Introduction
Autosomal dominant neurological disorders include
paroxysmal kinesigenic dyskinesia [PKD; Online Mendelian
Inheritance in Man (OMIM) 128200] and benign familial infantile
epilepsy (BFIE; OMIM 605751), which are related and may occur
either in combination or separately (1,2). PKD
is one of the most common hereditary paroxysmal movement disorders.
The typical clinical manifestations of PKD are recurrent attacks of
choreic and dystonic movements, such as dystonic postures, chorea
and athetosis triggered by localized or generalized transient
involuntary movements (3,4). The symptoms generally develop in
adolescence or early adulthood and last for <2 min, typically or
up to 5 min on occasion (5).
However, usually they spontaneously disappear with age.
Anticonvulsant drugs such as carbamazepine and phenytoin may
effectively halt the disease's progression and improve the symptoms
of patients (6). BFIE is an
autosomal dominant epilepsy, which is characterized by focal
seizures or may progress to secondarily generalized seizures. The
seizures are clustered and have an onset at the age of 3-7 months,
and they usually alleviate prior to the age of 2 years with a good
prognosis (7). Certain members of
families with BFIE may develop PKD during childhood or adolescence
(8,9).
It has been reported that gene variants in
proline-rich transmembrane protein 2 (PRRT2; NM_001256442.2),
potassium voltage-gated channel subfamily A member 1 (KCNA1),
cholinergic receptor nicotinic alpha 4 subunit, calcium-activated
potassium channel subunit alpha-1 (KCNMA1) and sodium voltage-gated
channel alpha subunit 8 (SCN8A) may cause PKD and BFIE. PRRT2 is
the dominant pathogenic gene in PKD and BFIE (10-14).
The PRRT2 gene is predominantly expressed in the nervous system,
particularly in the cerebral cortex, hippocampus, basal ganglia and
cerebellum (15). According to the
Human Genome Variation Society (HGVS; http://varnomen.hgvs.org/), almost 100 variants have
been reported in the PRRT2 gene, including missense, nonsense,
frameshift, splice site, deletion and insertion variants.
In the present study, clinical information was
collected from three large families with PKD and/or BFIE, and
whole-exome sequencing was performed to screen the genetic
variants. One novel c.324_334del(p.Val109Argfs*21) frameshift
variant and two reported variants c.510_513del(p.Ser172Argfs*3) and
c.649dupC(p.Arg217Profs*8) in PRRT2 were identified from these
three Chinese families with hereditary PKD/BFIE. In the three
families, PRRT2 variants were observed in all family members with
PKD/BFIE but not in healthy family members.
Patients and methods
Patients
Three families with autosomal dominant disorders
were recruited at Central South University Xiangya Hospital
(Changsha, China) between May 2018 and August 2019. Clinical
information and 4-ml blood samples were collected from three
probands and their family members. Written informed consent was
obtained from the legal guardians of minors under the age of 18
years and all adult participants and approval was obtained from the
Ethics Committee of Xiangya Hospital of Central South University
(Changsha, China). The three probands were all diagnosed with PKD
for the first time. A detailed analysis of the family history
revealed that family B and family C had a history of BFIE.
Whole-exome sequencing
Genomic DNA was extracted from blood samples using
the QIAamp DNA Blood Mini kit (Qiagen GmbH) according to the
manufacturer's protocol and 0.6 µg genomic DNA of each proband was
used as input material. Exome capture was performed for each
proband using the Agilent SureSelect Human All Exon V6 kit (Agilent
Technologies, Inc.) according to the manufacturer's protocol. Using
an ultrasonoscope, DNA was sheared into fragments varying from 180
to 280 bp in size. Sequencing-capture DNA libraries were sequenced
on the Illumina Hiseq platform (Illumina, Inc.). The original
fluorescence image files were transformed to raw data by base
calling and these short reads were recorded in FASTQ format, which
contained corresponding sequencing quality information. Valid
sequencing data were aligned to the human reference genome sequence
from the National Center for Biotechnology Information (NCBI)
database using the Multi-Vision software package of Burrows Wheeler
Aligner (16-17). Small deletions and insertions were
determined by the GATK InDel Genotyper and single nucleotide
variants were identified using SOAPsnp. Reported variants were
filtered through the Single Nucleotide Polymorphism Database
(dbSNPs; https://www.ncbi.nlm.nih.gov/projects/SNP/), ExAC
browser (http://exac.broadinstitute.org/) and the 1,000 Genomes
Project database (www.internationalgenome.org). Previously identified
disease-causing mutations were obtained from the Human Gene
Mutation Database at the Institute of Medical Genetics in Cardiff
(HGMD; http://www.hgmd.cf.ac.uk/ac/index.php).
