Introduction
Hepatocellular carcinoma (HCC) is the leading cause
of cancer-associated deaths and is extremely resistant to
chemotherapy (1). Due to poor
prognosis and lack of effective drugs, the overall survival rate of
patients with HCC is below 30% (2,3). Most
patients go undiagnosed until the disease has progressed to an
advanced stage (4). Molecular
therapy targeted against specific molecules in tumor cells or their
niche is currently the standard treatment for patients with
advanced liver cancer (5).
Sorafenib, a multi-kinase inhibitor, is a Food and Drug
Administration (FDA)-approved chemical drug for treating patients
with HCC (6-8).
However, when treated with sorafenib, the average overall survival
of patients is only extended by 2.8 months compared with that of
untreated patients. Patients treated with sorafenib either suffer
from severe side-effects or show disease progression after the
initial response (4,9). Previous studies have shown that
sorafenib is no longer effective in patients after months of
treatment, which suggests that the shortcomings of sorafenib are
associated with the development of drug resistance (10-12).
ATP-binding cassette (ABC) transporters are involved in tumor cell
multidrug resistance (MDR), such as ABCB1 and ABCG2. ABC proteins
can transport a wide variety of anticancer drugs, including
inhibitors of tyrosine kinases (13). In HCC cells, ABC proteins are
upregulated, which is associated with the activation of survival
pathways (14,15). ABCB1 has been associated with
decreased median survival time in patients with HCC and ABCG2
contributes towards the MDR phenotype in HCC (16,17).
Wh-4, a heat shock protein 90 (Hsp90) inhibitor, was
synthesized in the laboratory and was derived from the existing
inhibitor, SNX-2112 (18-23).
Hsp90 is a member of a highly conserved family of molecular
chaperones present in all eukaryotes (24). Although Hsp90 accounts for only 1-2%
of total cellular protein content, it is responsible for regulating
several activities, including client proteins activity, stability,
conformation and function (25,26).
Hsp90 facilitates metastasis, rapid cell division, resistance and
evasion of apoptosis in cancer cells (27). Many kinases, including PI3K, ERK,
vascular endothelial growth factor receptor (VEGFR) and
insulin-like growth factor receptor, are Hsp90 client proteins
(26). These functionally important
kinases depend on Hsp90 to achieve an active conformation or to
gain increased stability (25).
Cancer cells are dependent on Hsp90, and thus Hsp90 has been
successfully used as a target in tumor therapeutics in many
clinical trials for Hsp90 inhibitors in multiple tumor types
(28). For example, the
benzoquinone ansamycin Hsp90 inhibitors, including geldanamycin and
its derivative 17-AAG (26,29,30),
induced cancer cell apoptosis and disrupted the transcriptional
function of HIF1α. Moreover, 17-AAG decreased the colony-formation
capacity of lymphoma stem cells (31). In addition, SNX-2112, a novel Hsp90
inhibitor, decreased the cell viability and tumorigenicity of
multiple myeloma cells (32).
Although sorafenib, a tyrosine kinase inhibitor,
remarkably suppresses the Raf/Ras/MEK/ERK signaling pathway and
inhibits receptor tyrosine kinases, including VEGFR,
platelet-derived growth factor receptor (PDGFR), and fibroblast
growth factor receptor, HCC cells are resistant to sorafenib and
its side effects are also severe (33-35).
Hsp90 is a molecular chaperone that stabilizes the folding and
conformation of proteins in cancer cells. Hsp90 client proteins
play an important part in cancer cell proliferation, resistance,
and other important cellular processes. According to previous
studies, sorafenib interferes with the unfolded protein response
(36,37). For example, sorafenib interacts with
Hsp90/Hsp70 inhibitors to disrupt the folding of nascent proteins
(38,39), which suggests that the combination
of sorafenib with wh-4 can effectively inhibit cancer cell
proliferation.
Here, we investigated the effect of sorafenib in
combination with wh-4 on liver cancer cells. In addition, we
examined the anti-tumor efficacy of a novel Hsp90 inhibitor, wh-4,
in liver cancer cells.
Materials and methods
Cell lines
Liver cancer cell lines SK-HEP-1, which are liver
sinusoidal endothelial cells of tumorigenic origin (cat. no.
FH0072), Huh7 (cat. no. FH0873) and HepG2 (cat. no. FH0076) were
purchased from the FUHENG Biotechnology in Shanghai. The SK-HEP-1
and HepG2 cell lines were authenticated using short-tandem repeat
profiling (FUHENG Biotech). The cells were seeded in DMEM (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 1%
penicillin-streptomycin and incubated in a humidified atmosphere of
5% CO2 at 37˚C. Sorafenib (cat. no. Y0002098), Tris,
glycine, SDS and dimethyl sulfoxide (DMSO) were purchased from
Sigma-Aldrich (Merck KGaA). Wh-4 (purity >98.0%) was a kind
donation from Professor Huang (Serenex, Inc.; Durham, USA). Wh-4 is
a benzamides derivative and was designed to inhibit proteins with
purine binding sites, which yielded a novel benzamide hit for
Hsp90. Synthetic and modeling analyses of this chemical scaffold
prompted effort to combine the benzamide with a
1,2,3,9-tetrahydro-4H-carbazol-4-one moiety. The
1,2,3,9-tetrahydro-4H-carbazol-4-one ring system was established by
means of combining 1,3-cyclohexanedione and phenyl hydrazine via
the Fischer indole synthesis in a Personal Chemistry microwave
apparatus. Use of dimedone or the mono-methyl reagent instead of
1,3-cyclohexanedione yielded the related analogs. The purified
tetrahydro-4H-carbazol-4-one was then reacted with the desired
4-fluorobenzonitrile in the presence of sodium hydride (40). Wh-4 was dissolved in DMSO, and a
10-mM stock solution in DMSO was prepared. For further use, the
stock was diluted in cell culture medium.
MTT assay
The effect of sorafenib and wh-4 on cell
proliferation was determined by the MTT (cat. no. KGT525500;
Nanjing KeyGen Biotech Co., Ltd.) uptake method. Approximately
3x103 cells were seeded in each well of a 96-well plate
and incubated for 12 h. On the next day, the cells were exposed to
the following treatments: Various concentrations of wh-4 only, 10
µM sorafenib only, or a combination of both drugs. The treatment
was carried out at 37˚C for 48 h. Finally, MTT (5 mg/ml; cat. no.
96992; Sigma-Aldrich; Merck KGaA) was added to each well and
incubated at 37˚C for 4 h. The absorbance was measured using a
Shimadzu reader (Thermo Fisher Scientific, Inc.) at 570 nm.
Western blotting
Cells were lysed in ice-cold 1% SDS buffer and
centrifuged at 8,000 x g at 4˚C for 10 min. The protein
concentration was determined using the BCA method (Beyotime
Institute of Biotechnology, Inc.). Then, 20 µg protein was
separated by 12% SDS-PAGE and transferred to 0.20-µm polyvinylidene
fluoride membranes (cat. no. ISEQ00010; EMD Millipore). The
polyvinylidene fluoride membranes were blocked with 5.0% milk in
0.1% TBST (0.1% Tween-20 in Tris-base buffer, pH 7.0) at room
temperature for 1.5 h. Then, the membrane was incubated with
primary antibodies at 4˚C for 16 h. The primary antibodies used in
this study (all purchased from Cell Signaling Technology, Inc.)
included anti-Bcl2 (1:1,000; cat. no. 15071), anti-Bax (1:1,000;
cat. no. 5023), STAT3 (1:1,000; cat. no. 12640), phosphorylated
(p)STAT3Y705 (1:1,000; cat. no. 9145), caspase-3
(1:1,000; cat. no. 9668), caspase-9 (1:1,000; cat. no. 9508), ABCB1
(1:1,000; cat. no. 13342), ABCG2 (1:1,000; cat. no. 42078) and
GAPDH (1:1,000; cat. no. 5174;). The membrane was washed with 0.1%
TBST buffer three times and subsequently incubated with horseradish
peroxidase-conjugated secondary antibody (1:8,000; cat. no. 7074;
Cell Signaling Technology, Inc.) for 1 h at 37˚C before being
treated with the chemiluminescence reagent (EMD Millipore) and
exposed to Kodak film.
