Introduction
The terminal complement components, which comprise
complement components 5 (C5) to C9, participate in the assembly of
the membrane attack complex (MAC) (1). The MAC is responsible for the lytic
action of complement, which is essential in both the innate and
adaptive immune responses against invading pathogens (2). Genetic deficiencies of any of the
terminal complement components lead to failure to form the MAC and
susceptibility to Neisseria meningitides (Nm) infections,
the so-called terminal complement component deficiencies (TCCD)
(3).
As one of five terminal complement components, the
sixth complement component (C6) is a constituent of the MAC.
Deficiency of C6 (C6D; Online Mendelian Inheritance in Man #612446)
is associated with increased susceptibility to Nm infections and
patients with C6D usually present with recurrent meningococcal
disease (MD) (4). According to
patients' C6 levels, C6D is divided into complete genetic
deficiency of C6 (C6Q0) and subtotal genetic deficiency of C6
(C6SD). Patients with C6Q0 present with a complete lack of
functional C6, as opposed to C6SD patients, in whom a small amount
of C6 activity remains (5).
Similar to most other complement protein
deficiencies, C6D results from a mutation in the C6 gene, which
maps to chromosome 5p13. The C6 gene spans 80 kbp and is composed
of 18 exons (4). To date, only 15
mutations of the C6 gene have been identified in patients with C6D.
However, the genetic factors of C6D remain largely unknown and the
detailed molecular mechanisms involved require to be further
investigated.
Materials and methods
Subjects
The patient who participated in the present study
was diagnosed at the Third Xiangya Hospital of Central South
University (Changsha, China) in September 2019. The diagnosis of
C6SD was made according to the standard formulated previously
(6). Blood was collected from the
patient and the patient's parents. A total of 200 unrelated healthy
subjects were collected from the general population of Hunan in
China as control subjects to exclude polymorphisms. The healthy
controls (males/females, 100/100; age, 36.7±8.6 years) lacked C6D
diagnostic features. The baseline characteristics of these 200
healthy controls were reported in a previous study by our group
(7).
Biochemical analyses
The levels of CH50 (cat. no. NK095.OPT), C5 (cat.
no. RN027.3), C6 (cat. no. RN102.38), C7 (cat. no. RN103.3), C8
(cat. no. RN089.3), C9 (cat. no. RN028.3) were measured by radial
immunodiffusion assay (RID) kits (BINDARID™; The Binding Site Group
Ltd.) according to the manufacturer's instructions.
DNA extraction
Genomic DNA was prepared from peripheral blood of
the patient and all other participants using a DNAeasy Blood &
Tissue kit (Qiagen GmbH) on the QIAcube automated DNA extraction
robot (Qiagen GmbH).
PCR
Each exon of the C6 gene was amplified by PCR as
previously described (7). The
sequences of the primers we used were obtained from Hobart et
al (8). All of the coding exons
and exon-intron boundaries of the C6 gene were amplified by PCR in
a 25-µl reaction mixture, which consisted of 0.3 mM
deoxyribonucleotide triphosphates, 1X PCR buffer (10 mM Tris-HCl pH
9.0, 50 mM KCl, 0.1% Triton X-100 and 0.01% w/v gelatin), 2.0 mM
MgCl2, 0.5 µM of each primer (forward and reverse), 1.5
U of Taq polymerase and 50 ng of genomic DNA. The thermal cycling
consisted of an initial denaturation at 95˚C for 4 min, followed by
35 cycles of amplification consisting of denaturation at 95˚C for 1
min, primer annealing at desired temperature as previously
described (7) for 30 sec and primer
extension at 72˚C for 1 min. A final extension step was performed
at 72˚C for 7 min.
Mutation sequencing
The sequences of the PCR products of C6
(NM_001115131) from the patient and other subjects, including the
parents of the patient and the 200 controls, were determined with
the ABI 3100 Genetic Analyzer (Applied Biosciences; Thermo Fisher
Scientific, Inc.) as previously described (7).
Bioinformatics analysis
MutationTaster software (http://www.mutationtaster.org/) was used to predict
the effect of the mutation on the function of the protein.
SWISS-MODEL software (https://swissmodel.expasy.org/interactive) was used to
determine the function of the mutation. The multiple C6 protein
sequences across species were aligned using the program MUSCLE
(http://www.ebi.ac.uk/Tools/msa/muscle/).
