Introduction
Gardner's syndrome is also known as familial
multiple colon polyposis-osteoma-soft tissue tumor syndrome. In
addition to the development of intestinal polyposis and colorectal
adenocarcinoma, which are key features of Gardner's syndrome,
Gardner's syndrome also exhibits extra-colonic presentation of the
familial adenomatous polyposis syndrome, which include dental
abnormalities, osteomas, soft-tissue tumors (including desmoid
tumors) and epidermoid cysts (1,2).
Dental abnormalities include impacted and additional teeth, and
osteomas typically occur in the mandible, but can also present in
the skull and long bones (3).
Osteoma and dental abnormalities may be early sensitive indicators
for the diagnosis of Gardner's syndrome (4,5).
Affected individuals also have an increased risk for extra-colonic
malignancies, including gastric (6)
and papillary tumors of the duodenum (7).
Gardner's syndrome is an autosomal-dominant disorder
that is caused by germline mutations in the adenomatous polyposis
coli (APC) gene (3). There
is no significant difference in the incidence of Gardner's syndrome
in various regions of the world, and its worldwide incidence ranges
between 1 in 4,000 and 1 in 12,000(8). Mutational analysis of the APC
gene indicates that the majority of germline variants include
nonsense mutations, which leads to the formation of a truncated
protein (9). According to genotypic
and phenotypic correlation studies involving APC mutations,
the mutation site associated with Gardner's syndrome mainly occurs
in the 3' end of the APC gene (10-14).
For example, in patients with Gardner's syndrome, truncating
mutations between codons 1403 and 1578 are associated with an
increase in mandibular lesions (10), while mutations beyond codon 1444 are
associated with a 2-fold increased risk in osteomas (11).
The majority of patients carrying APC
mutations have a family history of colorectal polyps and cancers
(9). However, 25-30% of APC
mutations are de novo and lack clinical or genetic
indications in unaffected family members (15,16).
It is now recognized that this can be partially explained as a
result of mutational mosaicism (17). The incidence of Gardner's syndrome
in China may be low, since it is currently very rare in the Han
Chinese population. The majority of current studies about Chinese
Gardner's syndrome are sporadic case reports or family studies. In
the current study, mutation analysis of the APC gene in a
Chinese patient with Gardner syndrome was performed. The mutational
analysis included determining whether the variant was de
novo or chimeric.
Materials and methods
Research subjects
The current study was approved by the Southern
Medical University Institutional Review Board. Informed consent was
obtained from all research participants. The proband, a 38-year-old
male, was from the Chaoshan area of Guangdong Province. A period of
5 years prior to diagnosis, pus was repeatedly present in the
individuals left lower posterior tooth. In 2016, after diagnosis of
fully impacted extraneous teeth and following treatment, the left
lower posterior tooth was extracted. The tooth extraction wound
underwent a prolonged period of healing, with postoperative gum
restoration being absent. Upon further examination, the mandibular
CT plain scan indicated that multiple osteomas were present in the
upper and lower jaw, part of the skull, paranasal sinus and
cervical vertebra. Multiple embedded teeth and a partially lost
tooth were observed. The left lower jaw indicated the presence of
an odontogenic infection. These results suggested that the patient
may have Gardner's syndrome. In 2018, a tumor was identified in the
right forearm of the proband and was surgically removed. During
consultation on the familial history of disease, it was revealed
that the patient's 62 year old mother had developed mandibular
hyperplasia when middle-aged. The patient's mother died from
colorectal cancer 3 months after the collection of a peripheral
blood sample.
Clinical examination
An X-ray dental panorama was performed on the oral
cavity and an X-ray examination was performed on the right forearm
of the proband. Pathological sections were taken from
hyperosteogeny tissue of the mandibular bone, and the right tibial.
Hyperosteogeny tissues were harvested and stored at formalin for
further analyses. Biochemical tests were performed on the 3 ml
peripheral blood obtained from the proband and his mother. After
collecting peripheral blood, the serum was separated via
centrifugation at 1,610 x g for 5 min at room temperature. The
electrochemiluminescence immunoassay and the kits for the
instrument was performed using Elecsys Cobas e 601 (Roche
Diagnostics) to detect the serum levels of carcinoembryonic antigen
(CEA; Elecsys CEA kit) and carbohydrate antigens including CA15-3
(Elecsys CA15-3 kit), CA19-9 (Elecsys CA19-9 kit) and CA72-4
(Elecsys CA72-4 kit). All Elecsys kits were all obtained from Roche
Diagnostics GmbH.
Hematoxylin and eosin staining
Bone tissues were pre-fixed in 10% neutral formalin
solution for 24 h at room temperature and rinsed with running tap
water for 24 h. After washing, decalcification was performed at 4˚C
under continuous shaking. The decalcifying solution (3% nitric
acid) was changed daily. When the bone was easily penetrated
through by a needle without any force, the decalcification process
was concluded. Decalcification lasted for a total of 8 days.
Finally, samples were neutralized with 0.1% aqueous ammonia
solution for 30 min at room temperature. After decalcification,
samples were washed in running tap water for 24 h and dehydrated
with an ascending ethanol gradient (70, 80, 90, 95 and 100%). The
fixed samples were embedded in paraffin and sliced to a thickness
of 3 µm. H&E staining was conducted according to routine
protocols (18). The section was
stained at room temperature with hematoxylin (cat. no. G4070;
Beijing Solarbio Science & Technology, Co., Ltd.) for 5 min and
eosin Y (cat. no. E8080; Beijing Solarbio Science & Technology,
Co., Ltd.) for 1 min.
Molecular analysis
All genomic DNA was extracted from peripheral blood
using the phenol-chloroform isoamyl alcohol (PCI) method (19). DNA samples were stored at -20˚C
until subsequent use. DNA integrity was evaluated by 1% agarose gel
electrophoresis and NanoDrop 2000 (Thermo Fisher Scientific, Inc.).