Reverse transcription-quantitative
(RT-q)PCR analysis
Total RNA (tRNA) was extracted using the TRIzol
Reagent kit (cat. no. DP424; Tiangen Biotech Co., Ltd.) and treated
with RNAse-free DNAase and diethyl pyrocarbonate (Nanjing KeyGen
Biotech Co., Ltd.). The extracted tRNA was reverse-transcribed into
cDNA using PrimerScript Master mix (Bio-Rad Biotechnology, Inc.)
according to the manufacturer's instructions. qPCR was used to
evaluate the expression level of genes in the RT-PCR system (CFX96
Real-Time System; Bio-Rad Laboratories, Inc.) using primers
(Shanghai GeneChem Co., Ltd.). Primer sequences were as follows:
ABCB1, forward, 5'-AGGTGGCGTGGAAGGTCCGGTCC-3', and reverse,
5'-GGTGAGGCCGTGGTAATCGGTGA-3'; ABCG2, forward,
5'-GGTCGGACCTGGTAGGTAATG-3', and reverse,
5'-AATGTTGACCGGTGGCAAGTTA-3'; GAPDH, forward,
5'-AGCCACATCGCTCAGACAC-3, and reverse, 5'-GCCCAATACGACCAAATCC-3.
The following PCR conditions were used on the Light Cycler: 95˚C
for 5 sec, 60˚C for 5 sec, followed by 40 cycles of 94˚C for 20 sec
and 60˚C for 1 min in a 25-µl reaction volume. Relative expression
levels were analyzed by the 2-ΔΔCq method with GAPDH as
the reference gene (41). All
experiments were performed three times.
Colony-formation assay
Approximately 5x103 cells were seeded in
each well of a 6-well dish. On the next day, the cells were treated
with different concentrations of wh-4 only, sorafenib only, or a
combination of both drugs (5 µM sorafenib, 5 µM wh-4) and incubated
at 4˚C for another 48 h. The plates were subsequently incubated at
37˚C in a humidified incubator for 21 days. Culture media was
replenished every 3 days. The colonies of more than 40 cells was
visualized as positive and stained with 0.5% crystal violet for 30
min at 37˚C (cat. no. KGA229; Nanjing KeyGen Biotech Co., Ltd.).
The numbers of positive colonies were counted under a light
microscope (Nikon Corporation) and calculated. The number of more
than 40 cells was divided by 5,000, in order to obtain the
colon-forming efficiencies. The experiments were repeated three
times.
Scratch wound healing assay
The combined effects of sorafenib and wh-4 treatment
on cell migration were examined using a scratch wound healing
assay. Cells were counted, and 2x105 cells were seeded
into 60-mm cell culture plates (Corning, Inc.) in DMEM medium
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal
bovine serum (Hangzhou Sijiqing Biological Engineering Materials
Co., Ltd.). Upon reaching 80% confluence, the bottom of the plates
was scratched gently and slowly with a sterile pipette tip, and the
gap created in the attached monolayer of cells was photographed
(Nikon Corporation). Then, the cells were cultured in in DMEM
medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with
10% fetal bovine serum (Hangzhou Sijiqing Biological Engineering
Materials Co., Ltd.). After 48 h, the migration distance of the
cells was captured under a light microscope (Nikon Corporation) and
calculated by subtracting the gap distance recorded at 0 h from the
current gap distance. Data were collected from three independent
experiments.
Cell apoptosis analysis
The cells were exposed to the following treatments:
Various concentrations of wh-4 only, sorafenib only, or a
combination of both drugs. The cells were incubated with the drugs
at 37˚C for 48 h. Subsequently, the cells were harvested and washed
three times with phosphate buffer, followed by the addition of 0.5
ml binding reagent and 5 µl Annexin V-FITC (cat. no. KGAV116;
Nanjing KeyGen Biotech Co., Ltd.). After 30 min, the cells were
stained with 5 µl 7-AAD (cat. no. KGAV116; Nanjing KeyGen Biotech
Co., Ltd.) for 15 min at room temperature according to the
manufacturer's instructions. Apoptosis in the cells was examined
using flow cytometry (BD FACSCalibur). All data were analyzed using
the FlowJo 10 software (FlowJo, Becton, Dickinson &
Company).
Cell Ki-67 analysis
A total of 50,000 cells were seeded in 6-cm plates
(Corning, Inc.). They were exposed to the following treatments at
37˚C: wh-4 only, sorafenib only, or a combination treatment of both
drugs. After 48 h, the cells were collected, washed, and incubated
with Alexa Fluor 488-conjugated Ki-67 antibody (1:100; cat. no.
ab197234; Abcam) at room temperature for 1.5 h. Images were
captured (Nikon Corporation). Image-Pro Plus was used to calculate
the fluorescence intensity of Ki-67 cells (Media Cybernetics).
Plasmid construct, siRNA sequence and
transient transfection
STAT3 mRNA was extracted from HepG2 cells and cloned
into the pcDNA3.1 vector (Shanghai GenePharma Co., Ltd.). STAT3 DNA
sequencing was performed by Sangon Biotech Co., Ltd. The vectors
were purified using a plasmid filter maxiprep kit (cat. no.
K210027; Thermo Fisher Scientific Inc.). The STAT3 recombinant
plasmid (pcDNA3.1-STAT3) was transfected using the 5 µl
Lipofectamine® 3000 reagent (cat. no. L300-015; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. pcDNA3.1-STAT3 recombinant was transfected using a
serum-free medium, and after 4 h, the medium was replaced with
normal medium. Synthetic small interfering RNA (si)-GFP, si-ABCB1
and ABCG2 had the following sequences: GFP sense,
5'-GCAUCAAGGUGAACUUCAA-3'; GFP antisense,
5'-UUGAAGUUCACCUUGAUGC-3'; ABCB1 sense, 5'-GCGGUUAACCAUCGAGUUA-3';
ABCB1 antisense, 5'-UAACUCGAUGGUUAACCGC-3'; ABCG2 sense,
5'-GCAAUCAGACCUGGAACAAUU-3'; ABCG2 antisense,
5'-AAUUGUUCCAGGUCUGAUUGC-3'. Then, 100 pmol siRNA were transfected
using the 5 µl Lipofectamine® 3000 reagent (cat. no.
L300-015; Thermo Fisher Scientific, Inc.) in 5% CO2 at
37˚C according to the manufacturer's instructions. Also, 8 µg/ml
polybrene (cat. no. G04001; Shanghai GenePharma Co., Ltd.) was used
to improve the transfection efficiency. After 4 h the medium were
replaced with normal medium. 72 h later, the cells were
harvested.
Statistical analysis
Statistical analysis was performed using SPSS 19.0
software (SPSS, Inc.). All data are presented as the mean ± SD for
at least three independent experiments. Student's t-test was used
for two-group comparisons, whilst comparisons among multiple groups
were performed using a one-way ANOVA followed by Tukey's test.
Results with P<0.05 were considered to indicate a statistically
significant difference.
Results
Combination of sorafenib with wh-4
demonstrates an inhibitory effect on the proliferation of liver
cancer cells
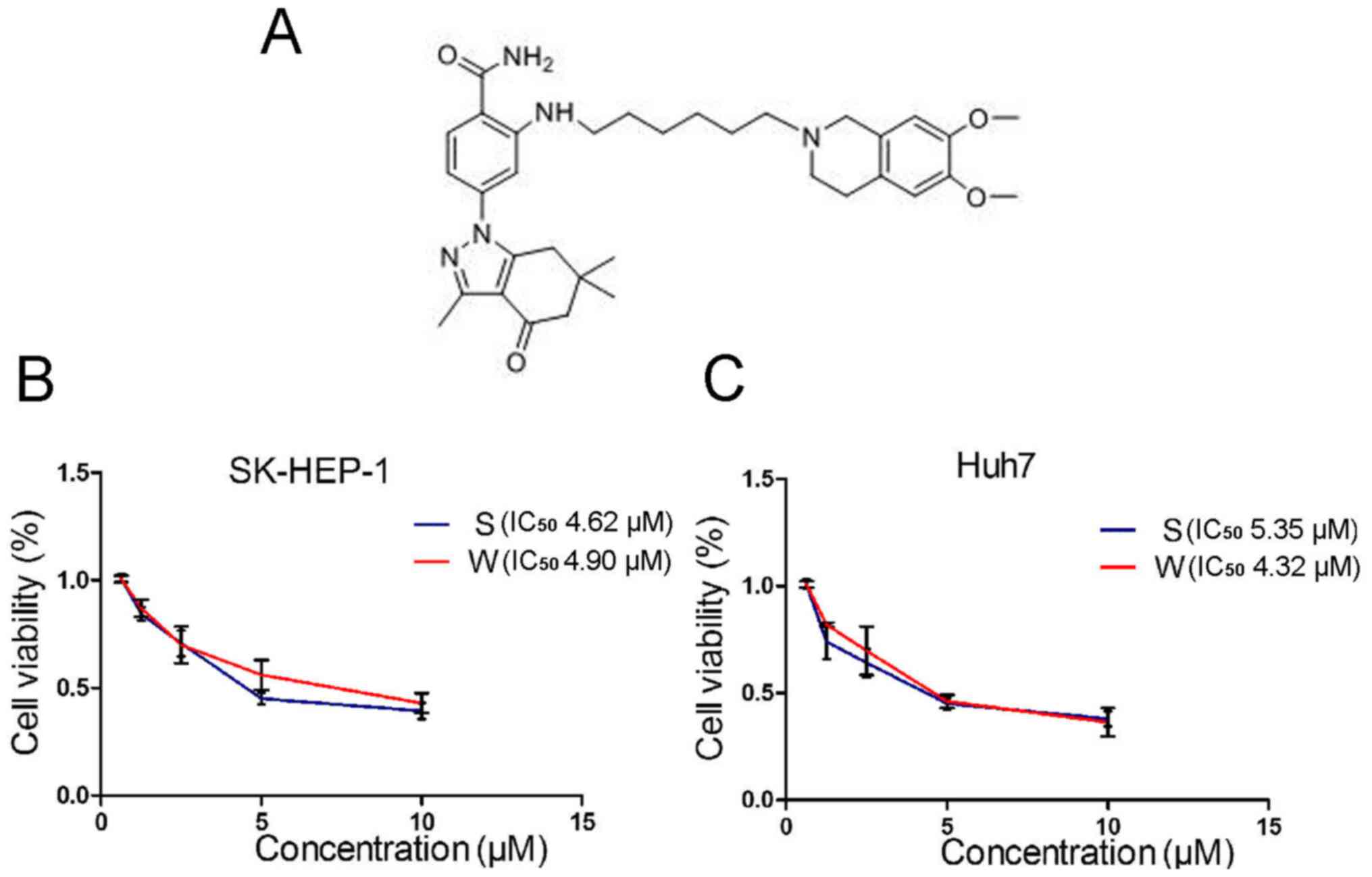
The chemical structure of wh-4 is shown in Fig. 1A. The MTT assay was used to evaluate
the inhibitory effect of the drugs on the proliferation of liver
cancer cells. Liver cancer cells were treated with various
concentrations of sorafenib or wh-4 for 24 h. It was found that the
IC50 values for wh-4 and sorafenib at 24 h were 4.90 and
4.62 µM, respectively, in SK-HEP-1 (Fig. 1B). In addition, the IC50
values for wh-4 and sorafenib at 24 h in Huh7 cells were 4.32 and
5.35 µM, respectively (Fig. 1C).