Results
Clinical features
In the present study, a Chinese trio family with
C6SD was enrolled. The proband (II-1), a 6-year-old male from Hunan
Province in China, was admitted to the Third Xiangya Hospital of
Central South University (Changsha, China) due to high-grade fever.
The patient had generalized muscle pain and had developed a
purpuric rash on the lower extremities. The cerebrospinal fluid and
the blood culture were positive for Nm. To provide protection
against Nm, the patient was treated with ceftriaxone for 10 days,
resulting in complete resolution of the fever. The patient's muscle
pain resolved fully on day 3. Repeated blood cultures were
negative. The patient's skin returned to normal within a month.
Medical history investigation revealed that the patient had no
history of previous meningococcal infection. Since the
immunodeficiency workup suggested that all of the immune parameters
were normal except for a low level of serum C6 (17.7 mg/dl, far
below the mean normal level of 45 mg/dl; Table I), the patient was primarily
diagnosed with C6SD (6). No other
malformations were observed in this patient. After the patient's
diagnosis, complement screening of the proband's father (I-1)
revealed that the latter had a low level of C6 (25.2 mg/dl).
However, the patient's father had never been diagnosed as
meningococcal infection. C6 levels of the patient's mother (I-2)
were in the normal range (56.0 mg/dl). Information on other members
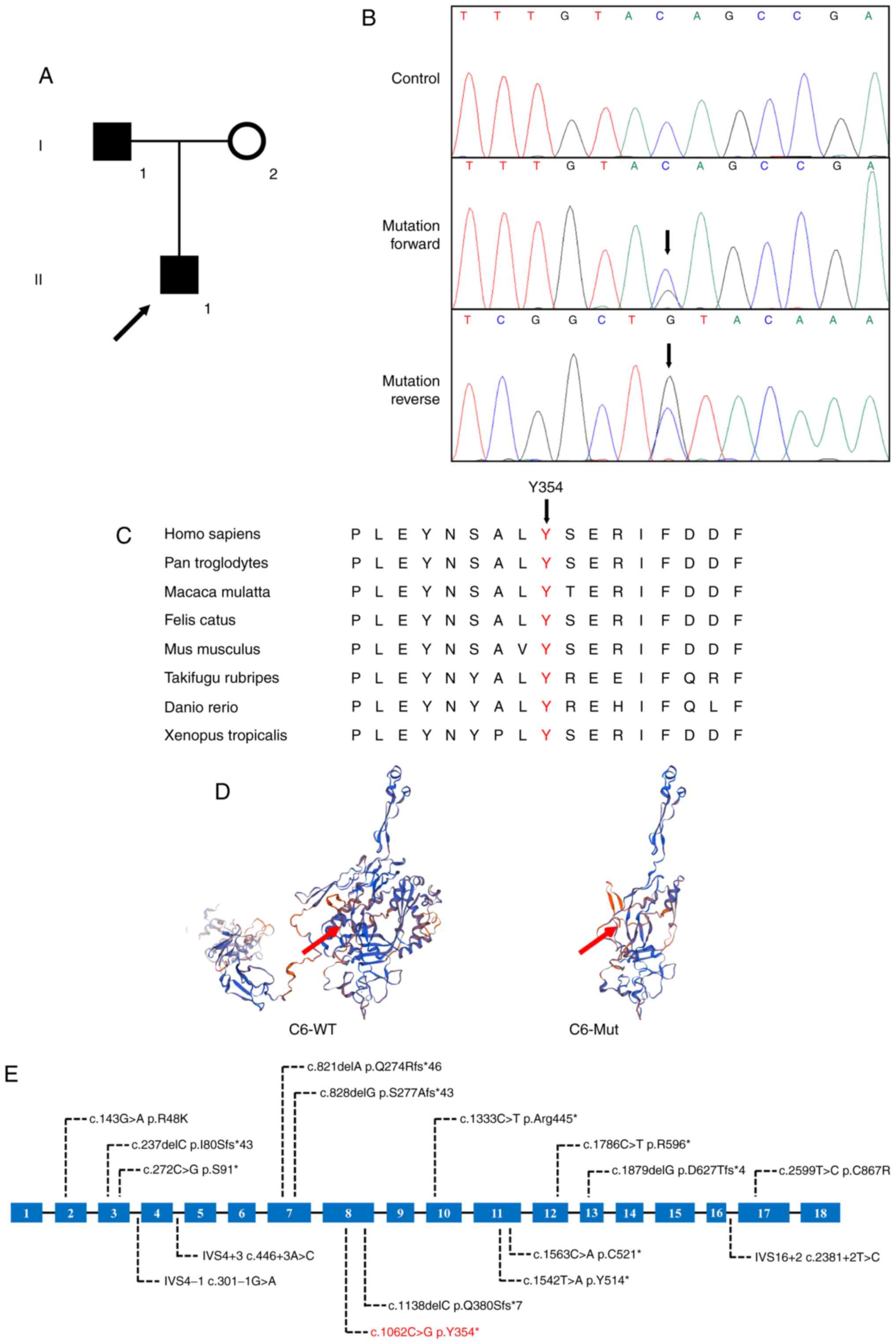
of the family was not available (Fig.