Based on the nucleotide sequence of the APC gene (RefSeq:
NM_000038, OMIM: 611731), 39 pairs of primers for 15 exons and
untranslated regions were designed (Table I). There were seven members of the
family that were available for DNA screening, except for II-1 and
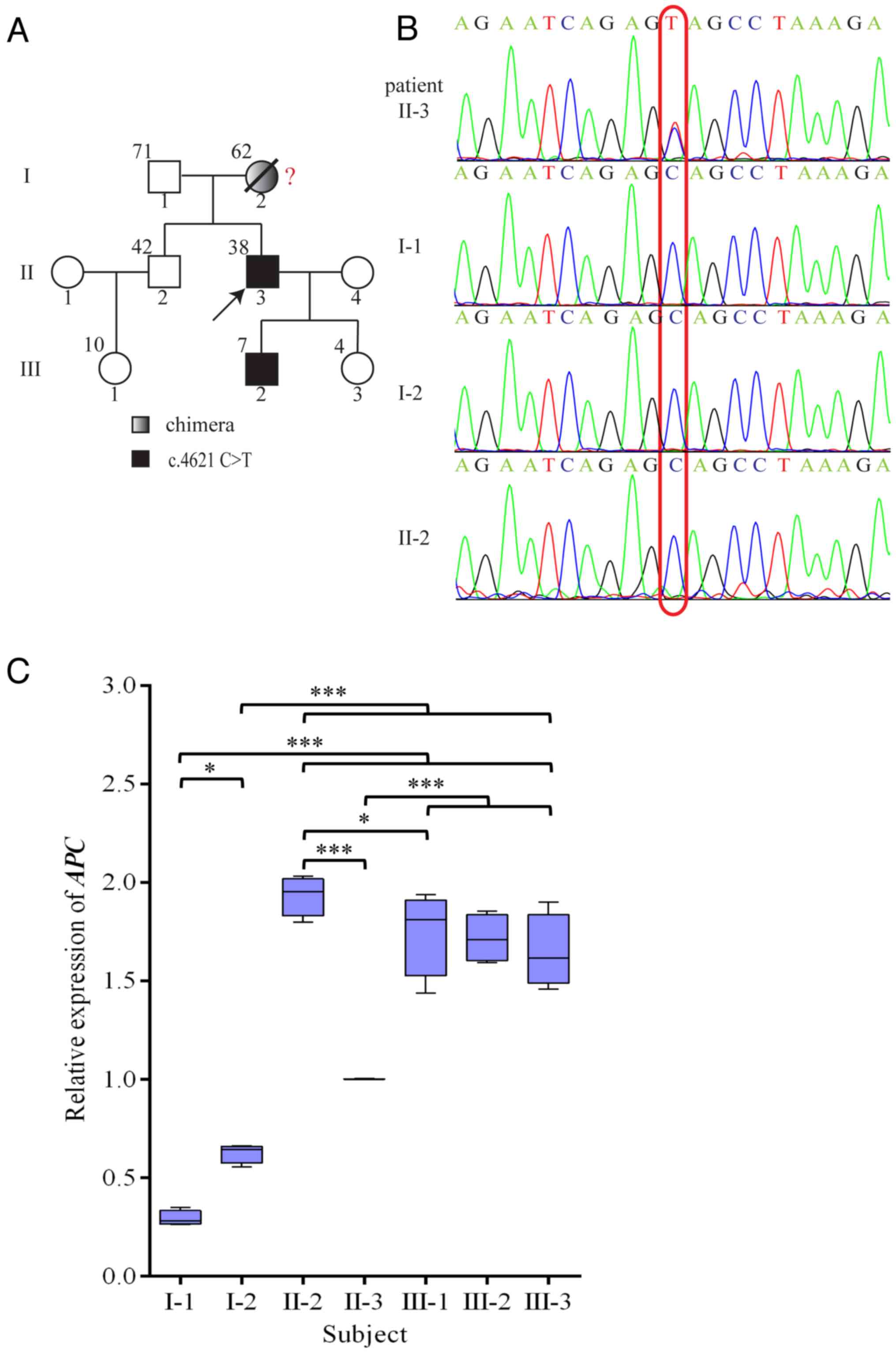
II-4 (Fig. 2A). The APC gene
was amplified using reverse-transcription quantitative PCT
(RT-qPCR) and sequenced using APC primers. RT-qPCR was
performed using GoTaq qPCR Master Mix (Promega Corporation) and the
CFX96™ Real-Time System (Bio-Rad Laboratories, Inc.). The
thermocycling conditions were as follows: 5 min at 95˚C, followed
by 40 cycles of denaturation for 20 sec at 95˚C, annealing for 20
sec at 60˚C and extension for 20 sec at 72˚C. Final melting curve
analysis was used to monitor the purity of the PCR product. The
2-∆∆Cq method was used to quantify mRNA abundance
(20). Relative gene expression
levels were normalized to GAPDH. SeqMan software (Lasergene,
Version 7.1.0; DNASTAR, Inc.) was used to view and align the
sequencing results. Total cellular RNA was isolated using TRIzol™
reagent (Gibco; Thermo Fisher Scientific Inc.), according to the
manufacturer's protocol. Complementary DNA (cDNA) synthesis was
performed by using the First-Strand cDNA Synthesis Kit (Toyobo Life
Science). Based on the complete mRNA sequence of APC gene
(GenBank: M74088.1; http://www.ncbi.nlm.nih.gov/nuccore/M74088.1), primers
forward, 5'-AGGGTCCAGGTTCTTCCAGA-3' and reverse,
5'-AGGCTGCTCTGATTCTGTTTCA-3' were designed to amplify the extensive
APC mRNA, and primers forward, 5'-GTGAAGGTCGGAGTCAACG-3' and
reverse, 5'-TGAGGTCAATGAAGGGGTC-3' to amplify the extensive
GAPDH mRNA as a control. Each study sample was analyzed in
triplicate, and this assay was performed repeatedly four times. The
results were expressed as the average (mean ± SD) of four
independent experiments. A panel of 25 heterozygous short tandem
repeat (STR) chromosomal markers were used to confirm maternity and
paternity (21) (Huaxia Platinum
PCR Amplification kit; Applied Biosystems; Thermo Fisher
Scientific, Inc.). As presented in Table II, these included D3S1358, vWA,
D16S539, CSF1PO and TPOX. Linkage equilibrium was assumed between
alleles on the same chromosome. At each locus, non-maternal and
non-paternal probabilities for each offspring were calculated as
the likelihood that unknown individuals could have been the
parents. The prevalence of each detected STR allele, for which
published allele frequencies are available online (http://www.ncbi.nlm.nih.gov/projects/SNP/). For each
child, the likelihood of parentage was calculated to be
(1-likelihood of non-parentage) and was 99.999%. Genetic marker PCR
products were determined using an Applied Biosystems 3130xl Genetic
Analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.).
