Introduction
Isolated tracheoesophageal fistula (or H-type
tracheoesophageal fistula) is a rare congenital disease described
for the first time in a 7-week-old infant by Lamb in 1873(1). It is one of the main five anatomical
types of congenital esophageal malformations, as it appears in both
Vogt and Gross classifications, which are still the most commonly
used today (2). Medical literature
shows that both ear, nose and throat (ENT) specialists and
pediatric surgeons are involved in H-TOF management.
H-TOF accounts for less than 5% of all congenital
tracheoesophageal malformations (3). The most common presentation includes
choking and coughing during feeding, repeated episodes of
pneumopathy, recurring cyanosis and abdominal distension (4). Even though these are recognized early
in the first days of life, the final diagnosis is often delayed.
This is a consequence of the broad spectrum of differential
diagnosis in infancy, taking into account more frequent neonatal
pathology such as perinatal asphyxia, with similarities in clinical
presentation, the rareness of the anomaly and, moreover, the low
sensitivity and specificity of the available evaluation tools whose
results are also dependent on the experience of the health
professional, mainly the surgeon or the radiologist. Often,
repeated evaluations are reported to be required until H-TOF is
recognized since it is difficult to diagnose after a single test
(4,5).
Imperatori reported the first repair of a H-TOF in
1939 in a 6-year-old child via a trans-tracheal route. Haight,
reported in 1948 the first thoracic approach in H-TOF repair and
Miller, the first trans-cervical division of an H-TOF located close
to the thyroid (1,3). The main goal in the surgical treatment
of H-TOF, is division of the fistula. This may be achieved through
a thoracic or cervical access with simple ligation or excision of
the fistula tract followed by tracheal and esophageal repair
(3). Fistula identification, with a
clear preoperative and intraoperative picture over the fistula's
position and-therefore, choosing the right approach-is a
centerpiece of H-TOF management. The aim in surgical planning is to
avoid extensive and unnecessary dissection and thus potential
complications taking into account the hazardous anatomy of the
cervical region or the posterior mediastinum, especially in
newborns or infants (4,6).
Patients and methods
Six patients were treated for H-TOF at the Pediatric
Surgery Clinic, Emergency Clinical Hospital for Children ‘Maria
Sklodowska Curie’, Bucharest, Romania, between 2005 and 2019.
Informed consent was obtained from the patient's parents. The
medical records of these patients were analyzed and the following
information was summarized into short case series: history and
clinical presentation, preoperative evaluation tools and
observations, age at surgery, surgical approach and intraoperative
comments. By compiling our data and correlating it to the current
knowledge, we wish to highlight ‘error traps’ or challenges that we
have encountered and other possible ‘red flags’ and establish a
relevant view of the practical safety in the diagnosis and
management of this rare congenital malformation.
Results
The sex distribution of our series describes 4 male
and 2 female patients. The youngest patient was diagnosed at 15
days of life, while surgery was performed at 38 months in the
oldest one. The median age of our series was 19 months and the mean
age was 12 months. Feeding difficulties (choking, especially during
liquid swallowing) and functional respiratory syndrome were the
most frequently noted clinical aspects. One case presented with
associated tracheomalacia. No other associated congenital
malformations were noted in this series. Regarding other
comorbidities, failure to thrive was noted in two cases and two
cases of associated gastroesophageal reflux disease (GERD). We also
report one case of H-TOF with matrilineal heredity with a 2nd
degree relative (mother's sister) with history of esophageal
atresia and distal tracheoesophageal fistula. Esophagogram was
performed in all cases with a true positive rate of 66% (4 out of
6). Esophagoscopy was conducted in three cases, indicating fistula
in only one case, while tracheobronchoscopy was performed in three
cases identifying the fistula in each of them. The fistula was
approached through open right thoracotomy in three cases and via
cervicotomy in the other three (7).
Case report 1
A 38-month-old female patient was referred to the
pediatric surgery department for occasional choking during feeding,
especially liquid foods. History revealed periodic episodes of
pneumopathies with repeated admissions for special care. The
performed esophagogram revealed an H-TOF of T3-T4 level which was
further repaired by right open thoracic access.
Case report 2
An 18-month-old male infant was referred for feeding
difficulties (choking during meal times) and associated cyanosis.
No relevant history was noted excepting occasional cough paroxysms,
ignored by the family. Esophagogram was performed showing a C7-T1
level H-TOF which was operated via right cervicotomy.