Validation of variants
In order to verify the DNA variants generated by
whole-exome sequencing, the target sites and their flanking
sequences were detected by PCR-Sanger DNA sequencing. Genomic DNA
reference sequences of candidate gene PRRT2 (NM_001256442.2) were
obtained from the University of California Santa Cruz (UCSC) Genome
Browser database (http://genome.ucsc.edu/). The PCR primers were
designed via online primer 3.0 (http://primer3.ut.ee/) and their sequences and
properties are presented in Table
I. Sanger sequencing was then performed for all probands and
family members. The PCR mixture contained genomic DNA,
deoxynucleoside triphosphate, GC buffer I (Sangon Biotech Co.,
Ltd.), primer, LA Taq DNA polymerase (Sangon Biotech Co., Ltd.) and
deionized water (18). The PCR
program was as follows: 95˚C for 3 min; 36 cycles of 94˚C for 30
sec, 58˚C for 30 sec and 72˚C for 45 sec; and finally, 72˚C for 8
min. Agarose gel electrophoresis was used to separate PCR products
and the QIAquick Gel Extraction kit (Qiagen GmbH) was used to
purify the target fragment. The sequencing data from the Applied
Biosystems 3730xl DNA Analyzer (Thermo Fisher Scientific, Inc.)
were aligned to the human reference sequence by the CodonCode
Aligner to confirm the variant (18).
 | Table IPrimers used for amplification of
potential mutations of proline-rich transmembrane protein 2 gene,
transcript NM_145239.3. |
Table I
Primers used for amplification of
potential mutations of proline-rich transmembrane protein 2 gene,
transcript NM_145239.3.
| Mutation | Mutation
location | Forward primer
(5'-3') | Reverse primer
(5'-3') | Tm (˚C) | Product size
(bp) |
|---|
| c.324_334del | Exon 2 |
CGCTGTCTCTGCTATTCCAT |
TTTTGAGGGTGGTGAGTGAG | 58 | 676 |
| c.510_513del | Exon 2 |
GCTCCAGAAACCACAGAGAC |
GGATGGCAAGGATGATGTAG | 58 | 619 |
| c.649dupC | Exon 2 |
GCCAAGAAACAGTGTCCAAA |
TGCTTAAGAGCTTGGCTACC | 58 | 639 |
Bioinformatics analysis
The protein sequences of the human PRRT2
(NP_660282.2) and the homologous sequences of different species in
FASTA format were obtained from the NCBI protein database.
Conservation analysis and sequence alignment were performed by MEGA
software (version 7) (19). The
pathogenicity of three frameshift variants was predicted by
MutationTaster (http://www.mutationtaster.org), and classified
according to the American College of Medical Genetics (ACMG)
criteria (20). The 1,000 Genomes
Project and Exome Aggregation Consortium (ExAC) are important
resources on human genetic variation supporting studies relating
genetic variation to health and disease
Results
Clinical findings
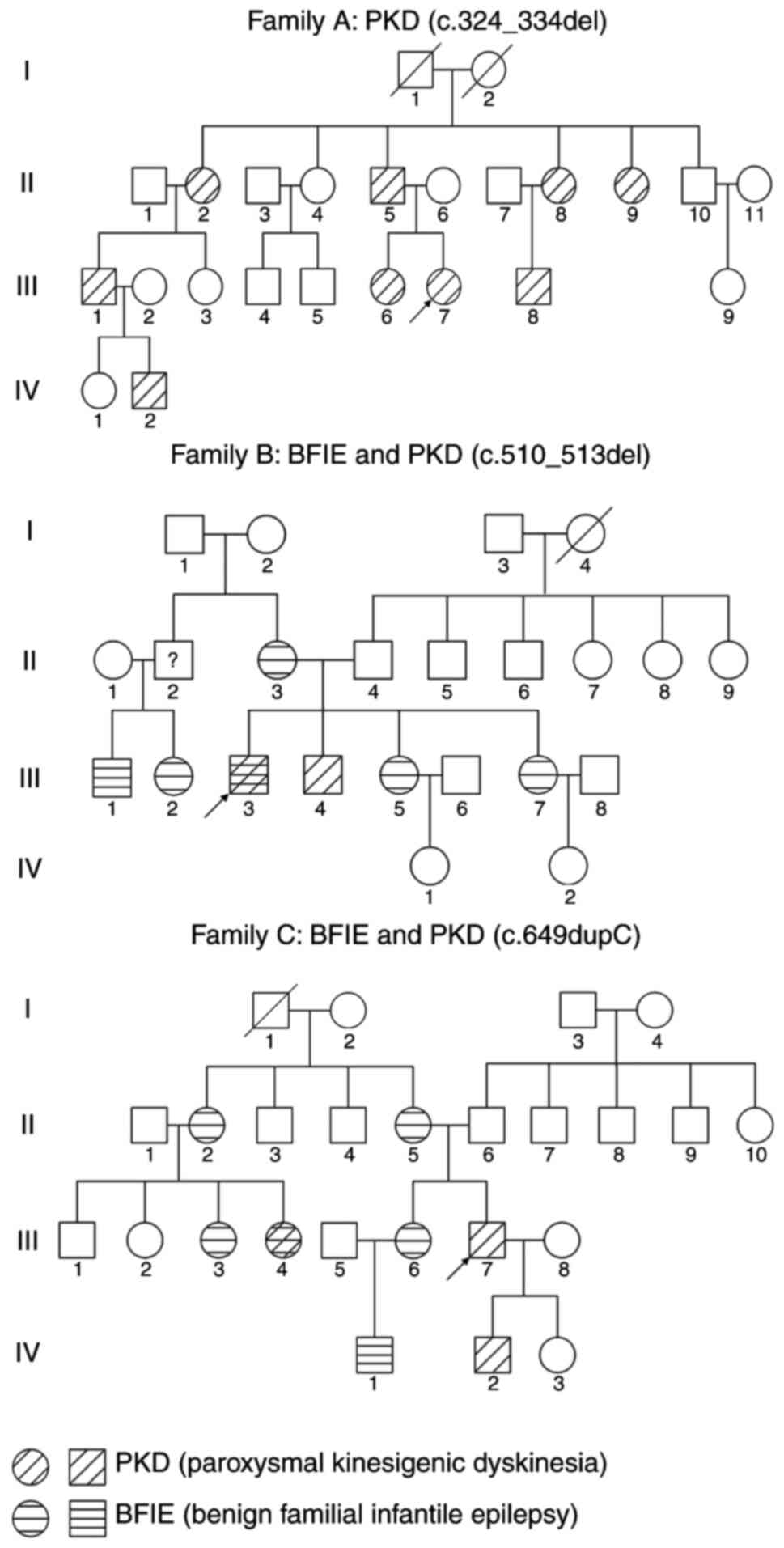
A total of 24 affected members from the three
families with PKD/BFIE were collected for the present study,
exhibiting extensive clinical heterogeneity (Fig. 1; Table
II). Family A was a fourth-generation Chinese pedigree
including nine affected cases who were all diagnosed with
idiopathic PKD (Fig. 1, Family A).
The proband (subject III7) was a 16-year-old female who had started
to have episodic dyskinesia attacks with a frequency of three or
four times per month at the age of 8 years. The main clinical
