a plant-derived polyphenol exerts
vasoprotection via enhancing KLF2 expression in endothelial cells.
Sci Rep. 7(6686)2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Novodvorsky P and Chico TJ: The role of
the transcription factor KLF2 in vascular development and disease.
Prog Mol Biol Transl Sci. 124:155–188. 2014.PubMed/NCBI View Article : Google Scholar
|