Introduction
Diabetes mellitus (DM) is frequently complicated by
cardiac dysfunction. Diabetes can affect cardiac structure and
function in the absence of high blood pressure and coronary artery
disease, a condition known as diabetic cardiomyopathy (DCM). DCM
increases the risk of heart failure and is one of the major causes
of death in both type 1 DM (T1DM) and type 2 DM (T2DM) (1). The pathogenesis of DCM is closely
related to hypoxia. Hyperglycemia-induced overproduction of
superoxides by the mitochondrial election-transport chain is
considered to be the common pathway. Mitochondrial dysfunction,
oxidative stress and the resulting increased ROS generation appear
to be key players in the development of DCM (2).
Sestrins (Sesns) family proteins are highly
conserved stress-induced proteins, which are widely expressed in
mammals and respond to a variety of environmental stresses
(3). Sestrin2 (Sesn2, also termed
Hi95) is an important member of the Sesn proteins, whose expression
is relatively low in resting cells. High abundance of Sestrin2 is
mainly induced by metabolic stresses such as hypoxia, DNA lesions,
oxidative stress and endoplasmic reticulum stress (4). Previously, research on Sestrin2
mainly focused on obesity-associated metabolic diseases and
age-related diseases. In addition, the role of Sestrin2 in
atherosclerotic and cardiac diseases is becoming a concern
(4). Nevertheless, research in DCM
is rarely seen. It was previously reported that Sestrin2 helped to
protect whole cellular energy metabolism and maintain mitochondrial
function (5). Overexpression of
Sestrin2 could reduce ROS accumulation, maintain mitochondrial
membrane potential, reduce ATP depletion, restore mitochondrial
DNA, and ultimately, reduce cellular apoptosis (5).
Considering the protective role of Sestrin2 in
maintaining mitochondrial function, especially under stress and the
pathogenesis mechanism including hypoxia, enhanced oxidative stress
and mitochondrial dysfunction in DCM, it has been speculated that
Sestrin2 and the related pathways may play important roles in the
development of DCM. The aim of this study was to investigate the
effects of Sestrin2 in DCM and to explore the underlying
mechanisms.
Materials and methods
Cell culture
The rat cardiomyoblast cell line, H9c2 cells (ATCC:
CRL-1446), and 293T cells (ATCC: CRL-3216) were purchased from The
Cell Bank of Type Collection of Chinese Academy of Sciences. Cells
were cultured with low-glucose Dulbecco's modified essential medium
(DMEM; Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10%
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.), 1%
penicillin-streptomycin solution (Gibco; Thermo Fisher Scientific,
Inc.) and incubated with a humidified atmosphere of 5%
CO2 at 37˚C. The cells were sub-cultured every 2-3 days
at a ratio of 1:3 or subjected in subsequent experiments at a
70-80% confluence.
Construction of Sestrin2 shRNA
lentivirus
To construct a Sestrin2 silencing lentiviral vector,
three candidate sequences of Sestrin2 small hairpin RNA (shRNA)
lentiviral vectors targeting rat Sestrin2 mRNA were designed and
synthesized by Invitrogen; Thermo Fisher Scientific, Inc. The
shRNAs were constructed into the lentivirus expression vector using
a lentivirus expressing system (HanBio Biotechnology Co., Ltd.)
according to the manufacturer's instructions. Recombinant
lentiviruses were produced by transfecting 293T cells with the
lentiviral expression plasmid. At 24 h prior to transfection, 293T
cells were cotransfected with plasmid containing Sestrin2 shRNA (10
µg) and lentiviral packaging vector (15 µg). Cells were then
incubated in DMEM with 10% FBS for 48 h at 37˚C, followed by
harvesting, centrifugation (50,000 x g at 4˚C for 2 h) and
filtration. The Sestrin2 shRNA targeting sequence
(5'-GCAGAGACCCATTGAACAACT-3') was the most effective one and was
used in subsequent experiments. The amplified virus with a
multiplicity of infection of 50 was transfected to H9c2 cells. The
cells were treated with a reagent as indicated for further
experiments after 48 h. The empty vector was used as a negative
control. Puromycin at a final concentration of 5 µg/ml was added to
the medium 48 h after transfection to select for purely transfected
cells. Cells were subsequently cultured every 2-3 days at a ratio
of 1:3 for 2-3 generations for stable constructions and the
infection efficiency was assessed using western blot analysis.