symptoms included dystonia and choreoathetosis, which only occurred
during the daytime and were mostly triggered by sudden movements.
The attacks generally lasted for <60 sec. The patient then
received carbamazepine monotherapy, which significantly reduced the
attacks. The patients in this family exhibited PKD disease alone
and the age of seizure onset in this family was 8-13 years (median,
9.4 years). The other patients in family A did not receive any
treatment but the symptoms disappeared prior to the age of 17
years. The clinical characteristics of family A are provided in
Table II.
| Table IIClinical information of patients in
the three families with PKD/BFIE. |
Table II
Clinical information of patients in
the three families with PKD/BFIE.
| A, Family A |
|---|
| Patient no. | Sex | Age (years) | Age at seizure
onset | Duration of attacks
(sec) | Frequency of
attacks | Diagnosis | Symptoms |
|---|
| II2 | Female | 69 | 10 y | 10 | 1 attack/d | PKD | Disappeared after age
of 12 years |
| II5 | Male | 63 | 8 y | 5 | 3-4 attacks/m | PKD | Disappeared after age
of 16 years |
| II8 | Female | 58 | 9 y | NA | NA | PKD | Disappeared after
age of 15 years |
| II9 | Female | 55 | 9 y | NA | 2-3 attacks/w | PKD | Disappeared after
age of 13 years |
| III1 | Male | 47 | 10 y | NA | 1 attack/d | PKD | Disappeared after
age of 15 years |
| III6 | Female | 28 | 13 y | NA | 3-4 attacks/d | PKD | Were relieved by
oral administration of carbamazepine |
| III7 | Female | 16 | 8 y | NA | 3-4 attacks/m | PKD | Disappeared after
age of 9 years without treatment |
| III8 | Male | 22 | 8 y | NA | 3-4 attacks/w | PKD | Disappeared after
age of 13 years |
| IV2 | Male | 16 | 10 y | NA | 3-4 attacks/w | PKD | NA |
| B, Family B |
| Patient no. | Sex | Age (years) | Age at seizure
onset (years) | Duration of attacks
(sec) | Frequency of
attacks | Diagnosis | Symptoms |
| II3 | Female | 67 | 6-7 m | 10 | 1 attack/d | BFIE | Disappeared after
age of 30 years without treatment |
| III1 | Male | 33 | 7-8 m | 10 | 2-3 attacks/w | BFIE | Improved after age
of 1 year |
| III2 | Female | 30 | 7-8 m | 10 | 2-3 attacks/w | BFIE | Disappeared after
age of 1 year |
| III3 | Male | 46 | 6-7 m | NA | 3-4 attacks/w | PKD/BFIE | Still present |
| III4 | Male | 39 | 8 y | 20 | 4-5 attacks/w | PKD | Improved after age
of 1 year |
| III5 | Female | 43 | 6-7 m | NA | NA | BFIE | NA |
| III7 | Female | 36 | 6-7 m | 30 | NA | BFIE | NA |
| C, Family C |
| Patient no. | Sex | Age (years) | Age at seizure
onset | Duration of attacks
(sec) | Frequency of
attacks | Diagnosis | Symptoms |
| II2 | Female | 56 | 5 m | 60 | NA | BFIE | Disappeared after
age of 13 months |
| II5 | Female | 57 | 6m | 30 | NA | BFIE | Disappeared after
age of 16 months |
| III3 | Female | 20 | 6 m | NA | NA | BFIE | NA |
| III4 | Female | 20 | 6 m | 180-240 | 5 attacks/d | PKD/BFIE | Still present |
| III6 | Female | 41 | 5-6 m | NA | NA | BFIE | Disappeared after
age of 18 months |
| III7 | Male | 42 | 16 y | 240 | NA | PKD | Disappeared after
age of 18 years |
| IV1 | Male | 3 | 8 m | NA | NA | BFIE | Disappeared after