Case report 3
A 3-month-old male infant presented to the emergency
room for severe coughing and choking which worsened at feeding
attempts leading to cyanosis. Esophagogram was inconclusive. T2-T3
H-TOF fistula was confirmed by esophagoscopy at 11 cm from the
incisor line. Identification was possible after the
anesthesiologist was asked to assist by hyperventilation of the
patients so that air bubbles could be identified in the esophagus.
Right thoracotomy was performed in order to ligate and divide the
fistula.
Case report 4
A 2-month-old male infant was transferred to our
hospital for constant coughing and choking precipitated by feeding
attempts. Clinical examination revealed functional respiratory
syndrome and poor weight gain (200 g since birth). Previous history
showed a prolonged hospitalization since birth for neonatal sepsis
associated with functional respiratory syndrome and choking at
feeding attempts for which neurological swallowing disorder was
considered and nasogastric tube feeding was indicated. Fluoroscopic
swallowing exam and esophagogram were conducted shortly after
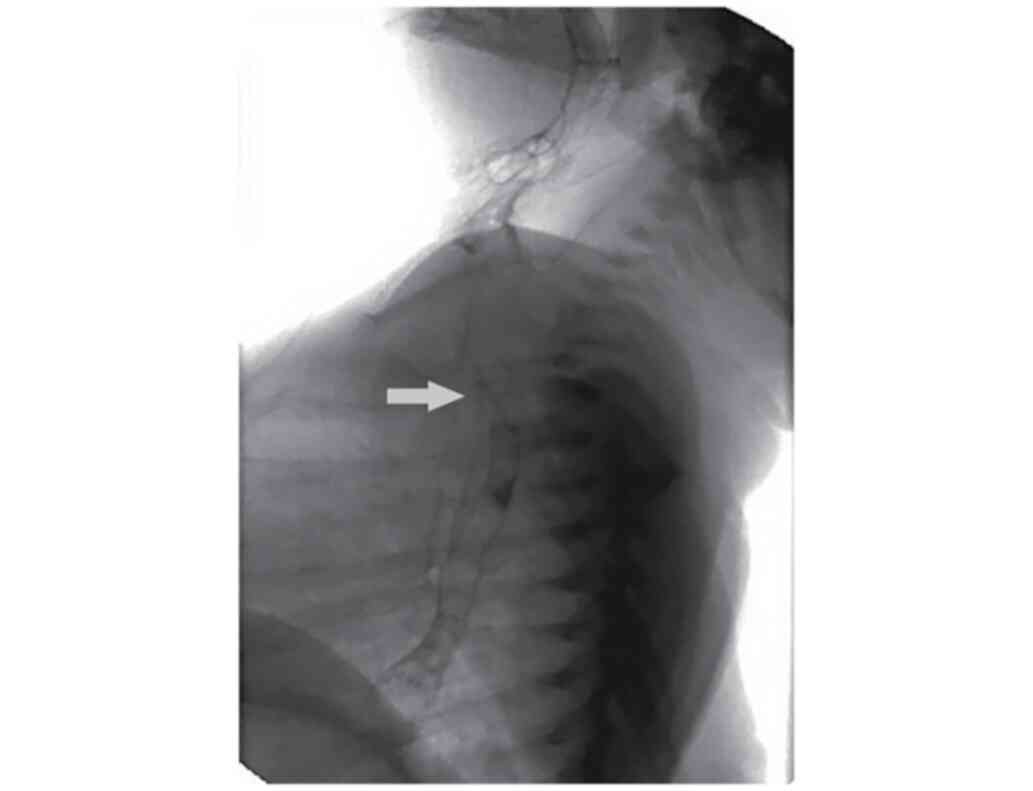
admission to our clinic showing a T3 level H-TOF (Fig. 1) and invasion of barium solution
into trachea and larynx which was later confirmed by
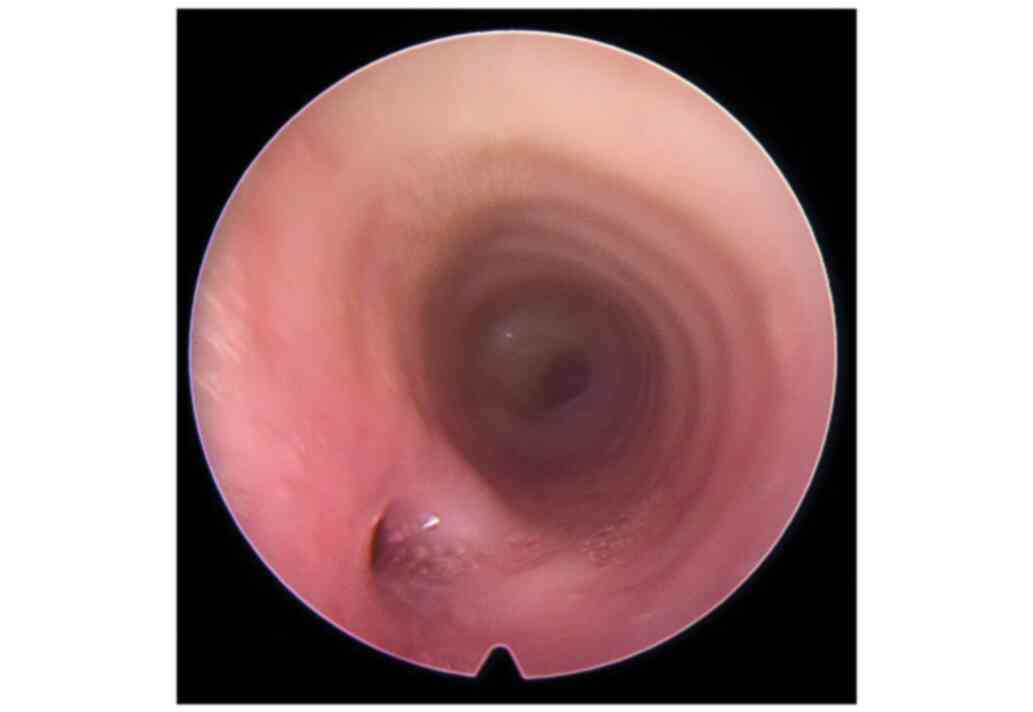
tracheobronchoscopy (Fig. 2). Right
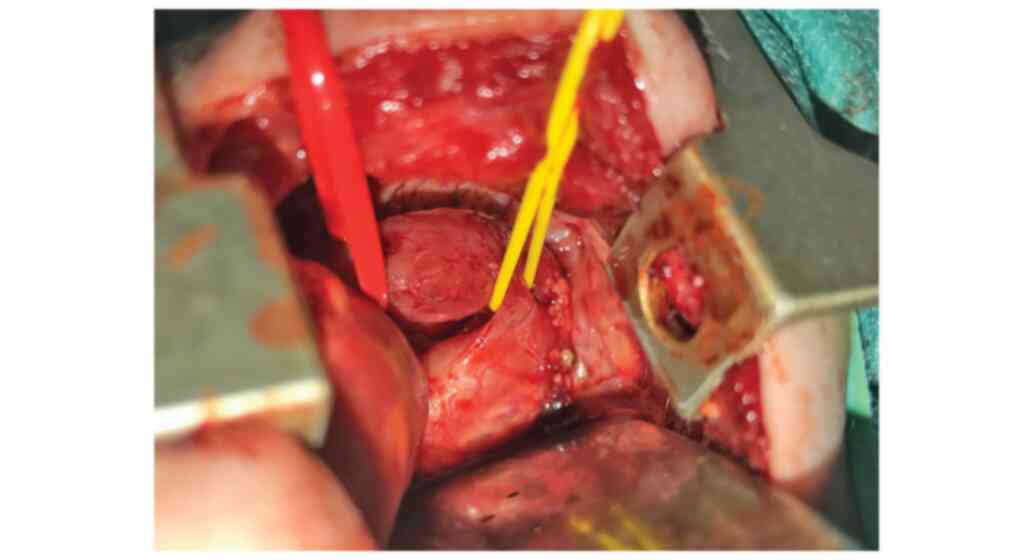
thoracic open approach was performed in this case (Fig. 3).