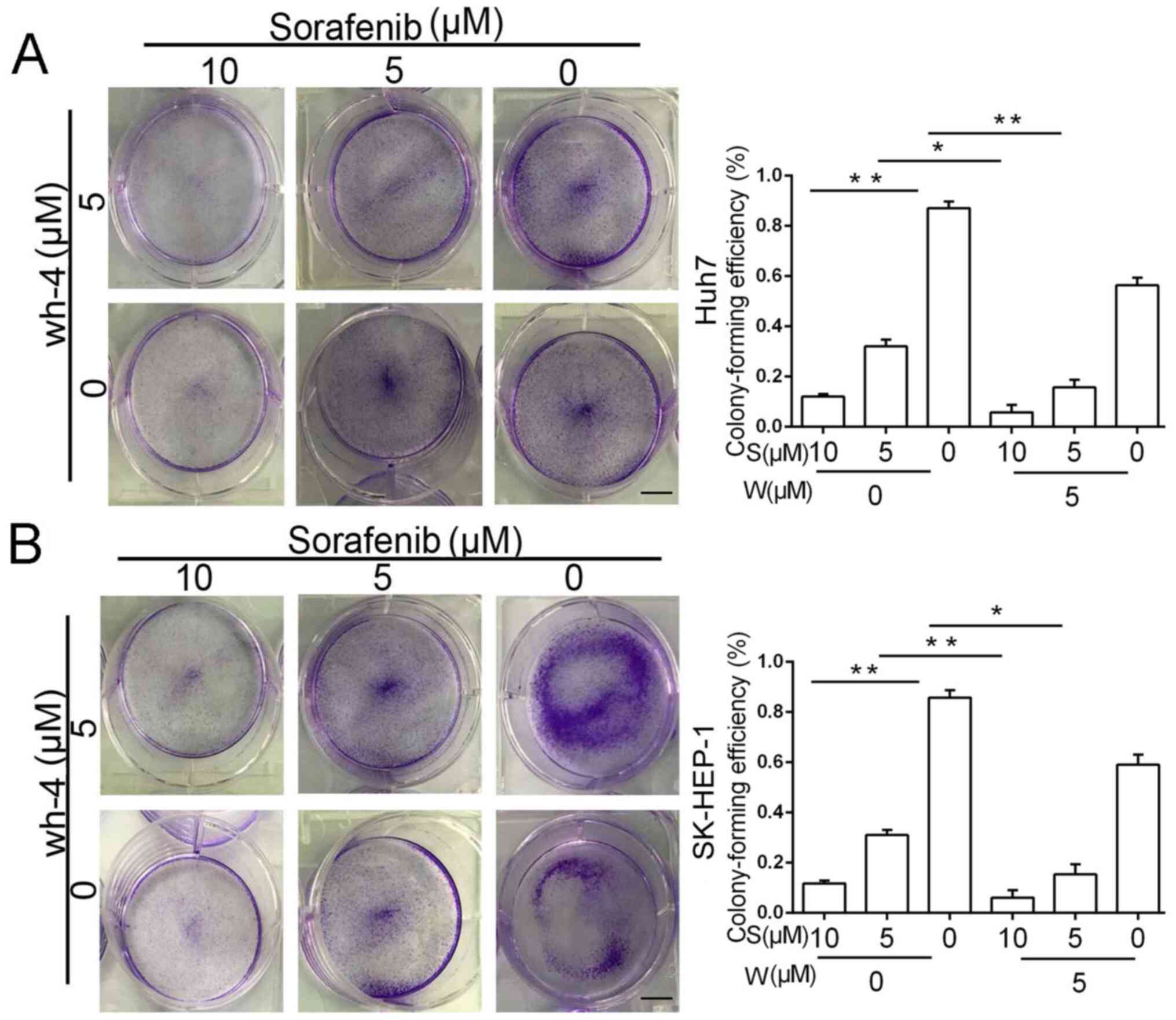
Furthermore, a colony-formation assay was performed to evaluate the
anti-tumor effect of the drugs, and a similar outcome was observed
(Fig. 2A and B). The colony-formation experiments showed
that the combination of 5 µM sorafenib and 5 µM wh-4 significantly
inhibited colony formation of liver cancer cells. The combination
treatment decreased the efficiency of the colony formation more
significantly than with sorafenib or wh-4 alone.