 | Table IAPC primers. |
Table I
APC primers.
| Primer | Exon position | Forward
(5'-3') | Reverse
(5'-3') |
|---|
|
APC-5'UTR | 5'UTR |
GGAAGCGGAGAGAGAAGCAG |
TGACACTGGTATCTGTTTGCCAC |
| APC-1 | Exon 1 |
AGGCAAATGTATTCAGACAC |
GTACTTGCCAAATAAGACAAC |
| APC-2 | Exon 2 |
GTTTCTAATACCTTGCACAG |
AGATATCCTTTTAAAACTGCT |
| APC-3 | Exon 3 |
TGATAATAATTGAAGCCAGAC |
ATAGGTCTTCAGAATCCCAG |
| APC-4 | Exon 4 |
AGCCTTTGGTGAAGTGTAAG |
CCTATAGAGTATGTTTGGCAATTC |
| APC-5 | Exon 5 |
TTTAAGTGAAATAGGCCAATC |
TAGTCATTAGCTACCGGAAG |
| APC-6 | Exon 6 |
CAATTTGTTATTAAAGGGTG |
GTTATCACTTGTTTACCTGCT |
| APC-7 | Exon 7 |
TGCAGCTCCATTAAATGTCA |
CATAATCAAAATGCAGCTTAGAGT |
| APC-8 | Exon 8 |
TTCATACTTAGTTTTCTGGCAAT |
TCCAACTATTCTCATGCCTC |
| APC-9 | Exon 9 |
GGCCACTCATACTATTTACTCAC |
CTTTGAAACATGCACTACGAT |
| APC-10 | Exon 10 |
TGATCCACTAAAATTCCGTG |
ATAATAATTCCCTCTGATGCTC |
| APC-11 | Exon 11 |
AAGAGTGTTTATAAAGCCCTA |
ACAAATGAGTAAAGATAAGCG |
| APC-12 | Exon 12 |
ATCATTTCTCACCACTTATTC |
ACTAAATACTGAGCAACAATC |
| APC-13 | Exon 13 |
TTATGGCTCACAGTAACCTCA |
AAAATACAAATAGCCGGGAG |
| APC-14 | Exon 14 |
TTGTGTTCTGCTTGTTTTATAGAG |
TCTCTGGATCACGCTCATAG |
|
APC-15-1 | Exon 15 |
AGAGTGGCACCCAACCATAG |
TCCCATAATGCTTCCTGGTC |
|
APC-15-2 | Exon 15 |
CAGGCAAATCCTAAGAGAGAACA |
CTTGATGAAGAGGAGCTGGG |
|
APC-15-3 | Exon 15 |
GCTCAAGCTTGCCATCTCTT |
TATGGGCAGCAGAGCTTCTT |
|
APC-15-4 | Exon 15 |
CCAGGAACTTCTTCAAAGCG |
GTGAAGGACTTTGCCTTCCA |
|
APC-15-5 | Exon 15 |
GTCAATACCCAGCCGACCTA |
AGGCTGATCCACATGACGTT |
|
APC-15-6 | Exon 15 |
TTCCAACCACATTTTGGACA |
GAGCTGATTCTGCCTCTTGG |
|
APC-15-7 | Exon 15 |
AACGTCATGTGGATCAGCCT |
TGCTGGATTTGGTTCTAGGG |
|
APC-15-8 | Exon 15 |
CAGACGACACAGGAAGCAGA |
GCAGCTTGCTTAGGTCCACT |
|
APC-15-9 | Exon 15 |
GTGAACCATGCAGTGGAATG |
TGTTGGCATGGCAGAAATAA |
|
APC-15-10 | Exon 15 |
TTTGCCACGGAAAGTACTCC |
TATCATCCCCCGGTGTAAAA |
|
APC-15-11 | Exon 15 |
CTGTGGCAAGGAAACCAAGT |
TGATTTTTGTTGGGTGCAGA |
|
APC-15-12 | Exon 15 |
CCCAAAGGGAAAAGTCACAA |
GCTGATTGTTGGTTGGAGGT |
|
APC-15-13 | Exon 15 |
TCACCTCATCATTACACGCC |
GGTTCTCCCTGTGAGTCAGG |
|
APC-15-14 | Exon 15 |
ACTCCGGTTTGCTTTTCTCA |
AGCAGCAGCAGCTTGATGTA |
|
APC-15-15 | Exon 15 |
GCCTTCAAGACTCAAGGGTG |
TTGTCCTGCCTCGAGAGATT |
|
APC-15-16 | Exon 15 |
GCTGCTGCTGCATGTTTATC |
TGGCAACAGGGCTTAATTCT |
|
APC-15-17 | Exon 15 |
AATCTCTCGAGGCAGGACAA |
TCCTTTGGAGGCAGACTCAC |
|
APC-15-18 | Exon 15 |
CAGGTTTATCCAAGAATGCCA |
TTCAGAATGAGACCGTGCAA |
|
APC-15-19 | Exon 15 |
CCCACCTAATCTCAGTCCCA |
CAATCACCGGGGGAGTATTA |
|
APC-15-20 | Exon 15 |
AAATGGCACCTGCTGTTTCT |
TTCCACTGGATTCTGTGCTG |
|
APC-15-21 | Exon 15 |
TTGGAAAATCGCCTGAACTC |
TGGCTTCCAGAACAAAAACC |
|
APC-3’UTR-1 | 3'UTR |
ACAAAGAAGCGAGATTCCAA |
CCACTGTAGCTATCTCTATGCAC |
|
APC-3’UTR-2 | 3'UTR |
CAGTAATATGGTTCCCGATG |
CCAATGCTTAGTCTGTGCTAG |
|
APC-3’UTR-3 | 3'UTR |
GAAGACTGTTGCCACTTAACC |
TATTTGGCCTGCTATCGATT |
| Table IIGenotyping. |
Table II
Genotyping.