1A).
 | Table IImmunological findings in the
patient. |
Table I
Immunological findings in the
patient.
| Item | Value | Reference range |
|---|
| CD3+ (%) | 71 | 57-81 |
| CD4+ (%) | 35 | 26-48 |
| CD8+ (%) | 28 | 20-42 |
| CD16+56+ (%) | 23 | 8-28 |
| CD20+ (%) | 11 | 10-27 |
| In vitro
response to PHA (%) | 59.1 | 65.8±9.2 |
| IgG (mg/dl) | 1,069 | 745-1,804 |
| IgA (mg/dl) | 197 | 57-282 |
| IgM (mg/dl) | 222 | 78-261 |
| Anti B titer | 1/64 | >1/10 |
| CH50a | Present | Present |
| C5a (mg/dl) | 115 | 90-172 |
| C6a (mg/dl) | 17.7 | 45-95 |
| C7a (mg/dl) | 81 | 55-85 |
| C8a (mg/dl) | 121 | 112-172 |
| C9a (mg/dl) | 237 | 125-265 |
Genetic analysis
Sanger sequencing analysis of the C6 gene was
performed and a heterozygote nonsense mutation
(c.1062C>G/p.Y354*) was detected (Fig. 1B). Segregation analysis suggested
that the mutation was also present in the patient's father, while
it was excluded in the patient's mother. This newly identified
c.1062C>G mutation was not present in the 200 ethnically
matched, healthy controls and was also not present in the dbSNP144
(http://www.ncbi.nlm.nih.gov/projects/SNP/index.html)
and Exome Variant Server (EVS; http://evs.gs.washington.edu/EVS/) databases.
Alignment of C6 amino acid sequences from humans, chimpanzees,
monkeys, cats, mice, globefish and zebrafish revealed that the
affected amino acid was evolutionarily conserved (Fig. 1C). The MutationTaster tool predicted
that the p.Y354* variant is a disease-causing mutation. Analysis
with SWISS-MODEL software for exploring the spatial configuration
of the protein suggested a loss of more than half of the total
protein in the mutated C6 protein compared with that in the
wild-type protein (Fig. 1D).
Discussion
In the present study, the genetic lesion of a Han
Chinese pediatric patient with C6SD who was diagnosed due to having
MD was investigated. A heterozygote nonsense mutation
(c.1062C>G/p.Y354*) of C6 was identified by Sanger sequencing.
The nonsense mutation is located in exon 8 and alters the tyrosine
codon at position 354 to a termination codon, leading to a
truncated protein. Although the dysmorphic C6 protein still has
certain residual activity (9), the
truncated C6 protein in the present study is expected to fail to
produce a functional MAC, as it lacks more than half of the normal
protein. Thus, it was indicated that the nonsense mutation
(c.1062C>G/p.Y354*) identified in the C6 gene may be a potential
candidate factor for the development of C6D, which is consistent
with previous findings (4,10).