Combination of sorafenib with wh-4
induces apoptosis in liver cancer cells
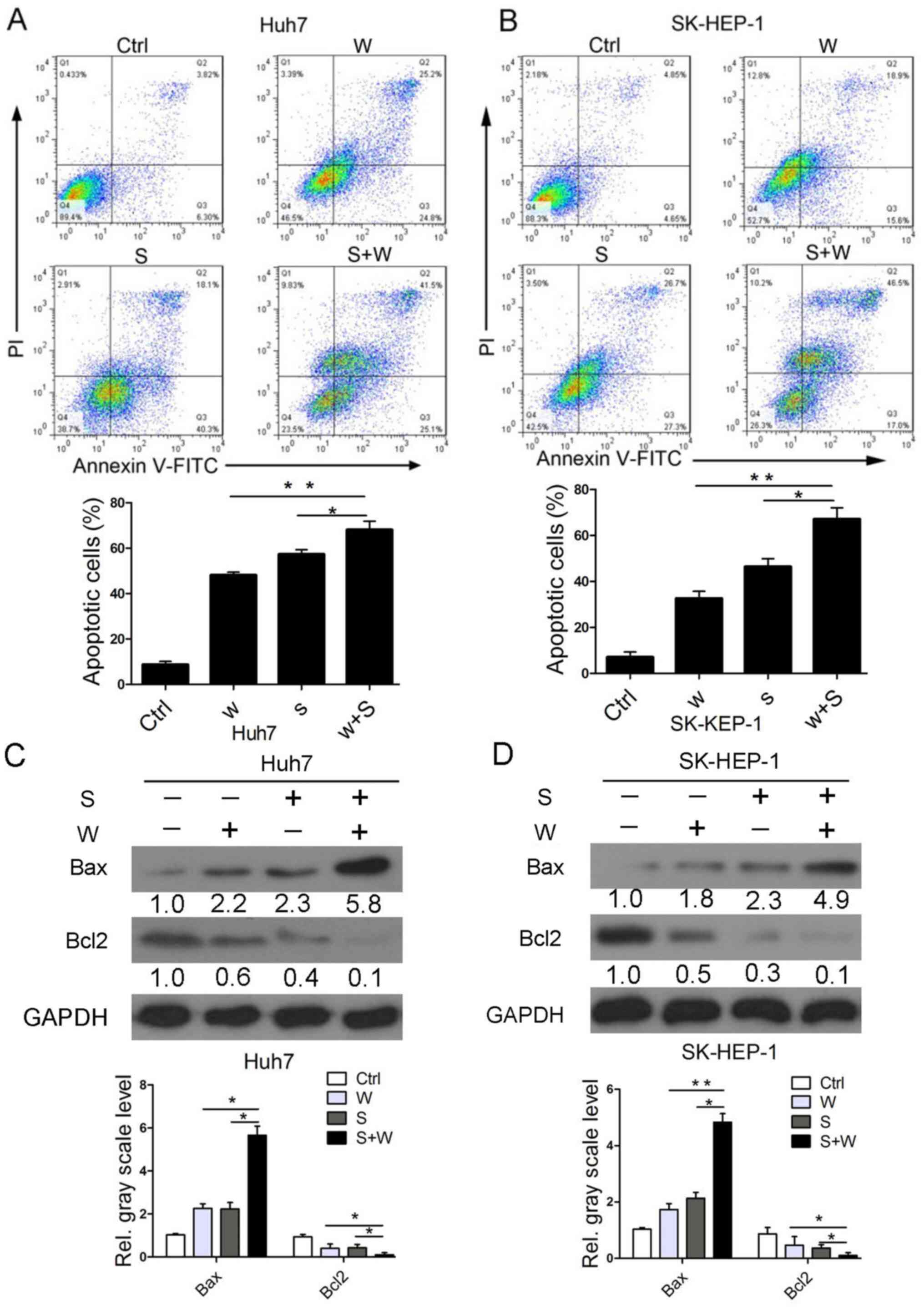
Flow cytometry results demonstrated that the
percentage of Huh7 cells undergoing apoptosis was (54.4±5.64)% when
treated with 5 µM wh-4 and (58.7±6.51)% when treated with 5 µM
sorafenib (Fig. 3A). The fraction
of apoptotic cells after the combination treatment of sorafenib and
wh-4 was (66.6±6.22)%, which was higher than that after single-drug
therapy (Fig. 3A). Furthermore the
effect of combination treatment with the two drugs were examined in
SK-HEP-1 cells. The percentage of SK-HEP-1 cells undergoing
apoptosis after the combination treatment was (63.5±5.85)%, which
was higher than that after single-drug therapy with sorafenib or
wh-4. The fraction of apoptotic SK-HEP-1 cells after treatment with
sorafenib or wh-4 alone was (54.0±6.34)% and (34.5±4.89)%,
respectively (Fig. 3B). The
aforementioned results demonstrated that the Bax levels in Huh7 and
SK-HEP-1 cells notably increased when subjected to combination
treatment compared to those with either drug alone (Fig. 3C and D). The levels of Bcl2 in Huh7 and SK-HEP-1
cells were significantly decreased after combination treatment with
the two drugs (Fig. 3C and D). In addition, the caspase-3 and
caspase-9 levels were not significantly different (Fig. S1). Collectively, the aforementioned
results suggested that combination treatment with sorafenib and
wh-4 increased apoptosis in liver cancer cells.
Sorafenib with wh-4 suppresses liver
cancer cell proliferation and migration
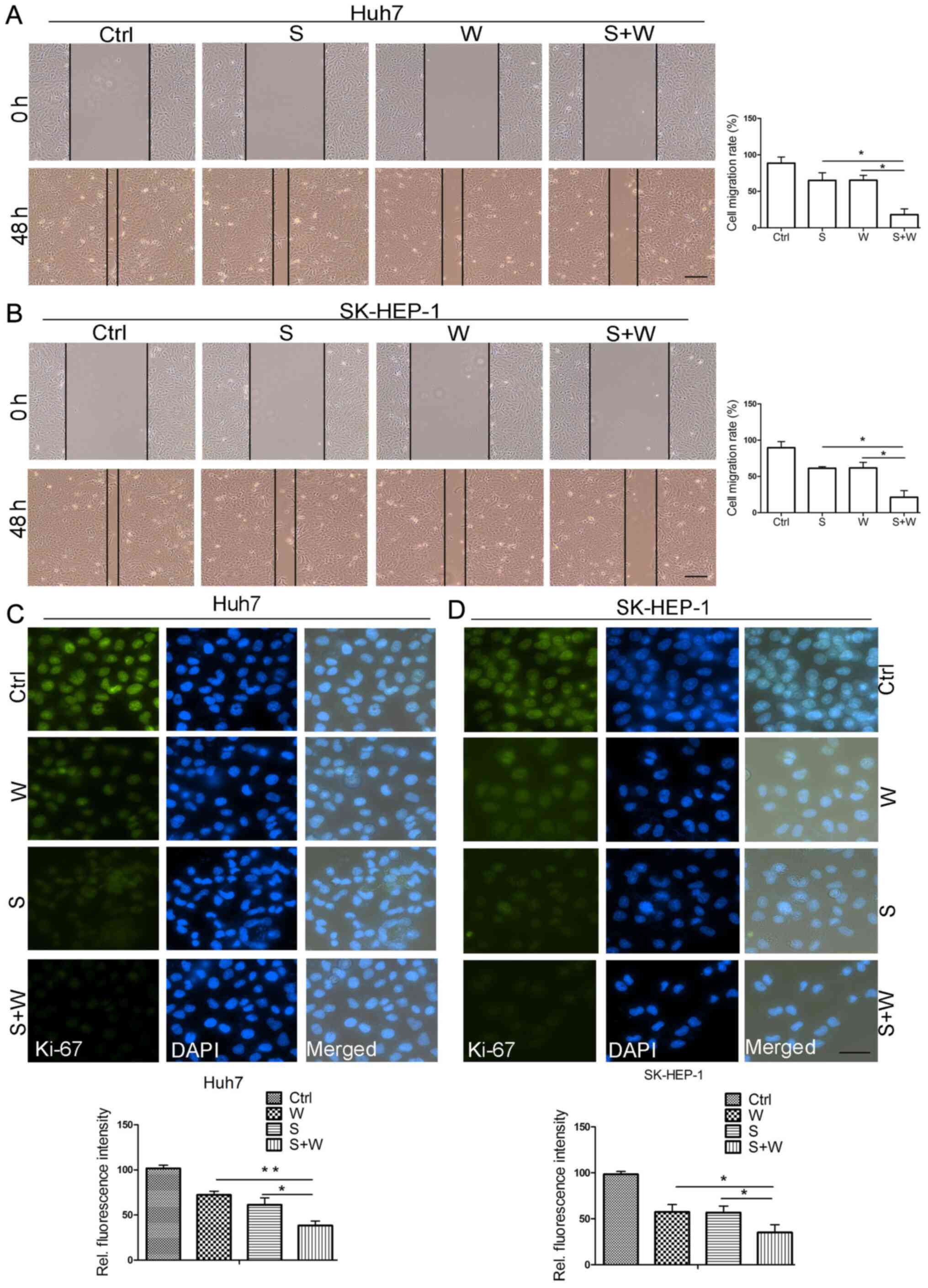
The scratch wound healing assay demonstrated that
both sorafenib and wh-4 inhibited the migration of liver cancer
cells. The data suggested that the combination treatment with
sorafenib and wh-4 significantly inhibited Huh7 migration (Fig. 4A). A similar additive effect of
sorafenib and wh-4 was also observed in SK-HEP-1 cells (Fig. 4B). In addition, the Ki-67 assay
demonstrated that sorafenib and wh-4 combination treatment
remarkably decreased the proliferation of Huh7 and SK-HEP-1 cells
(Fig. 4C and D). The fluorescence intensity level in
sorafenib with wh-4 combination treatment was notably decreased
(Fig. 4C and D). The aforementioned observations
indicated that sorafenib with wh-4 suppressed liver cancer cell
proliferation.
Combination treatment with sorafenib
and wh-4 decreases the levels of ABC transporter genes
ABCB1 and ABCG2 play an important part in liver
cancer cells proliferation (42,43).