| | Individual |
|---|
| Short tandem
repeat | I-1 | I-2 | II-3 (proband) |
|---|
| D3S1358 | 15 | 16 | 15 | 16 | 15 | 16 |
| vWA | 15 | 17 | 17 | 18 | 17 | 17 |
| D16S539 | 11 | 13 | 9 | 11 | 11 | 13 |
| CSF1PO | 12 | 12 | 10 | 10 | 10 | 12 |
| TPOX | 8 | 9 | 8 | 11 | 8 | 9 |
| Yindel | 1 | 0 | 0 | 0 | 1 | 0 |
| AMEL | X | Y | X | X | X | Y |
| D8S1179 | 10 | 15 | 14 | 16 | 14 | 15 |
| D21S11 | 29 | 30 | 30 | 31 | 29 | 31 |
| D18S51 | 13 | 16 | 17 | 19 | 16 | 17 |
| Penta E | 11 | 14 | 16 | 21 | 11 | 21 |
| D2S441 | 10 | 11 | 11 | 11.3 | 11 | 11 |
| D19S433 | 14 | 15.2 | 14.2 | 15.2 | 15.2 | 15.2 |
| TH01 | 9 | 9 | 9 | 9 | 9 | 9 |
| FGA | 22 | 24 | 21 | 22 | 22 | 22 |
| D22S1045 | 16 | 17 | 15 | 16 | 16 | 16 |
| D5S818 | 12 | 12 | 11 | 12 | 11 | 12 |
| D13S317 | 11 | 12 | 11 | 12 | 11 | 11 |
| D7S820 | 8 | 12 | 11 | 11 | 8 | 11 |
| D6S1043 | 13 | 14 | 10 | 17 | 14 | 17 |
| D10S1248 | 14 | 16 | 15 | 16 | 14 | 15 |
| D1S1656 | 11 | 14 | 15 | 15 | 14 | 15 |
| D12S391 | 19 | 23 | 17 | 23 | 17 | 19 |
| D2S1338 | 17 | 19 | 24 | 24 | 19 | 24 |
| Penta D | 9 | 9 | 9 | 9 | 9 | 9 |
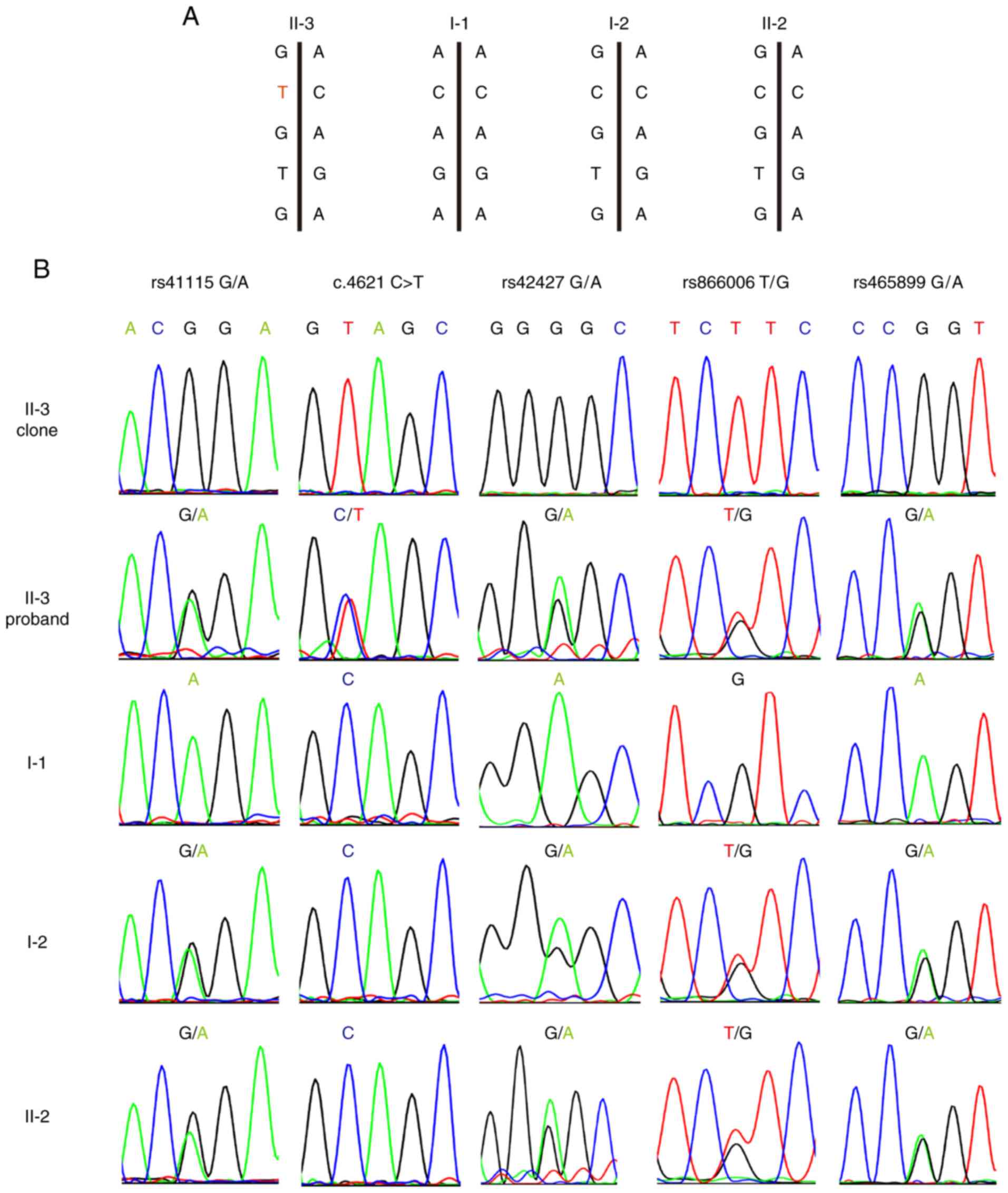
In order to avoid the interference of homologous
recombination exchange, primers forward, 5'-TCAAACAGCTCAAAC CAAG-3'
and reverse, 5'-AAATGATTTAGGAGCATAGCC-3' were designed for an
APC fragment that included the c.4621C>T variant and four
single nucleotide polymorphisms (SNPs). The SNPs included rs41115
G>A, rs42427 G>A, rs866008 T>G and rs465899 G>A, which
are 142 base-pairs upstream, 413 base-pairs downstream, 647
base-pairs downstream and 1259 base-pairs downstream of the
mutation site, respectively.