Case report 5
A 14-day-old male newborn was referred to our
neonatal intensive care unit for a unique episode of pneumonia
shortly after birth. Excessive secretions in the oro-pharynx,
dysphonic cry and mild respiratory functional syndrome were noted
in regards to the clinical observations. Esophagogram was performed
with a false-negative result. Therefore, tracheobronchoscopy was
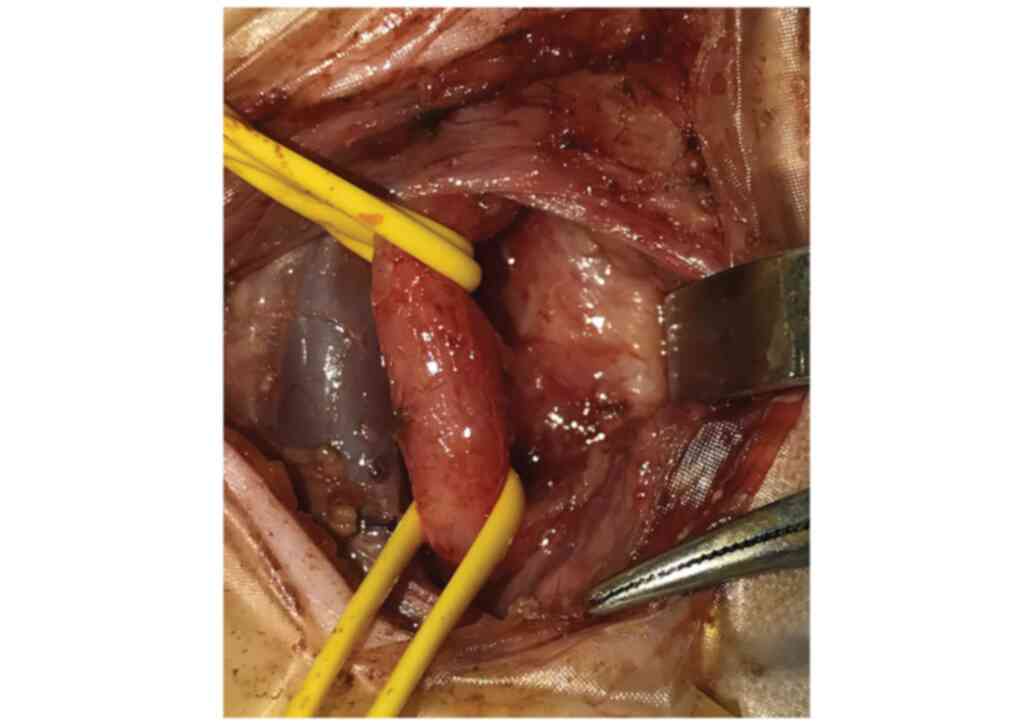
performed revealing the H-TOF 2 cm below the larynx and
tracheomalacia. Left cervicotomy access permitted ligation and
division of the fistula under safe conditions (Fig. 4).
Case report 6
A 10-month-old infant was admitted for medical care
for aspiration pneumonia. Recurrent episodes of upper respiratory
tract infections and GERD were outlined in the patient's history.
No other associated anomalies were noted. Clinical checkup
highlighted poor weight gain (4,500 g since birth). Esophagogram
imagery was achieved and high GER was described. Upper endoscopic
assessment did not reveal any signs of esophagitis. Therefore, the
patient underwent Nissen fundoplication surgery considering the
high-grade gastro-esophageal reflux complicated by repeated
aspirations. The patient had a poor outcome in the early
postoperative period in regards to respiratory function recovery
and therefore tracheobronchoscopy was performed revealing H-TOF
which was repaired using a right cervical approach.
Discussion
Generally, the treatment of rare pathological
conditions in the pediatric population is concentrated in
highly-specialized tertiary centers. The expertise of the health
professionals in these centers is an important factor for a better
outcome of these patients.
Congenital H-type (isolated) tracheoesophageal
fistula (H-TO) has an incidence of about 1:50,000-100,000
individuals and a slightly higher prevalence in males (3). It is sporadically reported in the
current literature in small series (8). Almost all patients are diagnosed
within the first three years of life, most of the cases being
recognized before the age of 12 months, respectively in the
neonatal period (4,9), while H-type TOF is rarely diagnosed
beyond childhood and even more limited in adult surgical experience
(10,11).
The clinical key points for H-TOF diagnosis are
classical and they are represented by coughing or paroxysmal
choking during feeding, pneumopathy or history of pneumopathy,
repeated cyanosis and, infrequently, abdominal distension (4,5), which
are often intermittent and they are related to fistula size,
co-existence of respiratory effort or endotracheal intubation
anesthesia (3,9).
Isolated tracheoesophageal fistulas are a very rare
group of congenital malformations. Continuity of esophagus pictures
H-TOF as an exclusive presentation over the anatomical variants of
congenital tracheoesophageal anomalies met in the clinical practice
(5,12). A noticeable fraction of H-TOF
patients (19-50%) present with other malformations or comorbidities
such as cardiac defects, anorectal malformations, cleft lip,
laryngeal cleft or may present multiple malformations such as
VACTERL associations, CHARGE syndrome, Goldenhar syndrome, Down or
Opitz syndrome (13-15).