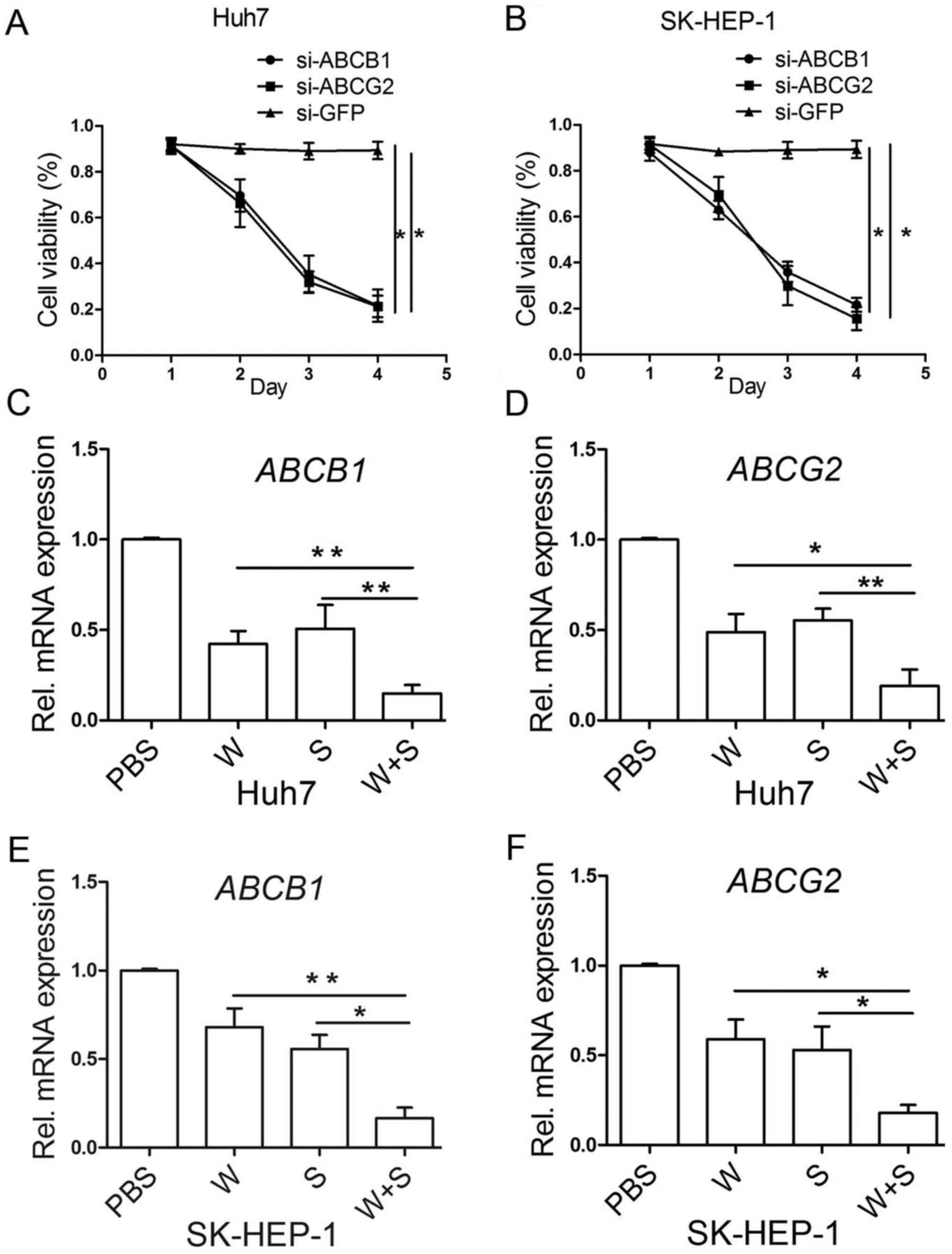
The si-ABCB1 and ABCG2 silencing efficiency was
evaluated (Fig. S2). Knockdown of
ABCB1 and ABCG2 inhibited the proliferation of liver
cancer cells (Fig. 5A and B). However, the mechanism by which
sorafenib leads to the development of resistance remains unclear.
After examining the effects of the drugs on cell proliferation, the
expression of ABC transporter genes that are responsible for drug
resistance were further investigated. The results demonstrated that
the combination treatment with sorafenib and wh-4 significantly
decreased the expression levels of ABCB1 and ABCG2 in
Huh7 cells (Fig. 5C and D). Next, the levels of ABCB1 and
ABCG2 were examined in SK-HEP-1 cells. The levels of
ABCB1 and ABCG2 were also decreased after combined
treatment with sorafenib and wh-4 (Fig.
5E and F). However, the changes
in the expression level of P-glycoprotein (P-gp)-encoded and breast
cancer resistance protein-encoded ABCG2 were not observed after the
liver cancer cells were treated with the chemicals (Fig. S3). The aforementioned results
demonstrated that the combination treatment decreased the
resistance level in treated cells.
Sorafenib and wh-4 additively inhibit
liver cancer cell proliferation by suppressing the STAT3 signaling
pathway
The potential molecular mechanism of the additive
inhibition of liver cancer cell proliferation by combination
treatment with sorafenib and wh-4 was further explored. Individual
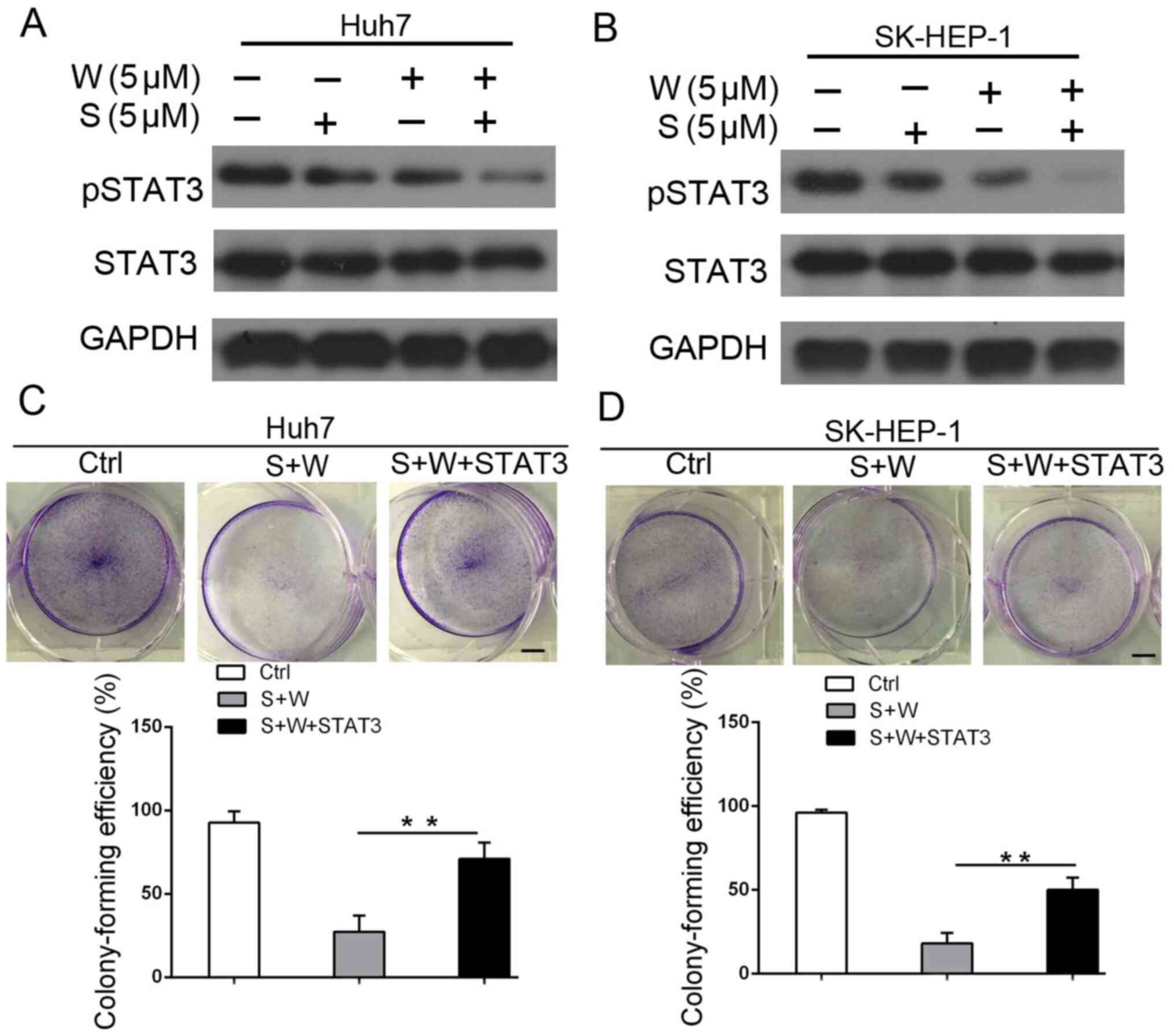
treatment with sorafenib and wh-4 decreased the phosphorylation
level of p-STAT3Y705 in both Huh7 and SK-HEP-1 cells
(Fig. 6A and B). To investigate whether STAT3 mediates
the proliferation of cells treated with a combination of sorafenib
and wh-4, a STAT3 overexpression vector was constructed. The
pcDNA3.1-STAT3 vector efficiency was analyzed (Fig. S4). As shown in the soft agar assay
in Fig. 6C, STAT3 overexpression
remarkably reversed the apoptosis induced by combination treatment
with sorafenib and wh-4. Similar effects were observed in SK-HEP-1
cells (Fig. 6D), indicating that
this reversal of apoptosis was not a cell-line-specific effect. The
aforementioned results suggested that sorafenib with wh-4 may
suppress the proliferation of liver cancer cells by the STAT3
pathway.