The APC PCR product from lymphoblast DNA was
cloned into the pMD19-T vector (Takara Bio, Inc.) and transformed
into chemically competent E. coli strain DH5α (Vazyme
Biotech Co., Ltd.). PCR amplification of the cloned APC
fragment amplicon was performed on isolated colonies, which were
selected using 100 µg/ml ampicillin (Sigma-Aldrich; Merck KGaA).
The products were then sequenced to determine which SNPs alleles
were in cis with the APC c.4621C>T variant (22). Three bacterial colonies were
sequenced per subject in order to obtain a consensus sequence.
Statistical analysis
Statistical analysis was performed with software
package of SPSS 20.0 (IBM Corp.). Figures were produced with Adobe
Photoshop/Illustrator CS6 imaging processing and drawing system
(Adobe, Inc.) and GraphPad Prism 6.0 software (GraphPad Software,
Inc.). Relative expression of APC gene among individuals in
the research family were compared using a one-way ANOVA and
Least-significant Difference (LSD) test. Bonferroni correction was
performed for further pairwise comparisons. Data were presented as
the mean ± SD from four independent experiments with a two-sided
P-value <0.05 for the difference was considered to be
statistically significant.
Results
Clinical outcomes
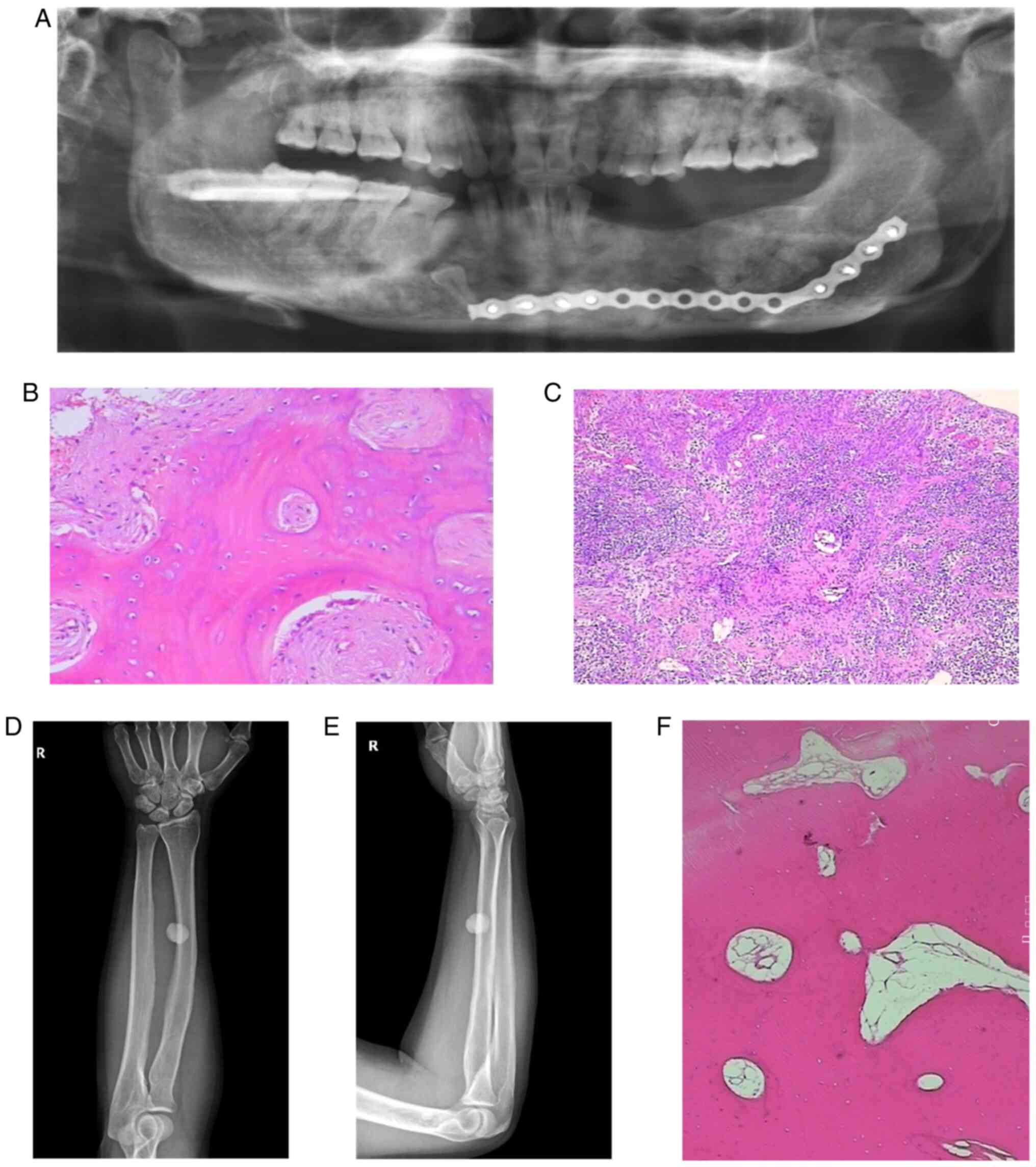
The postoperative X-ray dental panorama (Fig. 1A) of the proband showed that there
were impacted teeth in the right lower jaw, and that most of the
left mandibular teeth were absent. Regions of dense radiopacity
were observed around the teeth. Radiological reporting of these
scans confirmed the dense radio opacities to be consistent with the
presence of osteomas.
Pathological sections were obtained from gray-brown
bone tissue of the left and right mandible (Fig. 1B and C). The results confirmed that the
mandibular osteoma tissue consisted of a well-differentiated
lamellar bone. The trabeculae were connected with each other with
scattered, loose connective tissue was indicated between them.
There was no obvious abnormal shape in the bone cells with
osteoblasts being visible. A large number of acute and chronic
inflammatory cells had localized to the site of injury. The left
mandibular alveolar had lesioned with hyperplastic spindle cells
growing with a spiral shape in this area. The trabeculae in this
region were curved, and osteoblasts were visible around them.
Osteoblasts showed no obvious abnormalities.