age of 19 months |
| IV2 | Male | 17 | 15 y | NA | 10 attacks/d | PKD | NA |
A total of 7 patients in family B had BFIE and the
proband (subject III3) had both BFIE and PKD. The patients of
family B had seizures at about seven months of age and presented
with eye deviation to one side with consciousness, lasting about 10
sec. The proband of family B had symptoms of binocular roll-up at
six months without incentive, and duration was about 1 min. His
condition improved after the patient turned 1 years old. The
proband exhibited weakness in the legs when he was running at the
age of 10 years and had suffered falls when the condition was
severe. The patient's right hand unconsciously tremored. The
symptoms that were triggered by sudden movements have not improved
so far, and he was 46 years old upon symptom evaluation. The
probands and other members of family B had similar symptoms.
Excluding the proband, they occurred at 6-8 months of age and
symptoms disappeared after one year of age.
In family C, three patients exhibited PKD, five
patients had BFIE and one patient (individual III-4) had both of
these diseases. The proband (subject III7) was a 17-year-old male
who experienced weakness in both lower limbs when attempting to
perform dancing exercise at the age of 15 years. The patient's
symptoms improved significantly after taking oxcarbazepine.
Among the affected members of families B and C, 2
had PKD only, 13 had BFIE only and 2 patients had BFIE and PKD
combined. The onset age of seizures in the 13 patients with BFIE
was 5-8 months (mean age, 6.5 months). Seizures in patients were
either resolved spontaneously or with antiepileptic drug treatment.
All patients in families B and C had normal results of 24 h video
electroencephalography and brain MRI. The pedigrees of the two
families are presented in Fig. 1
and the clinical features of affected individuals were collected
and listed in Table II.
Genetic analysis
In order to identify the pathogenic variant
responsible for the disease in the three families, whole-exome
sequencing was performed on the probands. According to the exome
sequencing data, the average coverage was ~99.5% and the average
median depth reached 162-fold. First, the study focused on
identifying deleterious variants in known related genes of
paroxysmal movement disorders, mainly related to PKD/BFIE, such as
the PRRT2, KCNA1, KCNMA1 and SCN8A genes. The variants that
presented with a high frequency in the dbSNP database or the 1000
Genome database were also excluded. A total of three potential
pathogenic variants in the PRRT2 gene were confirmed in the three
probands associated with PKD/BFIE. They included the novel
heterozygous PRRT2 variant c.324_334delAGTGTCCAAAC in pedigree A
with a family history of PKD. Furthermore, variant
c.510_513del(p.Ser172Argfs*3) in family B and variant
c.649dupC(p.Arg217Profs*8) in family C were identified, which had
been reported in the previous literature (8).
Bioinformatics analysis of the
variants
The PRRT2 variant c.324_334delAGTGTCCAAAC in family
A was predicted to produce a truncated protein (p.Val109Argfs*21).