Discussion
The present study reported that treatment with
sorafenib suppressed the proliferation of live cancer cells. This
inhibitory effect of sorafenib was significantly enhanced in
combination with the Hsp90 inhibitor wh-4, suggesting an additive
mechanism of action of these drugs on the inhibition of liver
cancer cell proliferation.
Patients with HCC are diagnosed at intermediate or
advanced stages when therapies are no longer effective (44). Sorafenib is the first anti-tumor
drug approved by the FDA for treating patients with HCC (45). Clinical trials demonstrated that
sorafenib prolonged the median overall survival time of patients by
about 3-5 months (9,46,47).
However, the side effects of this treatment, including anorexia,
diarrhea, vomiting and squamous cell carcinoma are apparent
(48). In addition, the drug is not
effective for all patients with liver cancer, and some patients
develop resistance (9). The ABC
transporters play a key role in liver cancer cells proliferation
(49). In the present study, it was
found that the expression of ABC transporter genes was decreased in
Huh7 and SK-HEP-1 cells (Fig. 5),
suggesting that the chemoresistance was partially limited. In
addition, knockdown of ABCB1 and ABCG2 inhibited the
proliferation of liver cancer cells in the present study.
Combination treatment with sorafenib and wh-4 additively decreased
the resistance in liver cancer cells. In Fig. 5A and B, si-GFP was used as a control and the
Fig. S2 results demonstrated that
the si-ABCB1 partly decreased the expression level of ABCB1. Also,
si-ABCG2 demonstrated the same silencing effect.
The development of drug resistance is a major
challenge in the treatment of patients with HCC. Thus, a more
rational treatment plan should focus on combining two or more
therapeutic methods. Wh-4 is a derivative of SNX-2112, and SNX-2112
inhibits target proteins, such as Akt, p38, MAPK and Erk that play
a crucial role in regulating cell survival, proliferation,
resistance and homeostasis (25).
In addition, the anticancer activity of sorafenib is attributed to
its multi-kinase inhibitory function on several signaling pathways,
such as Raf-1, B-Raf, and the receptor tyrosine kinase activity of
VEGFRs and PDGFR-β (50). Induction
of apoptosis in HCC cells suggests that sorafenib might promote
apoptosis in other cancer cells such as prostate, breast and
colorectal cancer cells (51-53).
According to previous studies, the anti-tumor activity of sorafenib
on cancer cell proliferation and viability may be useful in
combination with other therapies or signaling transduction pathway
inhibitors (38). Therefore,
functional inhibition of Hsp90 target proteins in combination with
targets of sorafenib may be an effective cancer treatment strategy.
In the present study, it was found that combination treatment with
sorafenib and wh-4 additively inhibited the proliferation of liver
cancer cells. In addition, it was significant to investigate the
additive effect of sorafenib with wh-4 on liver cancer cells. Drug
concentration is critical for anti-tumor effects and it was found
the effect of one drug was different at different concentration
(54,55). It is, therefore, meaningful to
adjust the concentration the sorafenib and wh-4.
The changes in the levels of STAT3 were also
investigated. Among the STAT family members, STAT3 has received the
most attention because it plays a central role in many oncogenic
signaling pathways and controls signal transduction pathways in
several inflammatory cytokines and growth factors that are
implicated in liver damage and repair mechanisms (56). In normal cells, STAT signaling is
critical for embryonic development, organogenesis, regulation of
cell differentiation, proliferation, growth, and apoptosis, whereas
constitutive activation of STAT3 is found in many human types of
cancer cell lines and primary tumors including liver, prostate,
breast, lung, gastric and head and neck cancer (57-59).
STAT3 plays a key role in HCC initiation and progression, and it
has been found that its phosphorylation is highly positive in the
analysis of HCC biopsies (60-62).
Previous more studies demonstrated that STAT3 is an attractive
molecular target for the prevention of proliferation and treatment
of HCC (56,63). In the present study, the STAT3
pathway was found to mediate apoptosis induced by combination
treatment with sorafenib and wh-4. The results demonstrated that
STAT3 is implicated in signal transduction that induces apoptosis
in liver cancer cells upon combination treatment with sorafenib and
wh-4.
In addition, the limitation of the present study was
that in vivo experiments in animal were not conducted.
Resistance to sorafenib is a major obstacle for clinical treatment.
The present in vitro study demonstrated that sorafenib with
wh-4 combination treatment significantly inhibited liver cancer
cells proliferation and reduced ABCB1 and ABCG2 expression levels
which were responsible for liver cancer cells resistance partly.
However, it was not known whether sorafenib with wh-4 had the
antitumor effect in vivo. It was desirable to investigate
the effect of sorafenib with wh-4 treatment on liver cancer cells
in vivo.
The present study showed that combination treatment
with sorafenib and wh-4 inhibits the proliferation of liver cancer
cells and suppresses the development of drug resistance. A novel
treatment regimen was also identified to improve the efficacy of
sorafenib in patients with liver cancer by targeting the STAT3
pathway. This study demonstrates that combination treatment with
sorafenib and wh-4 may present a promising strategy for further
clinical therapy of patients with liver cancer.
Supplementary Material
Western blot analysis of Caspase-3 and
Caspase-9. Liver cancer cells were treated with 5 μM sorafenib, 5
μM wh-4 and the combination (5 μM sorafenib, 5 μM wh-4,
respectively) for 48 h. S, sorafenib; W, wh-4; Ctrl, control.
Silencing efficiency analysis of
si-ABCB1 and ABCG2. (A) Western blotting and (B) RT-qPCR assays
were used to evaluate the effect of si-ABCB1 and ABCG2 in Huh7. (C)
Western blotting and (D) RT-qPCR assays were used to evaluate the
effect of si-ABCB1 and ABCG2 in SK-HEP-1. Liver cancer cells were
transduced with siRNA, and the cells were collected after 72 h.
*P<0.05 and **P<0.01. si-, small
interfering RNA; Rel., relative; RT-qPCR, reverse
transcription-quantitative PCR.
Western blot analysis of P-gp and
BCRP. Liver cancer cells, (A) Huh7 and (B) SK-HEP-1, were treated
with 5 μM sorafenib, 5 μM wh-4 and the combination (5 μM sorafenib,
5 μM wh-4, respectively) for 48 h. BCRP, breast cancer resistance
protein; P-gp, p-glycoprotein; S, sorafenib; W, wh-4; Ctrl,
control.
Western blot and reverse
transcription-quantitative PCR analyses of STAT3 in liver cancer
cells transduced with pcDNA3.1-STAT3 vector. Liver cancer cells
were transduced with pcDNA3.1-STAT3 vector and the cells were
collected after 72 h. (A) Huh7 cell protein was analyzed using
western blotting and (B) RT-qPCR assay was used to evaluate the
gene level. (C) Western blotting was employed to test the protein
level in SK-HEP-1 and (D) RT-qPCR assay was used to evaluate the
gene level in SK-HEP-1. *P<0.05. Rel., relative;
RT-qPCRreverse transcription-quantitative PCR
Acknowledgements
Not applicable.
Funding
Funding: The author(s) disclosed receipt of the following
financial support for the research, authorship, and/or publication
of this article: This study was supported by grants from the
Guangzhou Science and Technology Plan Program (nos. 201904010050
and 202102021276), Medical Scientific Research Fund Project of
Guangdong Province (nos. A2018238, A2017312 and A2018540), and Fund
of Guangdong Food and Drug Vocational College (nos. 2016YZ001 and
2016YZ023).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SHC carried out most of the experiments and wrote
the manuscript. DDX and PJZ analyzed the data and results. YaW and
QYL read and revised the manuscript and contributed to data
collection and statistical analysis. ZR and ZL provided technical
assistance with several experiments. XW participated in the study
design and drafted the paper. HQH conceived the study. YiW and XX
participated in the design and coordination of the study. YFW
designed the study and revised the manuscript. All authors have
read and approved the final manuscript. YFW and SHC have confirmed
the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Villanueva A, Minguez B, Forner A, Reig M
and Llovet JM: Hepatocellular carcinoma: Novel molecular approaches
for diagnosis, prognosis, and therapy. Annu Rev Med. 61:317–328.
2010.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Vitale A, Volk ML, Pastorelli D, Lonardi
S, Farinati F, Burra P, Angeli P and Cillo U: Use of sorafenib in
patients with hepatocellular carcinoma before liver
transplantation: A cost-benefit analysis while awaiting data on
sorafenib safety. Hepatology. 51:165–173. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Reyes R, Wani NA, Ghoshal K, Jacob ST and
Motiwala T: Sorafenib and 2-Deoxyglucose synergistically inhibit
proliferation of both Sorafenib-Sensitive and -Resistant HCC cells
by inhibiting ATP production. Gene Expr. 17:129–140.