The radius and ulna of the right forearm on
anteroposterior and lateral radiographs (Fig. 1D and E) revealed the presence of a circular high
density shadow. The boundary of the shadow was clear with an
approximate size of 1.7 by 1.5 cm. The shape and density of the
ulna and tibia were normal. No definite hyperosteogeny or
destruction was indicated. No fracture line was observed and the
soft tissue was normal.
Pathological sections were obtained from
hyperosteogeny tissue of the right tibial (Fig. 1F). The results confirmed that the
tissue consisted of a well-differentiated lamellar bone, and that
the cells were not abnormal. These results were consistent with a
diagnosis of osteoma.
Serum biochemical test demonstrated that the proband
(Fig. 2A; II-3) was normal
(Table III), but that his
mother's (I-2) serum CEA was 30.060 ng/ml (reference value range:
0.000-3.400), while her carbohydrate antigen 19-9 (CA19-9) was
>1,000.000 U/ml (reference value range: 0.000-27.000).
| Table IIIResults of the biochemical tests of
the proband and his mother. |
Table III
Results of the biochemical tests of
the proband and his mother.
| Parameter | Proband | Proband's
mother | Reference value
range |
|---|
| CEA (ng/ml) | 0.389 | 30.060 | 0.000-3.400 |
| CA15-3 (U/ml) | 15.670 | 15.380 | 0.000-25.000 |
| CA19-9 (U/ml) | 7.910 | >1,000.000 | 0.000-27.000 |
| CA72-4 (U/ml) | 3.860 | 2.860 | 0.000-6.900 |
Laboratory outcomes
Genetic testing was conducted on each of the family
members. A nonsense variant, c.4621C>T (23), was indicated in the proband (II-3)
and his son (III-2) by sequencing of the APC gene from
peripheral blood. None of the other members of this family had this
mutation (Fig. 2A and B). The mutation causes the codon at
position 1541 to change from a CAG of the encoded glutamine to a
stop codon (UAG). This resulted in early termination of
translation.
The amplification level of APC cDNA in the
proband (II-3) was 1.0, while the relative amplification from the
proband's unaffected brother (II-2), who is 4 years older than the
proband, was ~2.0 (Fig. 2C). The
relative amplification from the proband's father and mother was
~0.3 and 0.6, respectively. The relative amplification from the
third generation was approximated between 1.65 and 1.75. Paternity
testing was used to confirm the genetic contributions of the mother
and father to their children. This was conducted using 25
chromosomal markers. Based on known frequencies of these alleles in
the general population, non-paternity can be ruled out with 99.999%
certainty (Table II).
Because maternal and paternal inheritance was not an
issue, haplotype analysis of the APC locus was performed to
determine if the mutant allele originated from the mother. The
rs41115 SNP was homozygous (AA) in the father (I-1), but
heterozygous (GA) in the mother (I-2). Sequence analysis confirmed
that each of the two children were heterozygous and carried the GA
alleles. DNA cloning and sequencing of individual alleles
demonstrated that the c.4621C>T variant was co-inherited with
the rs41115 G allele at position 4479. This showed that the
mutation must occur in the C allele of the maternal line at
nucleotide position 4621, thus indicating the maternal origin of
the chromosome (Fig. 3A and
B). In addition, rs42427 G>A,
rs866008 T>G and rs465899 G>A confirmed this result.
Furthermore, both affected children (II-3) and an unaffected
sibling (II-2) inherited the identical chromosome from the mother
(Fig. 3A and B).
Discussion
As an extra-colonic presentation of the familial
adenomatous polyposis syndrome, Gardner's syndrome has prominent
characteristics. These include dental abnormalities, soft-tissue
tumors (including desmoid tumors), osteomas and epidermoid cysts.
Clinically, the proband in the current study exhibited dental
abnormalities and osteoma, which was consistent with the early
signs of Gardner's syndrome. Through mutation screening of the
APC gene, the proband was indicated to carry a rare
c.4621C>T variant. The genotype and phenotype of this mutation
correlated with Gardner's syndrome. Consistent with a mechanism of
nonsense-mediated mRNA decay (NMD) (24), APC gene expression in the
blood of the proband was half that of his wild-type brother.
However, the APC gene expression of the proband's father and
mother were less than the proband, which may be due to the fact the
expression of mRNA decreases with age. In the third generation, the
APC gene expression of the child with the mutation was
similar to those who lacked the mutation, indicating that the
pathogenic mutation did not affect APC expression in
childhood. This was also consistent with the characteristic of
Gardner's syndrome in that lesions only present after middle age.
The specific reason for this requires further investigation. To the
best of our knowledge, no study on the decrease of expression level
with increasing age or the antagonistic assumption that during
childhood the level of APC gene expression was not affected
by NMD has been performed. Therefore, to verify this speculation,
further follow-up observations on the research family should be
performed to study whether the expression dose of APC gene
changes with age. The location of the conserved sites is important
as mutations in such regions usually result in a genetic disorder
(25). In the present study, the
results demonstrated that mutations at the conserved c.4621
position in the APC gene also caused Gardner syndrome.
Combining the clinical phenotype of Gardner's syndrome and genetic
analysis, the proband was confirmed to be a patient suffering from
Gardner's syndrome. Concerning the development of clinical
characteristics of Gardner’s syndrome, dental malformations and
mandibular tumors preceded the development of other forms of the
syndrome, including intestinal polyps. Therefore, dentists should
be aware that dental malformations and mandibular tumors serve an
important role in the development of Gardner's syndrome.
Interestingly, mutation screening in other members
of the family revealed that the parents of the proband did not
carry the c.4621C>T variant. As this mutation was only detected
in the peripheral blood of the proband and his son, the
c.4621C>T variant appeared to be a de novo variant.
However, the proband's mother exhibited similar oral symptoms and
clinical history to the proband. Having died from cancer, it was
therefore necessary to determine if the transmission of the
mutation to the progeny was through maternal mosaicism. Serum
biochemical tests indicated that the CEA and CA19-9 levels of the
proband's mother were greater than the reference range. Among
numerous tumor markers, CEA and CA19-9 are the most common markers
to perform clinicopathological investigations on within colorectal
carcinoma (26,27). The results of the current study
indicated that the mother suffered from colorectal cancer. The high
levels of CEA and CA19-9 markers were associated with advanced
tumors which was consistent with the mother's disease course
(28). Unfortunately, the mother of
the proband died before the collection and analysis of the tissue
specimen, which included screening intestinal polyps for cell
mosaicism (29). Consequently, a
paternity test and haplotype analysis was designed to obtain more
information on the mosaic nature associated with the identified
variant. The results of paternity testing supported the parental
relationship between the proband and his parents. Haplotype
analysis demonstrated that the chromosome with the c.4621C>T
variant originated from the mother. As reported in the literature,
~20% of apparently sporadic cases of familial adenomatous polyposis
in which APC mutations can be identified are known to
display mosaicism (30). Therefore,
combined with the clinical phenotype of the mother, haplotype
analysis and literary references, this mutation was more likely to
have arisen from maternal mosaicism than due to a de novo
event in the proband.