The terminal complement system is essential for
fighting MD. As one of the terminal complement components, C6D
predisposes individuals to infection with Nm (4). Most patients with MD in the setting of
C6D have undetectable levels of C6. Nm infection in C6SD has rarely
been described (9,11). Multiple family studies have
demonstrated examples of individuals with C6 <50% the normal
levels who had no meningococcal infection (12). The present study reported on a trios
family diagnosed as C6SD. The proband presented with a recent
history of meningitis. A heterozygote mutation
(c.1062C>G/p.Y354*) in C6 was identified. Of note, the patient's
father harbored the same variant but had no meningococcal
infection, which is consistent with the previous study. Although
the numbers are small, patients with C6SD and MD have also been
described. Compared to normal individuals, the remaining C6 protein
in a patient with C6SD is not sufficient to resist the invasion of
bacteria, having certain bactericidal activity but to a lesser
extent (5,12). The same is suspected in the proband
of the present study. A single episode of MD may result in serious
long-term sequelae and studies indicated that 73% of C6Q0 patients
who suffered recurrent MD developed serious illness or died
(3). To protect patients with C6Q0
from further episodes of MD, patients were prescribed long-term
monthly injections with long-acting penicillin (bicillin) and given
a conjugated meningococcal vaccination (3). The susceptibility to MD or other
infectious diseases between C6Q0 and C6SD is not uniform. There may
be additional controllable factors, such as infections and
accidents, affecting the susceptibility of patients with C6SD. No
clear information is available with respect to the treatment and
prognosis of C6SD. It is only by identifying C6SD patients and
investigating other susceptibility factors that the best means of
treatment and prophylaxis may be determined. Likewise, prophylactic
treatment and/or vaccination are recommended for patients at risk
of MD (13,14). It is crucial to test C6 levels
accurately and determine the C6 genetic defects responsible for
either C6Q0 or C6SD, which contributes to the optimal management of
patients with C6D (3). As part of
the management of C6D, the pediatric patient of the present study
was advised to immunize against meningococci ACYW and B.
Prophylactic oracillin was considered. The present study not only
proved the diagnosis of C6SD for this child, but also further
confirmed the association of meningococcal infections with
TCCD.
Previous research has indicated that homozygosity,
or compound heterozygosity of C6 defects, resulted in C6Q0(3). However, heterozygous carriers of C6D
have always been described as C6SD (11). The pediatric patient with a
heterozygote nonsense mutation in the present study had a low level
of C6, which is consistent with heterozygous C6
sufficiency/deficiency. To date, approximately 15 mutations of C6
have been reported in patients with C6D. All of these known
mutations of the C6 gene are briefly summarized in Fig. 1E, which may be conducive to the
genetic diagnosis of C6D and counseling of such patients. Of note,
the mutation (c.1062C>G/p.Y354*) identified in the present study
has not been previously published and is therefore considered
novel.
The incidence of C6D varies considerably within
populations. C6D has demonstrated a prevalence in Western Cape
South Africans and it has been diagnosed much more frequently in
South Africa than elsewhere (15).
Furthermore, affected individuals in Irish families were also
reported in countries where C6D appears to be sporadic and rare
(4). C6D also appears to be
sporadic and rare in East Asia. Population data on the prevalence
of C6D are only available for Japan, where it was determined to be
2-7 per 100,000(16). No data are
currently available for China. In the present study, a Han Chinese
pediatric patient with C6D was described. To the best of our
knowledge, the present study was the first to investigate the role
of the C6 gene in Han Chinese patients with C6D, and further
studies are essential to evaluate the association between the C6
gene and C6D in the Han Chinese population.
In conclusion, the present study identified a novel
nonsense mutation (c.1062C>G/p.Y354*) in the C6 gene as a
possible cause of C6SD in a Han Chinese pediatric patient who was
diagnosed due to having MD. The present study not only expanded the
spectrum of C6 mutations but also confirmed the clinical diagnosis
for this patient as C6SD.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Science and Technology
Major Project of the Ministry of Science and Technology of China
(grant no. 2017ZX10103005-006) and the National Natural Science
Foundation of China (grant no. 81970403).
Availability of data and materials
The datasets for this article are not publicly
available due to concerns regarding participant/patient anonymity.
The datasets used and/or analyzed during the current study are
available from the corresponding author on reasonable request.
Authors' contributions
AQZ designed the study. YXL performed the molecular
analysis and participated in manuscript writing. JYJ recruited and
examined the family, collected blood samples and extracted DNA. CYW
performed the bioinformatics analysis. DBX and LLF analyzed the
data, participated in manuscript preparation and acquired funding
for the study. YXL and LLF checked and confirmed the authenticity
of the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
the Third Xiangya Hospital of Central South University (Changsha,
China) and performed in accordance with the principles outlined in
the Declaration of Helsinki. Written informed consent was obtained
from all of the adult participants and legal guardians of minor
participants.
Patient consent for publication
The patients/participants provided written informed
consent to participate in this study. Written informed consent was
obtained from the individuals for the publication of any
potentially identifiable images or data included in this
article.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
DiScipio RG and Hugli TE: The molecular
architecture of human complement component C6. J Biol Chem.