2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Shen YC, Hsu C and Cheng AL: Molecular
targeted therapy for advanced hepatocellular carcinoma: Current
status and future perspectives. J Gastroenterol. 45:794–807.
2010.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Palmer DH: Sorafenib in advanced
hepatocellular carcinoma. N Engl J Med. 359:2498–2499.
2008.PubMed/NCBI
|
|
7
|
Johnson P and Billingham L: Sorafenib for
liver cancer: The horizon broadens. Lancet Oncol. 10:4–5.
2009.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Tejeda-Maldonado J, Garcia-Juarez I,
Aguirre-Valadez J, González-Aguirre A, Vilatobá-Chapa M,
Armengol-Alonso A, Escobar-Penagos F, Torre A, Sánchez-Ávila JF and
Carrillo-Pérez DL: Diagnosis and treatment of hepatocellular
carcinoma: An update. World J Hepatol. 7:362–376. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al: Sorafenib in advanced hepatocellular carcinoma. N Engl J
Med. 359:378–390. 2008.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Villanueva A and Llovet JM: Second-line
therapies in hepatocellular carcinoma: Emergence of resistance to
sorafenib. Clin Cancer Res. 18:1824–1826. 2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Xin HW, Ambe CM, Hari DM, Wiegand GW,
Miller TC, Chen JQ, Anderson AJ, Ray S, Mullinax JE, Koizumi T, et
al: Label-retaining liver cancer cells are relatively resistant to
sorafenib. Gut. 62:1777–1786. 2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Tai WT, Cheng AL, Shiau CW, Liu CY, Ko CH,
Lin MW, Chen PJ and Chen KF: Dovitinib induces apoptosis and
overcomes sorafenib resistance in hepatocellular carcinoma through
SHP-1-mediated inhibition of STAT3. Mol Cancer Ther. 11:452–463.
2012.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Marin JJ, Monte MJ, Blazquez AG, Macias
RI, Serrano MA and Briz O: The role of reduced intracellular
concentrations of active drugs in the lack of response to
anticancer chemotherapy. Acta Pharmacol Sin. 35:1–10.
2014.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Marin JJG, Macias RIR, Monte MJ, Romero
MR, Asensio M, Sanchez-Martin A, Cives-Losada C, Temprano AG,
Espinosa-Escudero R, Reviejo M, et al: Molecular bases of drug
resistance in hepatocellular carcinoma. Cancers.
12(1663)2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wang XQ, Ongkeko WM, Chen L, Yang ZF, Lu
P, Chen KK, Lopez JP, Poon RT and Fan ST: Octamer 4 (Oct4) mediates
chemotherapeutic drug resistance in liver cancer cells through a
potential Oct4-AKT-ATP-binding cassette G2 pathway. Hepatology.
52:528–539. 2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Gao B, Yang FM, Yu ZT, Li R, Xie F, Chen
J, Luo HJ and Zhang JC: Relationship between the expression of MDR1
in hepatocellular cancer and its biological behaviors. Int J Clin
Exp Pathol. 8:6995–7001. 2015.PubMed/NCBI
|
|
17
|
Nies AT, Konig J, Pfannschmidt M, Klar E,
Hofmann WJ and Keppler D: Expression of the multidrug resistance
proteins MRP2 and MRP3 in human hepatocellular carcinoma. Int J
Cancer. 94:492–499. 2001.PubMed/NCBI View
Article : Google Scholar
|
|
18
|
Wang S, Du Z, Luo J, Wang X, Li H, Liu Y,
Zhang Y, Ma J, Xiao W, Wang Y and Zhong X: Inhibition of heat shock
protein 90 suppresses squamous carcinogenic progression in a mouse
model of esophageal cancer. J Cancer Res Clin Oncol. 141:1405–1416.
2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Liu Y, Wang X, Wang Y, Zhang Y, Zheng K,
Yan H, Zhang L, Chen W, Wang X, Liu Q, et al: Combination of
SNX-2112 with 5-FU exhibits antagonistic effect in esophageal
cancer cells. Int J Oncol. 46:299–307. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Wang X, Wang S, Liu Y, Ding W, Zheng K,
Xiang Y, Liu K, Wang D, Zeng Y, Xia M, et al: The Hsp90 inhibitor
SNX-2112 induces apoptosis of human hepatocellular carcinoma cells:
The role of ER stress. Biochem Biophys Res Commun. 446:160–166.
2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Liu KS, Liu H, Qi JH, Liu QY, Liu Z, Xia
M, Xing GW, Wang SX and Wang YF: SNX-2112, an Hsp90 inhibitor,
induces apoptosis and autophagy via degradation of Hsp90 client
proteins in human melanoma A-375 cells. Cancer Lett. 318:180–188.
2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Liu KS, Ding WC, Wang SX, Liu Z, Xing GW,
Wang Y and Wang YF: The heat shock protein 90 inhibitor SNX-2112
inhibits B16 melanoma cell growth in vitro and in
vivo. Oncol Rep. 27:1904–1910. 2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Bachleitner-Hofmann T, Sun MY, Chen CT,
Liska D, Zeng Z, Viale A, Olshen AB, Mittlboeck M, Christensen JG,
Rosen N, et al: Antitumor activity of SNX-2112, a synthetic heat
shock protein-90 inhibitor, in MET-amplified tumor cells with or
without resistance to selective MET Inhibition. Clin Cancer Res.
17:122–133. 2011.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Borkovich KA, Farrelly FW, Finkelstein DB,
Taulien J and Lindquist S: hsp82 is an essential protein that is
required in higher concentrations for growth of cells at higher
temperatures. Mol Cell Biol. 9:3919–3930. 1989.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Trepel J, Mollapour M, Giaccone G and
Neckers L: Targeting the dynamic HSP90 complex in cancer. Nat Rev
Cancer. 10:537–549. 2010.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Cheng W, Ainiwaer A, Xiao L, Cao Q, Wu G,
Yang Y, Mao R and Bao Y: Role of the novel HSP90 inhibitor AUY922
in hepatocellular carcinoma: Potential for therapy. Mol Med Rep.
12:2451–2456. 2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Sarto C, Binz PA and Mocarelli P: Heat
shock proteins in human cancer. Electrophoresis. 21:1218–1226.
2000.PubMed/NCBI View Article : Google Scholar
|
|
28
|
McConnell JR and McAlpine SR: Heat shock
proteins 27, 40, and 70 as combinational and dual therapeutic
cancer targets. Bioorg Med Chem Lett. 23:1923–1928. 2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kamal A, Thao L, Sensintaffar J, Zhang L,
Boehm MF, Fritz LC and Burrows FJ: A high-affinity conformation of
Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature.
425:407–410. 2003.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Solit DB, Zheng FF, Drobnjak M, Münster
PN, Higgins B, Verbel D, Heller G, Tong W, Cordon-Cardo C, Agus DB,
et al: 17-Allylamino-17-demethoxygeldanamycin induces the
degradation of androgen receptor and HER-2/neu and inhibits the
growth of prostate cancer xenografts. Clin Cancer Res. 8:986–993.
2002.PubMed/NCBI
|
|
31
|
Newman B, Liu Y, Lee HF, Sun D and Wang Y:
HSP90 inhibitor 17-AAG selectively eradicates lymphoma stem cells.
Cancer Res. 72:4551–4561. 2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Okawa Y, Hideshima T, Steed P, Vallet S,
Hall S, Huang K, Rice J, Barabasz A, Foley B, Ikeda H, et al:
SNX-2112, a selective Hsp90 inhibitor, potently inhibits tumor cell
growth, angiogenesis, and osteoclastogenesis in multiple myeloma
and other hematologic tumors by abrogating signaling via Akt and
ERK. Blood. 113:846–855. 2009.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Eilard MS, Andersson M, Naredi P,
Geronymakis C, Lindnér P, Cahlin C, Bennet W and Rizell M: A
prospective clinical trial on sorafenib treatment of hepatocellular
carcinoma before liver transplantation. BMC Cancer.
19(568)2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Kim JB, Lee M, Park SY, Lee S, Kim HR, Lee
HS, Yoon JH and Kim YJ: Sorafenib inhibits cancer side population
cells by targeting cJun Nterminal kinase signaling. Mol Med Rep.
12:8247–8252. 2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Ha TY, Hwang S, Hong HN, Choi YI, Yoon SY,
Won YJ, Song GW, Kim N, Tak E and Ryoo BY: Synergistic effect of
sorafenib and vitamin K on suppression of hepatocellular carcinoma
cell migration and metastasis. Anticancer Res. 35:1985–1995.