A number of literature reports have indicated that
mosaicism of APC mutations can be identified through
sequence analysis of peripheral blood samples and/or adenomas
(17,29,31-34).
However, if only peripheral blood is inspected for the presence of
mutation, mosaicism may be missed due to very low, or even null
presentation of the mutated alleles (34). The present study presents an example
of this. If only the presence or absence of a mutation in the blood
is examined without considering the abnormality of the clinical
phenotype and further verification through haplotype analysis, such
a variant may be considered to be de novo in origin.
Therefore, it can be suggested that if de novo mutations are
detected in cases with Gardner's syndrome, the presence of
mutational mosaicism should be verified. Researchers should
carefully consider the clinical symptoms of the parents, and
collect colorectal polyps, or preferably, tumor tissue cells for
investigating the presence of mutational mosaicism (35). This strategy should be incorporated
in suspected familial adenomatous polyposis cases for which
APC mutation have been ruled out. Familial adenomatous
polyposis is an autosomal dominant genetic disorder that is
typically characterized by the development of hundreds to thousands
of adenomas in the colon and rectum during youth (36). If no APC mutations are
indicated in the DNA from peripheral blood of suspected familial
adenomatous polyposis cases, samples from tissues (such as colon
polyps and osteomas) should be collected to detect APC
mutations. If different members of the proband's family or
different specimen types have inconsistent APC mutation, the
possibility of mosaicism should be considered. According to the
human geneticist's point of view (37), there are three main types of
mosaicism. Mosaicism may exist in various parts of the body with
different mosaic proportions or only exist in certain parts of the
body. Detecting this will aid future in-depth research and prevent
missed diagnosis of familial adenomatous polyposis. If no tissue is
collected, haplotype analysis could be used as indirect evidence
for mutational mosaicism. The current study had certain limitations
that should be acknowledged. Firstly, the possibility of mosaicism
was not considered prior to experimentation, as the colon tissue
samples of the mother of the proband could not be collected in
time. Additionally, there was no direct evidence that could verify
the existence of mosaicism. Secondly, the detection of chimera is
challenging, which mainly depends on the type of chimera, the
location and the proportion of the chimera.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural Science
Foundation of Guangdong Province (grant no. 2018B030311033), the
Science and Technology Program of Guangzhou (grant no.
201707010301) and the National Natural Science Foundation of China
(grant no. 31970558).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DC designed and performed research, analyzed data,
and was a major contributor in writing the manuscript; DC and FH
performed experiments; XX analyzed the data and revised the
manuscript; FX and LZ designed and supervised the research, and
wrote the manuscript. All authors reviewed and edited the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Southern
Medical University Institutional Review Board. Informed consent was
obtained from all research participants.
Patient consent for publication
Informed consent for the publication of any
associated data and accompanying images was obtained from all
research participants.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kamel SG, Kau CH, Wong ME, Kennedy JW and
English JD: The role of Cone beam CT in the evaluation and
management of a family with Gardner's syndrome. J Craniomaxillofac
Surg. 37:461–468. 2009.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Dinarvand P, Davaro EP, Doan JV, Ising ME,
Evans NR, Phillips NJ, Lai J and Guzman MA: Familial adenomatous
polyposis syndrome: An update and review of extraintestinal
manifestations. Arch Pathol Lab Med. 143:1382–1398. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Gómez García EB and Knoers NV: Gardner's
syndrome (familial adenomatous polyposis): A cilia-related
disorder. Lancet Oncol. 10:727–735. 2009.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Yu D, Ng Cw B, Zhu H, Liu J and Lin Y:
Bone and dental abnormalities as first signs of familial Gardner's
syndrome in a Chinese family: A literature review and a case
report. Med Sci (Paris) null:. 34:20–25. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Adisen MZ, Okkesim A and Misirlioglu M:
The importance of early diagnosis of Gardner's syndrome in dental
examination. Niger J Clin Pract. 21:114–116. 2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
de Oliveira JC, Viana DV, Zanardo C,
Santos EMM, de Paula AE, Palmero EI and Rossi BM:
Genotype-phenotype correlation in 99 familial adenomatous polyposis
patients: A prospective prevention protocol. Cancer Med.
8:2114–2122. 2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yachida T, Nakajima T, Nonaka S, Nakamura
K, Suzuki H, Yoshinaga S, Oda I, Moriya Y, Masaki T and Saito Y:
Characteristics and clinical outcomes of duodenal neoplasia in
Japanese patients with familial adenomatous polyposis. J Clin
Gastroenterol. 51:407–411. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Basaran G and Erkan M: One of the rarest
syndromes in dentistry: Gardner syndrome. Eur J Dent. 2:208–212.
2008.PubMed/NCBI
|
|
9
|
Half E, Bercovich D and Rozen P: Familial
adenomatous polyposis. Orphanet J Rare Dis. 4(22)2009.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Juhn E and Khachemoune A: Gardner
syndrome: Skin manifestations, differential diagnosis and
management. Am J Clin Dermatol. 11:117–122. 2010.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Galiatsatos P and Foulkes WD: Familial
adenomatous polyposis. Am J Gastroenterol. 101:385–398.
2006.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wallis YL, Macdonald F, Hultén M, Morton
JE, McKeown CM, Neoptolemos JP, Keighley M and Morton DG:
Genotype-phenotype correlation between position of constitutional
APC gene mutation and CHRPE expression in familial adenomatous
polyposis. Hum Genet. 94:543–548. 1994.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Davies DR, Armstrong JG, Thakker N, Horner
K, Guy SP, Clancy T, Sloan P, Blair V, Dodd C, Warnes TW, et al:
Severe Gardner syndrome in families with mutations restricted to a
specific region of the APC gene. Am J Hum Genet. 57:1151–1158.