The variants co-segregated with the phenotype in each of the three
families (Fig. 1) and three
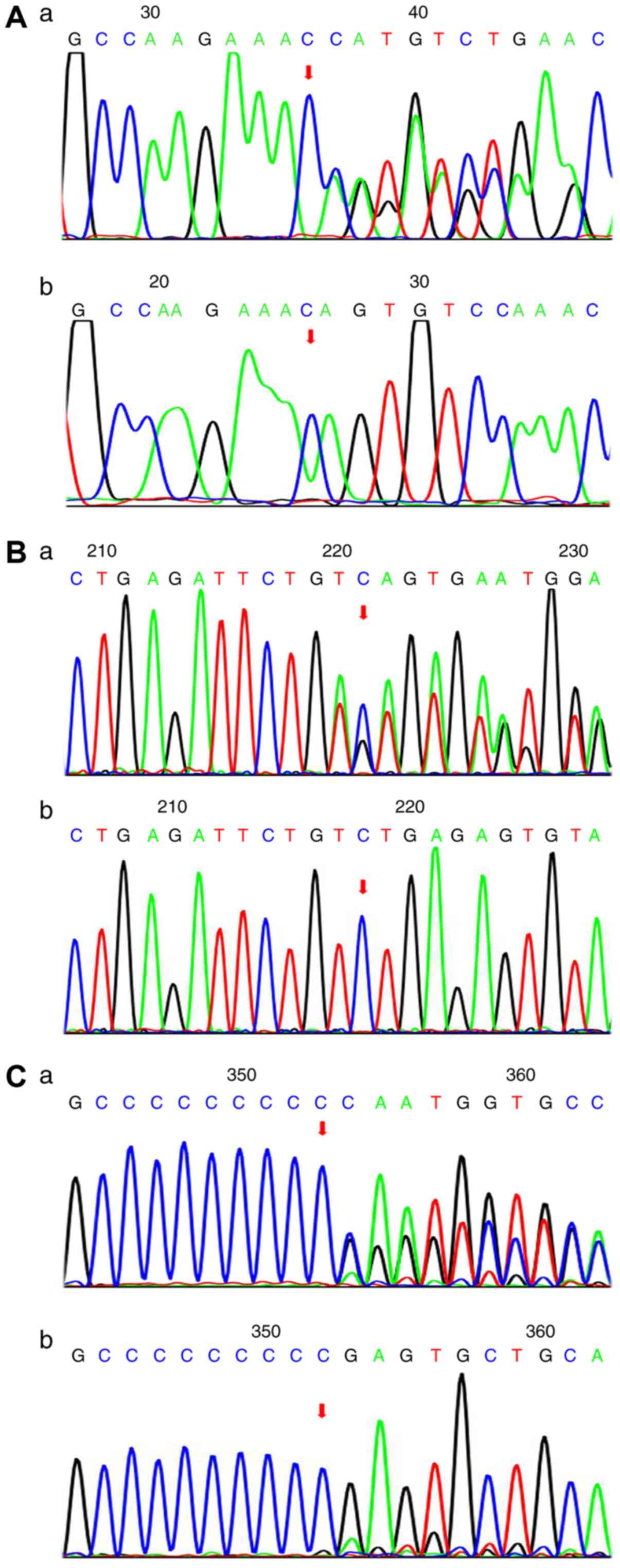
variants were further confirmed by Sanger sequencing (Fig. 2). Conservation analysis revealed
that the deleted base fragment was conserved in diverse species
(Fig. 3). This may corroborate the
pathogenicity of the mutant gene fragments. MutationTaster
predicted that the three frameshift variants are pathogenic
variants. Furthermore, the variant frequencies of c.324_334del and
c.510_513del were not recorded in the Exome Aggregation Consortium
(ExAC) browser and 1,000 Genomes Project database, indicating that
these variants are rare events in the human genome. The ACMG has
previously developed guidelines for the interpretation of sequence
variation and provided novel criteria for classifying pathogenic
variants in 2020(21). According to
the ACMG criteria, the variants of family A and family B are likely
pathogenic variants and the variant of family C is a pathogenic
variant (Table III).
| Table IIISummary and predicted effects of five
potentially disease-causing mutations in proline-rich transmembrane
protein 2 gene transcript NM_145239.3. |
Table III
Summary and predicted effects of five
potentially disease-causing mutations in proline-rich transmembrane
protein 2 gene transcript NM_145239.3.
| Family/proband | Nucleotide
change | Amino acid
change | Mutation type | Novel/
reported | ExAC | 1,000 Genomes
database | MutationTaster | Depth | Ref/alt | Evidence
(ACMG) | Pathogenicity
(ACMG) |
|---|
| A/III:7 | c.324_334del |
p.Val109Argfs*21 | Frameshift | Novel | - | - |
Disease-causing | 135 | 66/69 | PVS1+PM2 | Likely
Pathogenic |
| B/III:3 | c.510_513del |
p.Ser172Argfs*3 | Frameshift | Reported | - | - |
Disease-causing | 63 | 29/34 | PVS1+PM2 | Likely
Pathogenic |
| C/III:7 | c.649dupC |
p.Arg217Profs*8 | Frameshift | Reported | 0.0063 | - | Disease-
causing | 97 | 38/59 | PVS1+PS4 | Pathogenic |
Discussion
Paroxysmal and epilepsy disorders related to
involuntary movements have been known for a long time and these
disorders may be triggered by several types of stimuli (22,23).