Therefore, the symptoms may be easily misleading or diverted when
an accurate workup plan is not conducted (4,14). All
of these may act as a ‘smokescreen’ in the diagnostic pathway.
While Fallon et al identified 29% of H-TOF cases as preterm
(8), some authors advocate
prematurity-related conditions (such as respiratory distress)
motivating the delay in diagnosis (15). We would like to strengthen this idea
by adding the importance of neonatal dysphagia and swallowing
disorders which has an incidence of 13% in the general population
which is doubled (26%) in preterm babies (16).
A meaningful misleading element in the early
diagnosis of H-TOF is gastroesophageal reflux disease (GERD). The
extensive set of symptoms and complications that GERD may present
(both gastrointestinal-regurgitation, vomiting, dysphagia, failure
to thrive, or regarding the airways-wheezing, stridor, recurrent
pneumopathy) (17) may easily lead
to clinical judgement bias considering the rarity of H-TOF. More
than that, the diagnosis of H-TOF may be missed because of the low
specificity of esophagogram and esophagoscopy and furthermore, of
the masquerading symptoms if nasogastric tube feeding is initiated
(5). Fallon et al reports,
in his multicentric review (8),
three cases of fundoplication diagnosed prior to H-TOF repair
suggesting a pitfall in fistula diagnosis. Within our small period
of experience, one case out of six presented the same pattern. GERD
is alleged by Tarcan et al (15) in his isolated case for its
confounding symptoms in preterm babies. In addition, taking
previous statements into account and the predilection site of H-TOF
above the T2 vertebra level (4),
gastroesophageal reflux identified on esophagogram, especially
grade II-III or higher according to McCauley's classification
(18), has a high probability to
generate unexplained recurrent H-TOF symptoms (in the absence of
feeding) as a consequence of the gastroesotracheal reflux.
Work-up on any suspicion of H-TOF should begin with
a simple plain chest X-ray and this is usually performed
considering the practical generally agreed consensus upon the
initial evaluation of respiratory symptoms. This exam is not
helpful, but signs such as gaseous distension of the
gastro-intestinal tract and especially of the esophagus are elusive
(4,5) and might be easily missed. Taking these
facts into account, we would like to point out an article by
Boybeyi et al depicting 5 cases of megaesophagus on chest
X-ray indicating H-TOF in patients diagnosed late (between 12 and
22 years of age), invoking caution and the role of manometry before
deciding to perform an esophagomyotomy since the chronic effects of
H-TOF may be complicated (19).
Thus, it is important to raise a ‘red flag’ for any patient beyond
childhood with unexplained esophageal dilatation with an associated
history related to the H-TOF clinical picture. Therefore, an
esophagogram exam in a prone or supine position is usually
recommended. This is considered by many authors sufficient for
confirmation of the diagnosis and beneficial for the exclusion of
other diagnoses such as cleft larynx, gastroesophageal reflux,
swallowing discoordination (5).
Yet, this may be accompanied by a series of pitfalls, with the
sensitivity ranging from 50 to 73% in H-TOF (20). A series of arguments may be taken
into account: a) the ‘N’ aspect of the fistula in which the
esophageal opening is lower than the tracheal orifice creating a
tight space between the two lumens and causing fistula to clog most
of the time; b) normal active swallowing movements may not be
strong enough to generate sufficient esophageal distension and
consecutive contrast substance invasion to the trachea and the
contrast agent may need to be injected under pressure; c)
esophageal mucosal folds or esophageal muscular spasm which may
keep the digestive opening of the fistula closed. The contrast
substance may be transient in the respiratory tree during the
examination, coughing being able to eliminate it. For this reason,
radioscopy or continuous acquisition of films with image
amplification are indicated to boost the chances of fistula
identification. On the other hand, considering that the contrast
agent should be instilled up in the cervical region to delineate
the esophagus as much as possible and not to miss high fistulas,
laryngeal aspiration of the contrast agent is more possible. This
may be produced either directly through the H-TOF if positive, or
in the context of high GERD. In the later eventuality, the contrast
agent can be present in the respiratory tree without a visible
fistula path if the radiologist is not dynamically achieving
imagery. Accordingly, a false-negative esophagogram should be
considered as long as clinical doubt persists. Radiological studies
in the premise of H-TOF should be made with particular vigilance in
sites with quick access to intensive care units and professionals
prepared for prompt resuscitation and, preferably, by experienced
radiologists. Isotonic water-soluble contrast agents are
recommended (5,9,13). In
our experience, barium swallow has been used to obtain conclusive
images of H-TOF and the preparation of a low viscosity solution
(dilution) plays a significant role in obtaining adequate images of
the tracheoesophageal passage. This may lead to a more facile
aspiration or to lesser enhanced contrast pictures, and we support
the rapid obtaining of many frames during pharyngeal and esophageal
stages of deglutition.