264:16197–16206. 1989.PubMed/NCBI
|
|
2
|
Merle NS, Noe R, Halbwachs-Mecarelli L,
Fremeaux-Bacchi V and Roumenina LT: Complement system part II: Role
in immunity. Front Immunol. 6(257)2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Orren A, Owen EP, Henderson HE, van der
Merwe L, Leisegang F, Stassen C and Potter PC: Complete deficiency
of the sixth complement component (C6Q0), susceptibility to
Neisseria meningitidis infections and analysis of the frequencies
of C6Q0 gene defects in South Africans. Clin Exp Immunol.
167:459–471. 2012.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Parham KL, Roberts A, Thomas A, Wurzner R,
Henderson HE, Potter PC, Morgan BP and Orren A: Prevalence of
mutations leading to complete C6 deficiency (C6Q0) in the Western
Cape, South Africa and detection of novel mutations leading to C6Q0
in an Irish family. Mol Immunol. 44:2756–2760. 2007.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wurzner R, Orren A, Potter P, Morgan BP,
Ponard D, Spath P, Brai M, Schulze M, Happe L and Gotze O:
Functionally active complement proteins C6 and C7 detected in C6-
and C7-deficient individuals. Clin Exp Immunol. 83:430–437.
1991.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ikinciogullari A, Tekin M, Dogu F, Reisli
I, Tanir G, Yi Z, Garrison N, Brilliant MH and Babacan E:
Meningococccal meningitis and complement component 6 deficiency
associated with oculocutaneous albinism. Eur J Pediatr.
164:177–179. 2005.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Xiang R, Fan LL, Huang H, Cao BB, Li XP,
Peng DQ and Xia K: A novel mutation of GATA4 (K319E) is responsible
for familial atrial septal defect and pulmonary valve stenosis.
Gene. 534:320–323. 2014.PubMed/NCBI
|
|
8
|
Hobart MJ, Fernie BA, Fijen KA and Orren
A: The molecular basis of C6 deficiency in the western Cape, South
Africa. Hum Genet. 103:506–512. 1998.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Orren A, Wurzner R, Potter PC, Fernie BA,
Coetzee S, Morgan BP and Lachmann PJ: Properties of a low molecular
weight complement component C6 found in human subjects with
subtotal C6 deficiency. Immunology. 75:10–16. 1992.PubMed/NCBI
|
|
10
|
El Sissy C, Rosain J, Vieira-Martins P,
Bordereau P, Gruber A, Devriese M, de Pontual L, Taha MK, Fieschi
C, Picard C and Frémeaux-Bacchi V: Clinical and genetic spectrum of
a large cohort with total and sub-total complement deficiencies.
Front Immunol. 10(1936)2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wurzner R, Hobart MJ, Fernie BA, Mewar D,
Potter PC, Orren A and Lachmann PJ: Molecular basis of subtotal
complement C6 deficiency. A carboxy-terminally truncated but
functionally active C6. J Clin Invest. 95:1877–1883.
1995.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Rosenfield L, Cvetkovic A, Woodward K and
Quirt J: Late presentation of subtotal C6 deficiency in a patient
with recurrent Neisseria meningitides infections. Ann Allergy
Asthma Immunol. 120:432–433. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Westra D, Kurvers RA, van den Heuvel LP,
Wurzner R, Hoppenreijs EP, van der Flier M, van de Kar NC and
Warris A: Compound heterozygous mutations in the C6 gene of a child
with recurrent infections. Mol Immunol. 58:201–205. 2014.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Owen EP, Leisegang F, Whitelaw A, Simpson
J, Baker S, Wurzner R, Potter P and Orren A: Complement component
C5 and C6 mutation screening indicated in meningococcal disease in
South Africa. S Afr Med J. 102:525–527. 2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Ram S, Lewis LA and Rice PA: Infections of
people with complement deficiencies and patients who have undergone
splenectomy. Clin Microbiol Rev. 23:740–780. 2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Nishizaka H, Horiuchi T, Zhu ZB, Fukumori
Y, Nagasawa K, Hayashi K, Krumdieck R, Cobbs CG, Higuchi M,
Yasunaga S, et al: Molecular bases for inherited human complement
component C6 deficiency in two unrelated individuals. J Immunol.
156:2309–2315. 1996.PubMed/NCBI
|