PKD is the most common form of paroxysmal movement disorder, which
is characterized by recurrent and brief attacks of involuntary
movements whose onset may be triggered by sudden voluntary
movements (24). The involuntary
movements occur in adolescence or childhood and diminish with age;
they usually last several seconds, manifesting as chorea, dystonia
or their combination. PKD has a favourable response to relatively
low dosages of anticonvulsant medications (25). BFIE is a type of autosomal dominant
epilepsy, which begins in infancy (onset between 3 and 12 months of
age) and is characterized by brief seizures with motor arrest,
hypertonia, cyanosis and limb jerk. The clustered seizures respond
well to antiepileptic drugs and usually remit prior to the age of 2
years with a good prognosis (26).
The PRRT2 gene has been reported as a major
causative gene for PKD and BFIE. It is also responsible for several
familial or sporadic cases with paroxysmal non-kinesigenic
dyskinesia, paroxysmal exercise-induced dyskinesia and hemiplegic
migraine, but the underlying role of PRRT2 in various pathologies
has remained elusive (27,28). The PRRT2 protein is mainly expressed
in the central nervous system, regulates the release of synaptic
neurotransmitters and facilitates synaptic transmission. To date,
>100 types of PRRT2 gene variants have been identified. The
highest percentage of frameshift variants, which mainly occur in
exon 2, cause unstable truncation proteins of various lengths
(29). In the present study, the
three frameshift variants identified were predicted to result in
the production of a truncation protein and haploinsufficient state.
Therefore, the decrease of PRRT2 protein may affect synaptic
neurotransmitter release, causing dysregulated neuronal
excitability in various brain regions and eventually triggering
paroxysmal movement disorders and seizures.
The present study reported on a novel pathogenic
PRRT2 variant, c.324_334del, in family A, which was likely
responsible for the rare familial neurological disorder PKD. The
variant is novel and has not been reported in the 1,000 Genomes
Project, dbSNP, Exome Variant Server or ExAC database. Furthermore,
all affected cases in the family carried this variant, but it was
verified that the healthy individuals did not carry the variant.
Evidence of pathogenicity for ACMG's level of evidence of PVS1
should be considered when: The pathogenic mechanism of a disease is
loss of function (LOF) or if there are no functional mutations
including nonsense, frameshift, classical ±1 or 2 splicing, initial
codon or single/multiple exon deletion mutations. Furthermore, the
following should be considered: i) Whether the LOF of the gene is
the clear pathogenic mechanism of the disease; ii) a functional
deletion variation at the 3' end should be interpreted with
caution; iii) attention should be paid to whether the selective
deletion of exons affects the integrity of the protein; iv) a gene
may have multiple transcripts. Evidence of pathogenicity of ACMG's
level of evidence of PM2 should be considered if mutations or
low-frequency loci in recessive genetic diseases are not detected
in normal controls in the Exome Sequencing Project database, 1,000
Genomes Project database and ExAC database (27). In family A, c.324_334del
(p.Val109Argfs*21) was detected as a novel variant, and thus, the
evidence was ‘PVS1+PM2’. the variants of family A is likely
pathogenic variant.
The variant c.510_513del in PRRT2 has been
previously reported in one family with PKD (2), and it was detected in family B of the
present study, including proband III3 with BFIE, who developed
accompanying PKD with age. Chen et al (2) identified a heterozygous 4-bp deletion
(514delTCTG) in exon 2 of the PRRT2 gene in the proline-rich
domain, resulting in a frameshift and premature termination. The
mutation was not present in 1,000 genomes project. Expression of a
truncated form of PRRT2 in COS-7 cells led to loss of membrane
targeting and localization of the truncated protein in the
cytoplasm (28). The symptoms of
III3 in pedigree B worsened after taking carbamazepine; however,
the symptoms improved after switching to lamotrigine. Li et
al (29) reported that low
doses of carbamazepine, such as 100 or even 50 mg/day, were able to
effectively relieve the symptoms of 12 PRRT2 variant carriers.
However, in the present study, carbamazepine was not efficacious
for proband (III3) in family B. Thus, the patient required
follow-up of the treatment effects for a long time. c.649 is a
hotspot variant site: The deletion variant c.649delC, the c.649dupC
and a c.649C>T variant have been reported to affect the same
nucleotide (30,31). Among diverse frameshift variants,
the c.649dupC frameshift variant is the most common one and may be
observed in 80.5% of cases of PKD. Family C of the present study
with PKD/BFIE carried the hotspot pathogenic variant
c.649dupC(p.Arg217Profs*8). While the patients of the family C
carried the same variant, clinical manifestations varied widely
across families. For ACMG's level of evidence of PS4, the frequency
of the variation in the sick population is significantly higher
than that in the control population (18). According to the ACMG criteria, the
variants of family B are likely pathogenic variant and the variant
of family C is a pathogenic variant.
An optimal diagnosis of PKD and BFIE may be made by
combining clinical symptoms, genetic test results and the outcomes
of empirical treatment. As the disease was correctly diagnosed,
certain patients with PKD were treated with carbamazepine, the most
commonly reported anticonvulsant, and patients with BFIE were
treated with antiepileptic drugs such as valproate. After a period
of treatment, the symptoms of PKD/BFIE may be significantly
alleviated (26). Therefore,
accurate genetic testing may help doctors make a rapid and precise
judgment regarding various paroxysmal diseases and may help
patients receive timely treatment with a suitable medicine.