Esophagoscopy is less useful in H-TOF identification
since the esophageal ostium is smaller and the intraluminal
air-pressure during the digestive endoscopy may intermittently
close the opening with mucosal folds (3,5,13).
Tracheobronchoscopy represents the most reliable
tool in regards to clinical tracheoesophageal fistula suspicion
(4,20). This test can also discern other
associated malformations such as laryngeal cleft, laryngeal
stenosis or tracheomalacia (20).
In contrast with the esophagus, the firm aspect of the trachea
makes the tracheal opening of the fistula easier to be visualized.
In the case of doubt or difficulties in visualization a series of
applicable guidelines are suggested in previous studies: i) Passing
a suction catheter tip over the posterior tracheal wall may drop
into a blind fistula opening (3);
or ii) more complex mixed endoscopic technique such as injecting
saline solution into the esophagus along with positive pressure
ventilation may produce air bubbles at the fistula site (4) or esophageal instillation with
methylene blue (6). In addition to
its diagnostic value, tracheobronchoscopy is recommended
preoperatively or during surgery in order to localize the fistula
and to decide the best approach, avoiding long, meaningless,
hazardous dissections. In this order, a Fogarty catheter may be
inserted through the tracheal ostium, but this may migrate during
surgical or anesthetic procedures. Garcia et al (6) and Ko et al (21) introduced the idea of knot locked
guide wire passed through the fistula before orotracheal intubation
using both esophagoscopy and bronchoscopy. Recently, Goyal et
al (22) promoted the idea of
using intraoperative transillumination of the fistula using
flexible miniature bronchoscopy. Nevertheless, the value of
tracheobronchoscopy in order to exclude extraordinary cases of more
than one congenital tracheoesophageal fistula without esophageal
atresia in the same patient should not be ignored (23,24).
Other diagnostic and preoperative evaluation tools
including virtual bronchoscopy and direct sagittal CT scan have
been reported in the current literature, but their benefits are
limited (5).
The first milestone in H-TOF repair is providing
clear landmarks of its level. Most frequently, the fistula is
located at the thoracic inlet having an oblique course with the
esophageal ostium opening lower. Cervical approach is recommended
for fistulas above the T2 vertebra level and thoracic approach is
the procedure of choice in cases where the fistula is below this
level (3). Regarding the side of
the cervical incision, right-side is preferred by most pediatric
surgeons who claim a reduced risk of recurrent laryngeal nerve
damage which arises more superiorly on this flank. In contrast, ENT
surgeons, more involved in high fistulas, prefer the left side for
a better access over the cervical esophagus. Right-side is also
preferred for low cervical H-TOF presentations (4,25).
Concerning the thoracic approach, we prefer open right-side surgery
because of the position of the aorta, and the better access to the
vagus nerve on this flank. It is important to consider malposition
of the aortic arch to the right prior surgery. Moreover, in case of
high thoracic fistulas, cautious dissection should be conducted
keeping in mind the position of the sympathetic plexus which once
injured may cause unwanted disorders such as Horner's syndrome.
From our experience, when surgical access gets difficult, head and
neck repositioning of the patient may have an important role
relatively changing the fistula's position, keeping in mind a
potential endotracheal tube displacement. Minimally invasive access
has cosmetic and magnification advantages, does not seem to be less
beneficial in regards to postoperative morbidity and does not
present with worse outcomes than the open approach in children.
Yet, this procedure is complex and should be taken into
consideration by well-trained surgeons in properly equipped
facilities (20,25). Endoscopic approach using
electrocautery, laser or tissue glue to obliterate the fistula have
been attempted with arguable results (3,24);
therefore these techniques are not promoted by us.
The second milestone of H-TOF surgery is its
intraoperative identification. In extremely rare cases, but
possible as previously reported, the fistula may be associated with
a duplication cyst which may enable difficulty in its
identification and dissection (26). We discussed this above along with
the advantages of tracheobronchoscopy. In our experience, we did
not find any major difficulties in establishing the fistula site.