2015.PubMed/NCBI
|
|
36
|
Yi P, Higa A, Taouji S, Bexiga MG, Marza
E, Arma D, Castain C, Le Bail B, Simpson JC, Rosenbaum J, et al:
Sorafenib-mediated targeting of the AAA+ ATPase p97/VCP
leads to disruption of the secretory pathway, endoplasmic reticulum
stress, and hepatocellular cancer cell death. Mol Cancer Ther.
11:2610–2620. 2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Sauzay C, Louandre C, Bodeau S, Anglade F,
Godin C, Saidak Z, Fontaine JX, Usureau C, Martin N, Molinie R, et
al: Protein biosynthesis, a target of sorafenib, interferes with
the unfolded protein response (UPR) and ferroptosis in
hepatocellular carcinoma cells. Oncotarget. 9:8400–8414.
2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Vaishampayan UN, Burger AM, Sausville EA,
Heilbrun LK, Li J, Horiba MN, Egorin MJ, Ivy P, Pacey S and Lorusso
PM: Safety, efficacy, pharmacokinetics, and pharmacodynamics of the
combination of sorafenib and tanespimycin. Clin Cancer.
16:3795–3804. 2010.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Booth L, Shuch B, Albers T, Roberts JL,
Tavallai M, Proniuk S, Zukiwski A, Wang D, Chen CS, Bottaro D, et
al: Multi-kinase inhibitors can associate with heat shock proteins
through their NH2-termini by which they suppress chaperone
function. Oncotarget. 7:12975–12996. 2016.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Barta TE, Veal JM, Rice JW, Partridge JM,
Fadden RP, Ma W, Jenks M, Geng L, Hanson GJ, Huang KH, et al:
Discovery of benzamide tetrahydro-4H-carbazol-4-ones as novel small
molecule inhibitors of Hsp90. Bioorg Med Chem Lett. 18:3517–3521.
2008.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Xu WW, Li B, Guan XY, Chung SK, Wang Y,
Yip YL, Law SY, Chan KT, Lee NP, Chan KW, et al: Cancer
cell-secreted IGF2 instigates fibroblasts and bone marrow-derived
vascular progenitor cells to promote cancer progression. Nat
Commun. 8(14399)2017.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Wang J, Lian Y, Gu Y, Wang H, Gu L and
Huang Y, Zhou L and Huang Y: Synergistic effect of farnesyl
transferase inhibitor lonafarnib combined with chemotherapeutic
agents against the growth of hepatocellular carcinoma cells.
Oncotarget. 8:105047–105060. 2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Nishanth RP, Ramakrishna BS, Jyotsna RG,
Roy KR, Reddy GV, Reddy PK and Reddanna P: C-Phycocyanin inhibits
MDR1 through reactive oxygen species and cyclooxygenase-2 mediated
pathways in human hepatocellular carcinoma cell line. Eur J
Pharmacol. 649:74–83. 2010.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Kuzuya T, Ishigami M, Ito T, Ishizu Y,
Honda T, Ishikawa T, Hirooka Y and Fujishiro M: Clinical
characteristics and outcomes of candidates for second-line therapy,
including regorafenib and ramucirumab, for advanced hepatocellular
carcinoma after sorafenib treatment. Hepatol Res. 49:1054–1065.
2019.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Yurdacan B, Egeli U, Guney Eskiler G,
Eryilmaz IE, Cecener G and Tunca B: Investigation of new treatment
option for hepatocellular carcinoma: A combination of sorafenib
with usnic acid. J Pharmacy Pharmacol. 71:1119–1132.
2019.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Cheng AL, Guan Z, Chen Z, Tsao CJ, Qin S,
Kim JS, Yang TS, Tak WY, Pan H, Yu S, et al: Efficacy and safety of
sorafenib in patients with advanced hepatocellular carcinoma
according to baseline status: Subset analyses of the phase III
Sorafenib Asia-Pacific trial. Eur J Cancer. 48:1452–1465.
2012.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S,
Kim JS, Luo R, Feng J, Ye S, Yang TS, et al: Efficacy and safety of
sorafenib in patients in the Asia-Pacific region with advanced
hepatocellular carcinoma: A phase III randomised, double-blind,
placebo-controlled trial. Lancet Oncol. 10:25–34. 2009.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Morisaki T, Umebayashi M, Kiyota A, Koya
N, Tanaka H, Onishi H and Katano M: Combining celecoxib with
sorafenib synergistically inhibits hepatocellular carcinoma cells
in vitro. Anticancer Res. 33:1387–1395. 2013.PubMed/NCBI
|
|
49
|
Ceballos MP, Rigalli JP, Cere LI, Semeniuk
M, Catania VA and Ruiz ML: ABC Transporters: Regulation and
association with multidrug resistance in hepatocellular carcinoma
and colorectal carcinoma. Curr Med Chemistry. 26:1224–1250.
2019.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Wilhelm S, Carter C, Lynch M, Lowinger T,
Dumas J, Smith RA, Schwartz B, Simantov R and Kelley S: Discovery
and development of sorafenib: A multikinase inhibitor for treating
cancer. Nat Rev Drug Discov. 5:835–844. 2006.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Meyer A, Cygan P, Tolzien K, Galvez AG,
Bitran JD, Lestingi TM and Nabhan C: Role of sorafenib in
overcoming resistance of chemotherapy-failure castration-resistant
prostate cancer. Clin Genitourin Cancer. 12:100–105.
2014.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Decker T, Overkamp F, Rösel S, Nusch A,
Göhler T, Indorf M, Sahlmann J and Trarbach T: A randomized phase
II study of paclitaxel alone versus paclitaxel plus sorafenib in
second- and third-line treatment of patients with HER2-negative
metastatic breast cancer (PASO). BMC Cancer. 17(499)2017.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Gongora C: Sorafenib inhibits ABCG2 and
overcomes irinotecan resistance-response. Mol Cancer Ther.
13(764)2014.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Sun Y, Zhang J, Zhou J, Huang Z, Hu H,
Qiao M, Zhao X and Chen D: Synergistic effect of cucurbitacin B in
combination with curcumin via enhancing apoptosis induction and
reversing multidrug resistance in human hepatoma cells. Eur J
Pharmacol. 768:28–40. 2015.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Kim JE, Kim SG, Goo JS, Park DJ, Lee YJ,
Hwang IS, Lee HR, Choi SI, Lee YJ, Oh CH, et al: The
α-iso-cubebenol compound isolated from Schisandra chinensis induces
p53-independent pathway-mediated apoptosis in hepatocellular
carcinoma cells. Oncol Rep. 28:1103–1109. 2012.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Subramaniam A, Shanmugam MK, Perumal E, Li
F, Nachiyappan A, Dai X, Swamy SN, Ahn KS, Kumar AP, Tan BK, et al:
Potential role of signal transducer and activator of transcription
(STAT)3 signaling pathway in inflammation, survival, proliferation
and invasion of hepatocellular carcinoma. Biochim Biophys Acta.
1835:46–60. 2013.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Schindler C and Darnell JE Jr:
Transcriptional responses to polypeptide ligands: The JAK-STAT
pathway. Ann Rev Biochem. 64:621–651. 1995.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Zeidler MP, Bach EA and Perrimon N: The
roles of the drosophila JAK/STAT pathway. Oncogene. 19:2598–2606.
2000.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Hirano T, Ishihara K and Hibi M: Roles of
STAT3 in mediating the cell growth, differentiation and survival
signals relayed through the IL-6 family of cytokine receptors.
Oncogene. 19:2548–2556. 2000.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Yoshikawa H, Matsubara K, Qian GS, Jackson
P, Groopman JD, Manning JE, Harris CC and Herman JG: SOCS-1, a
negative regulator of the JAK/STAT pathway, is silenced by
methylation in human hepatocellular carcinoma and shows
growth-suppression activity. Nat Genet. 28:29–35. 2001.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Niwa Y, Kanda H, Shikauchi Y, Saiura A,
Matsubara K, Kitagawa T, Yamamoto J, Kubo T and Yoshikawa H:
Methylation silencing of SOCS-3 promotes cell growth and migration
by enhancing JAK/STAT and FAK signalings in human hepatocellular
carcinoma. Oncogene. 24:6406–6417. 2005.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Feng DY, Zheng H, Tan Y and Cheng RX:
Effect of phosphorylation of MAPK and Stat3 and expression of c-fos
and c-jun proteins on hepatocarcinogenesis and their clinical
significance. World J Gastroenterol. 7:33–36. 2001.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Lee C and Cheung ST: STAT3: An emerging
therapeutic target for hepatocellular Carcinoma. Cancers (Basel).
11(1646)2019.PubMed/NCBI View Article : Google Scholar
|