In summary, a genetic analysis in three families
with PKD/BFIE was conducted and three frameshift variants,
including the two deletion variants c.324_334del and c.510_513del
and the duplication variant c.649dupC were identified in PRRT2. The
present study expanded the variant spectrum of PKD/BFIE, provided
further evidence for the precise diagnosis of the disease and may
serve as a solid basis for genetic counselling and prenatal gene
diagnosis for members of families affected by PKD/BFIE.
Acknowledgements
Not applicable.
Funding
This study was supported by the Project from Hunan Provincial
Science and Technology Department (grant no. 2019SK1010), the
National Natural Science Foundation of China (grant nos. 81671299,
81974206 and 81671300), the Natural Science Foundation of Hunan
Province Project (grant no. 2020JJ5914) and the Central South
University Graduate Innovation Foundation (grant no.
1053320183945).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
The work was carried out in collaboration between
all authors. HL and LL co-designed methods. BX, JH and HT carried
out the experiments. CL, LT and WX analyzed the experimental data
and performed statistical analysis. JH drafted the manuscript. JH
and HT examined the manuscript. HL and LL revised the article. BX
and LL confirm the authenticity of all the raw data. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
All procedures performed in studies involving human
participants were in accordance with the ethical standards of the
research committee of Xiangya Hospital and with the 1964 Helsinki
declaration and its later amendments or comparable ethical
standards. Informed consent was obtained from all individual
participants or the legal guardians of participants included in the
study.
Patient consent for publication
Written informed consent was obtained from the
patients' family for publication of this article and the
accompanying images, and the use of data for both the patients and
the families.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lu JG, Bishop J, Cheyette S, Zhulin IB,
Guo S, Sobreira N and Brenner SE: A novel PRRT2 pathogenic variant
in a family with paroxysmal kinesigenic dyskinesia and benign
familial infantile seizures. Cold Spring Harb Mol Case Stud.
4(a002287)2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Chen WJ, Lin Y, Xiong ZQ, Wei W, Ni W, Tan
GH, Guo SL, He J, Chen YF, Zhang Q, et al: Exome sequencing
identifies truncating mutations in PRRT2 that cause paroxysmal
kinesigenic dyskinesia. Nat Genet. 43:1252–1255. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Bhatia KP: Paroxysmal dyskinesias. Mov
Disord. 26:1157–1165. 2011.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Albanese A, Bhatia K, Bressman SB, Delong
MR, Fahn S, Fung VS, Hallett M, Jankovic J, Jinnah HA, Klein C, et
al: Phenomenology and classification of dystonia: A consensus
update. Mov Disord. 28:863–873. 2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
van Vliet R, Breedveld G, de Rijk-van
Andel J, Brilstra E, Verbeek N, Verschuuren-Bemelmans C, Boon M,
Samijn J, Diderich K, van de Laar I, et al: PRRT2 phenotypes and
penetrance of paroxysmal kinesigenic dyskinesia and infantile
convulsions. Neurology. 79:777–784. 2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Vigevano F, Fusco L, Di Capua M, Ricci S,
Sebastianelli R and Lucchini P: Benign infantile familial
convulsions. Eur J Pediatr. 151:608–612. 1992.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Okumura A, Hayakawa F, Kato T, Kuno K,
Negoro T and Watanabe K: Early recognition of benign partial
epilepsy in infancy. Epilepsia. 41:714–717. 2000.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zeng Q, Yang X, Zhang J, Liu A, Yang Z,
Liu X, Wu Y, Wu X, Wei L and Zhang Y: Genetic analysis of benign
familial epilepsies in the first year of life in a Chinese cohort.
J Hum Genet. 63:9–18. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Okumura A, Shimojima K, Kubota T, Abe S,
Yamashita S, Imai K, Okanishi T, Enoki H, Fukasawa T, Tanabe T, et
al: PRRT2 mutation in Japanese children with benign infantile
epilepsy. Brain Dev. 35:641–646. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Jiang YL, Yuan F, Yang Y, Sun XL, Song L
and Jiang W: CHRNA4 variant causes paroxysmal kinesigenic
dyskinesia and genetic epilepsy with febrile seizures plus?
Seizure. 56:88–91. 2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Usluer S, Kayserili MA, Eken AG, Yiş U,
Leu C, Altmüller J, Thiele H, Nürnberg P, Sander T and Çağlayan SH:
Association of a synonymous SCN1B variant affecting splicing
efficiency with Benign Familial Infantile Epilepsy (BFIE). Eur J
Paediatr Neurol. 21:773–782. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wang J, Gao H, Bao X, Zhang Q, Li J, Wei
L, Wu X, Chen Y and Yu S: SCN8A mutations in Chinese patients with
early onset epileptic encephalopathy and benign infantile seizures.