Asking the anesthesiologist to induce positive tracheal pressure by
disconnecting the patient from mechanical ventilation and using
rebreathing bag for a while or by injecting air through a
nasogastric tube located in the esophagus was very useful.
The last event in H-TOF repair is division and
ligation of the fistula using interrupted sutures on both tracheal
and esophageal sides of it. Transection should be performed sharp
on the tracheal side in order to prevent diverticula formation
taking into consideration our overall experience in esophageal
atresia. We support the suggestions of other authors (11) to position a muscular flap between
the trachea and esophagus or keeping a small cuff of peritracheal
connective tissue to ensure the tracheal suture. It is considered
that these procedures will promote better healing and will prevent
leakage even though its effectiveness is not clearly demonstrated.
We also favor the use of a drain tube and we recommend Penrose-type
since it does not clog.
Postoperative complications in H-TOF are proven to
be precipitated by the coexistence of other comorbidities. Reported
events are suture dehiscence, edema, fistula recurrence,
stridor/dysphonia or vocal cord palsy/paresis, esophageal stenosis,
swallowing difficulties, prolonged intubation or pulmonary
distress. Vocal cord palsy/paresis seem to be related to technical
issues, while suture permeability complications seem to be more
prevalent in children with pre-existing GERD. Therefore, proper
pharmacological control is recommended preoperatively and
nevertheless, bearing in mind that GERD and esophageal dysmotility
are also known as postoperative complications. Extubating should be
carefully conducted in order to prevent complications or
reintubation by taking into account edema and associated conditions
(3,4).
In conclusion, H-TOF management is not easy,
therefore it must be dedicated to experienced and skillful surgeons
who should utilize a well-experienced team in an adequately
technically supplied facility. The diagnosis should not be easily
excluded (especially in newborns and infants) when respiratory
symptoms persist. Revision of previously performed work-up should
be reconsidered with a consonant tenacity including meticulous
identification of associated conditions. Moreover, the surgeon
should keep in mind that the H-TOF diagnosis process is often
crucial to ‘how’ the evaluation tools are used. A special place in
diagnosis and treatment of H-TOF is gastroesophageal reflux which
may be a significant factor of confusion or act as a booster for
postoperative comorbidities. Cautious preoperative preparation,
fully aware of the anatomical landmarks and dissection steps that
will be encountered, builds up the best strategy and leads to
favorable outcomes.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
available in this published article.
Authors' contributions
RIS, DAI and CC contributed directly to the patient
diagnoses, surgical and intensive care unit management. RIS and DAI
designed, conceived and wrote the manuscript. DS and MODL reviewed
the literature and edited the article. All authors approved the
final manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
the ‘Marie S. Curie’ Emergency Clinic Hospital for Children having
the hospital registry number 38689/21.10.2020. All parents signed
an informed consent, allowing the use of all medical data for
academic purposes, including possible scientific publication of
them, respecting their anonymous character.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lacquet A and Fransen G: Isolated
Tracheoesophageal Fistula. In: Diseases of the Esophagus. Handbuch
der Inneren Medizin (Verdauungsorgane). Vol 3. Springer, Berlin,
pp639-642, 1974.
|
|
2
|
Spitz L: Oesophageal atresia. Orphanet J
Rare Dis. 2(24)2007.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Al-Salem AH, Mohaidly MA, Al-Buainain HM,
Al-jadaan S and Raboei E: Congenital H- tracheoesophageal fistula:
A national multicenter study. Pediatr Surg Int. 32:487–491.
2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Genty E, Attal P, Nicollas R, Roger G,
Triglia JM, Garabedian EN and Bobin S: Congenital tracheoesophageal
fistula without esophageal atresia. Int J Pediatr Otorhinolaryngol.
48:231–238. 1999.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ng J, Antao B, Bartram J, Raghavan A and
Shawis R: Diagnostic difficulties in the management of H-type
tracheoesophageal fistula. Acta Radiol. 47:801–805. 2006.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Garcia NM, Thompson JW and Shaul DB:
Definitive localization of isolated tracheoesophageal fistula using
bronchoscopy and esophagoscopy for guide wire placement. J Ped
Surg. 33:1645–1647. 1998.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Bratu N, Spătaru RI and Iozsa DA: Isolated
tracheoesophageal fistula-a rare congenital malformation. J
Pediatr. 17:67–78. 2014.
|
|
8
|
Fallon SC, Langer JC, Peter SD, Tsao K,
Kellagher CM, Lal DR, Whitehouse JS, Diesen DL, Rollins MD,
Pontarelli E, et al: Congenital H-type tracheoesophageal fistula: A
multicenter review of outcomes in a rare disease. J Ped Surg.