1995.PubMed/NCBI
|
|
14
|
Nieuwenhuis MH and Vasen HFA: Correlations
between mutation site in APC and phenotype of familial adenomatous
polyposis (FAP): A review of the literature. Crit Rev Oncol
Hematol. 61:153–161. 2007.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Bisgaard ML, Fenger K, Bülow S, Niebuhr E
and Mohr J: Familial adenomatous polyposis (FAP): Frequency,
penetrance, and mutation rate. Hum Mutat. 3:121–125.
1994.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Rozen P, Samuel Z, Rabau M, Goldman G,
Shomrat R, Legum C and Orr-Urtreger A: Familial adenomatous
polyposis at the Aviv Medical Center: Demographic and clinical
features. Fam Cancer. 1:75–82. 2001.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Aretz S, Stienen D, Friedrichs N, Stemmler
S, Uhlhaas S, Rahner N, Propping P and Friedl W: Somatic APC
mosaicism: A frequent cause of familial adenomatous polyposis
(FAP). Hum Mutat. 28:985–992. 2007.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Guo Y, Wang L, Ma R, Mu Q, Yu N, Zhang Y,
Tang Y, Li Y, Jiang G, Zhao D, et al: JiangTang XiaoKe granule
attenuates cathepsin K expression and improves IGF-1 expression in
the bone of high fat diet induced KK-Ay diabetic mice. Life Sci.
148:24–30. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Sambrook J, Fritsch EF and Maniatis T:
Molecular Cloning: A Laboratory Manual. 2nd edition. Cold Spring
Harbor Laboratory Press, Plainview, NY, ppE3-E15, 1989.
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Schwab AL, Tuohy TM, Condie M, Neklason DW
and Burt RW: Gonadal mosaicism and familial adenomatous polyposis.
Fam Cancer. 7:173–177. 2008.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kaufmann A, Vogt S, Uhlhaas S, Stienen D,
Kurth I, Hameister H, Mangold E, Kötting J, Kaminsky E, Propping P,
et al: Analysis of rare APC variants at the mRNA level: Six
pathogenic mutations and literature review. J Mol Diagn.
11:131–139. 2009.PubMed/NCBI View Article : Google Scholar
|
|
23
|
De Rosa M, Scarano MI, Panariello L,
Morelli G, Riegler G, Rossi GB, Tempesta A, Romano G, Renda A,
Pettinato G, et al: The mutation spectrum of the APC gene in FAP
patients from southern Italy: Detection of known and four novel
mutations. Hum Mutat. 21:655–656. 2003.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Castellsagué E, González S, Guinó E,
Stevens KN, Borràs E, Raymond VM, Lázaro C, Blanco I, Gruber SB and
Capellá G: Allele-specific expression of APC in adenomatous
polyposis families. Gastroenterology. 139:439–447, 447.e1.
2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al: ACMG
Laboratory Quality Assurance Committee: Standards and guidelines
for the interpretation of sequence variants: A joint consensus
recommendation of the American College of Medical Genetics and
Genomics and the Association for Molecular Pathology. Genet Med.
17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Nozoe T, Rikimaru T, Mori E, Okuyama T and
Takahashi I: Increase in both CEA and CA19-9 in sera is an
independent prognostic indicator in colorectal carcinoma. J Surg
Oncol. 94:132–137. 2006.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Lech G, Słotwiński R, Słodkowski M and
Krasnodębski IW: Colorectal cancer tumour markers and biomarkers:
Recent therapeutic advances. World J Gastroenterol. 22:1745–1755.
2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wang J, Wang X, Yu F, Chen J, Zhao S,
Zhang D, Yu Y, Liu X, Tang H and Peng Z: Combined detection of
preoperative serum CEA, CA19-9 and CA242 improve prognostic
prediction of surgically treated colorectal cancer patients. Int J
Clin Exp Pathol. 8:14853–14863. 2015.PubMed/NCBI
|
|
29
|
Out AA, van Minderhout IJ, van der Stoep
N, van Bommel LS, Kluijt I, Aalfs C, Voorendt M, Vossen RH, Nielsen
M, Vasen HF, et al: High-resolution melting (HRM) re-analysis of a
polyposis patients cohort reveals previously undetected
heterozygous and mosaic APC gene mutations. Fam Cancer. 14:247–257.
2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Tuohy TM and Burt RW: Somatic mosaicism: A
cause for unexplained cases of FAP? Gut. 57:10–12. 2008.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Filipe B, Albuquerque C, Bik E, Lage P,
Rodrigues P, Vossen R, Tops C and Nobre Leitão C: APC somatic
mosaicism in a patient with Gardner syndrome carrying the E1573X
mutation: Report of a case. Dis Colon Rectum. 52:1516–1521.
2009.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Hes FJ, Nielsen M, Bik EC, Konvalinka D,
Wijnen JT, Bakker E, Vasen HF, Breuning MH and Tops CM: Somatic APC
mosaicism: an underestimated cause of polyposis coli. Gut.
57:71–76. 2008.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Necker J, Kovac M, Attenhofer M, Reichlin
B and Heinimann K: Detection of APC germ line mosaicism in patients
with de novo familial adenomatous polyposis: A plea for the protein
truncation test. J Med Genet. 48:526–529. 2011.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Urbanova M, Hirschfeldova K, Obeidova L,
Janosikova B, Lastuvkova J, Lukas M, Kotlas J and Stekrova J: Two
Czech patients with familial adenomatous polyposis presenting
mosaicism in APC gene. Neoplasma. 66:294–300. 2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Kanter-Smoler G, Fritzell K, Rohlin A,
Engwall Y, Hallberg B, Bergman A, Meuller J, Grönberg H, Karlsson
P, Björk J, et al: Clinical characterization and the mutation
spectrum in Swedish adenomatous polyposis families. BMC Med.
6(10)2008.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Short E and Sampson J: The role of
inherited genetic variants in colorectal polyposis syndromes. Adv
Genet. 103:183–217. 2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Spinner NB and Conlin LK: Mosaicism and
clinical genetics. Am J Med Genet C Semin Med Genet. 166C:397–405.
2014.PubMed/NCBI View Article : Google Scholar
|