BMC Med Genet. 18(104)2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yin XM, Lin JH, Cao L, Zhang TM, Zeng S,
Zhang KL, Tian WT, Hu ZM, Li N, Wang JL, et al: Familial paroxysmal
kinesigenic dyskinesia is associated with mutations in the KCNA1
gene. Hum Mol Genet. 27:625–637. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ebrahimi-Fakhari D, Saffari A,
Westenberger A and Klein C: The evolving spectrum of
PRRT2-associated paroxysmal diseases. Brain. 138:3476–3495.
2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Rossi P, Sterlini B, Castroflorio E, Marte
A, Onofri F, Valtorta F, Maragliano L, Corradi A and Benfenati F: A
Novel Topology of Proline-rich transmembrane protein 2 (PRRT2):
Hints for an intracellular function at the synapse. J Biol Chem.
291:6111–6123. 2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Pruitt KD, Tatusova T and Maglott DR: NCBI
Reference Sequences (RefSeq): A curated non-redundant sequence
database of genomes, transcripts and proteins. Nucleic Acids Res.
33:D501–D504. 2005.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Li H and Durbin R: Fast and accurate short
read alignment with burrows-wheeler transform. Bioinformatics.
25:1754–1760. 2009.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Verma M, Kulshrestha S and Puri A: Genome
Sequencing. Methods Mol Biol. 1525:3–33. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Kumar S, Stecher G and Tamura K: MEGA7:
Molecular evolutionary genetics analysis version 7.0 for bigger
datasets. Mol Biol Evol. 33:1870–1874. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Meds. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Riggs ER, Andersen EF, Kantarci S, Kearney
H, Patel A, Raca G, Ritter DI, South ST, Thorland EC,
Pineda-Alvarez D, et al: Response to Maya et al. Genet Med.
22:1278–1279. 2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Ma H, Feng S, Deng X, Wang L, Zeng S, Wang
C, Ma X, Sun H, Chen R, Du S, et al: A PRRT2 variant in a Chinese
family with paroxysmal kinesigenic dyskinesia and benign familial
infantile seizures results in loss of interaction with STX1B.
Epilepsia. 59:1621–1630. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chen GH: Five cases of paroxysmal
kinesigenic dyskinesia by genetic diagnosis. Exp Ther Med.
9:909–912. 2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Lee HY, Huang Y, Bruneau N, Roll P,
Roberson ED, Hermann M, Quinn E, Maas J, Edwards R, Ashizawa T, et
al: Mutations in the gene PRRT2 cause paroxysmal kinesigenic
dyskinesia with infantile convulsions. Cell Rep. 1:2–12.
2012.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhao G, Liu X, Zhang Q and Wang K: PRRT2
mutations in a cohort of Chinese families with paroxysmal
kinesigenic dyskinesia and genotype-phenotype correlation
reanalysis in literatures. Int J Neurosci. 128:751–760.
2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Heron SE, Grinton BE, Kivity S, Afawi Z,
Zuberi SM, Hughes JN, Pridmore C, Hodgson BL, Iona X, Sadleir LG,
et al: PRRT2 mutations cause benign familial infantile epilepsy and
infantile convulsions with choreoathetosis syndrome. Am J Hum
Genet. 90:152–160. 2012.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Marini C, Conti V, Mei D, Battaglia D,
Lettori D, Losito E, Bruccini G, Tortorella G and Guerrini R: PRRT2
mutations in familial infantile seizures, paroxysmal dyskinesia,
and hemiplegic migraine. Neurology. 79:2109–2114. 2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Ishii A, Yasumoto S, Ihara Y, Inoue T,
Fujita T, Nakamura N, Ohfu M, Yamashita Y, Takatsuka H, Taga T, et
al: Genetic analysis of PRRT2 for benign infantile epilepsy,
infantile convulsions with choreoathetosis syndrome, and benign
convulsions with mild gastroenteritis. Brain Dev. 35:524–530.
2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Li HF, Chen WJ, Ni W, Wang KY, Liu GL,
Wang N, Xiong ZQ, Xu J and Wu ZY: PRRT2 mutation correlated with
phenotype of paroxysmal kinesigenic dyskinesia and drug response.
Neurology. 80:1534–1535. 2013.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Cao L, Huang XJ, Zheng L, Xiao Q, Wang XJ
and Chen SD: Identification of a novel PRRT2 mutation in patients
with paroxysmal kinesigenic dyskinesias and c.649dupC as a mutation
hot-spot. Parkinsonism Relat Disord. 18:704–706. 2012.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Zhang Y, Li L, Chen W, Gan J and Liu ZG:
Clinical characteristics and PRRT2 gene mutation analysis of
sporadic patients with paroxysmal kinesigenic dyskinesia in China.
Clin Neurol Neurosurg. 159:25–28. 2017.PubMed/NCBI View Article : Google Scholar
|