52:1711–1714. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Gardella C, Toma P, Sacco O, Girosi D,
Panigada S, Battistini E, Mattioli G, Jasonni V and Rossi GA:
Intermittent gaseous bowel distension: Atypical sign of congenital
tracheoesophageal fistula. Pediatr Pulmonol. 44:244–248.
2009.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Hajjar WM, Iftikhar A, Al Nassar SA and
Rahal SM: Congenital tracheoesophageal fistula: A rare and late
presentation in adult patient. Ann Thorac Med. 7:48–50.
2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Suen HC: Congenital H-type
tracheoesophageal fistula in adults. J Thorac Dis. 10 (Suppl
16):S1905–S1910. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Riazulhaq M and Elhassan E: Early
recognition of H-type tracheoesophageal fistula. APSP J Case Rep.
3(4)2012.PubMed/NCBI
|
|
13
|
Crabbe DC, Kiely EM, Drake DP and Spitz L:
Management of the isolated congenital tracheo-oesophageal fistula.
Eur J Pediatr Surg. 6:67–69. 1996.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Fraga JC, Adil EA, Kacprowicz A, Skinner
ML, Jennings R, Lillehei C and Rahbar R: The association between
laryngeal cleft and tracheoesophageal fistula: Myth or reality?
Laryngoscope. 125:469–474. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Tarcan A, Gurakan B, Arda S and Boybat F:
Congenital H-type fistula: Delayed diagnosis in a preterm infant. J
Matern Fetal Neonatal Med. 13:279–281. 2003.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Mercado-Deane MG, Burton EM, Harlow SA,
Glover AS, Deane DA, Guill MF and Hudson V: Swallowing dysfunction
in infants less than 1 year of age. Pediatr Radiol. 31:423–428.
2001.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Rosen R, Vandenplas Y, Singendonk M,
Cabana M, DiLorenzo C, Gottrand F, Gupta S, Langendam M, Staiano A,
Thapar N, et al: Pediatric gastroesophageal reflux clinical
practice guidelines: Joint recommendations of the north american
society for pediatric gastroenterology, hepatology, and nutrition
and the european society for pediatric gastroenterology,
hepatology, and nutrition. J Pediatr Gastroenterol Nutr.
66:516–554. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
McCauley RG, Darling DB, Leonidas JC and
Schwartz AM: Gastroesophageal reflux in infants and children: A
useful classification and reliable physiologic technique for its
demonstration. AJR Am J Roentgenol. 130:47–50. 1978.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Boybeyi O, Kose M and Ersoz DD:
Achalasia-like findings in a case with delayed diagnosis of H-type
tracheoesophageal fistula. Pediatr Surg Int. 24:965–969.
2008.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Cuestas G, Rodriguez V, Millan C, Munzon
PB and Munzon GB: H-type tracheoesophageal fistula in the neonatal
period: Difficulties in diagnosis and different treatment
approaches. A case series. Arch Argent Pediatr. 118:56–60.
2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ko BA, Frederic R, DiTirro PA, Glatleider
PA and Applebaum H: Simplified access for division of the low
cervical/high thoracic H-type tracheoesophageal fistula. J Ped
Surg. 35:1621–1622. 2000.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Goyal A, Potter F and Losty PD:
Transillumination of H-type tracheoesophageal fistula using
flexible miniature bronchoscopy: An innovative technique for
operative localization. J Ped Surg. 40:e33–e34. 2005.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Mattei P: Double H-type tracheoesophageal
fistulas identified and repaired in 1 operation. J Ped Surg.
47:e11–e13. 2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Sim J and Hong J: Double H-type
tracheoesophageal fistulae: A case report. Adv Pediatr Surg.
24:94–99. 2018.
|
|
25
|
Parolini F, Morandi A, Macchni F,
Gentilino V, Zanini A and Leva E:
Cervical/thoracotomic/thoracoscopic approaches for H-type
congenital tracheo-esophageal fistula: A systematic review. Int J
Pediatr Otorhinolaryngol. 78:985–989. 2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Spătaru RI, Popoiu MC and Ivanov M:
Foregut duplication cyst associated with esophageal
atresia-one-stage neonatal surgical repair. Indian J Surg. 77
(Suppl 1):S52–S55. 2014.PubMed/NCBI View Article : Google Scholar
|