Cell treatments
H9c2 cells were cultured in 6-well plates and were
randomly divided into four groups: normal glucose control group (C;
5.5 mM glucose), high glucose (HG) group (G; 33 mM glucose),
Sestrin2 small hairpin RNA (shRNA) group (Sesn2 shRNA; 33 mM
glucose), and negative control group (NC; 33 mM glucose). The cells
were treated with the corresponding medium for 48 h.
Animals and treatments
Male mice (C57BL/6; 8 weeks old) were purchased from
Beijing Weitong Lihua Experimental Animal Technology Co., Ltd. Mice
were housed in a temperature- (22±1˚C) and humidity-controlled
(50±10%) environment and maintained in a 12-h light/dark cycle.
Free access to food and water was provided. After acclimatization
for 1 week, mice were randomly divided into two groups: Control
(n=5) and DM (n=8) groups. Diabetes was induced in mice in the DM
group by intraperitoneal injection of streptozotocin (STZ;
MilliporeSigma) dissolved in 0.1 ml citrate buffer (pH 4.5) at a
dose of 50 mg/kg for five consecutive days. The equivalent volume
of citrate buffer was administered to mice in the control group.
The blood sample, 0.1-0.2 ml each time, was obtained through the
tail vein. At one week after the course of STZ administration, mice
with random blood glucose levels >16.7 mmol/l were considered to
be diabetic. After 8 weeks, the mice were euthanized with 4%
isoflurane, and then sacrificed by cervical dislocation. The hearts
were immediately harvested for subsequent experiments. Blood
samples and myocardial tissues were collected.
All animal procedures were approved by the Ethics
Committee of the Second Affiliated Hospital of Guangzhou Medical
University (approval no.: 20200407). All animal studies were
performed in accordance with the National Institutes of Health
Guide for the Care and Use of Laboratory animals.
Cell viability
H9c2 cells were seeded in 96-well plates at a
density of 5,000 cells/well. Viability was measured using the
CellTiter 96® AQueous One Solution Assay kit (MTS)
(Promega Corporation). Culture medium was replaced with complete
medium (100 µl/well) following treatment. Then the medium was
replaced with 10 µl MTS assay reagent per well and incubated for 4
h at 37˚C. A microplate reader was used to detect the optical
density (OD) at 490 nm, according to the manufacturer's
instructions.
Enzyme-linked immunosorbent assay
(ELISA)
H9c2 cell samples were collected at 1,000 g for 10
min and used for measurement of caspase-3 and cytochrome c
by ELISA using commercial kits (Nanjing Jiancheng Biological
Engineering Research Institute), following the manufacturer's
protocols.
Western blot analysis
Briefly, total protein of the cell and heart tissue
were isolated and prepared, using the RIPA-PMSF lysis buffer
(Beijing Solarbio Science & Technology) and quantified with a
BCA kit (Servicebio), followed by loading ~40 µg protein per lane.
The samples were separated on 10% SDS-PAGE and then transblotted
onto polyvinylidene fluoride (PVDF) membranes. PVDF membranes were
blocked with 5% non-fat milk at room temperature for 2 h. Samples
were incubated with primary antibodies against Sestrin2 (catalog
no. DF12003; 1:800; Affinity Bioscience) and GAPDH (catalog no.
AF7021; 1:800; Affinity Bioscience) overnight at 4˚C. Following
washes, the membranes were incubated with the goat anti-rabbit
HRP-linked secondary antibody (catalog no. GB23303; 1:3,000; Wuhan
Servicebio Technology Co., Ltd.) for 1 h at room temperature. The
blots were visualized with an enhanced chemiluminescence detection
system (Hangzhou Fude Biological Technology). ImageJ software
(ImageJ 64-bit Java 1.8.0_112) was used to measure the gray-scale
value of each band.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the cultured cells with
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's instructions. The purity and
quantity of RNA samples were determined using Epoch™ microplate
spectrophotometer (BioTek Instruments, Inc.). Total RNA was reverse
transcribed into cDNA according to the instructions of PrimeScript™
RT master mix (Takara Bio, Inc.). qPCR was performed using Applied
Biosystems™ PowerUp™ SYBR™ Green mix (invitrogen). The reaction
volume total was 30 µl and PCR was performed with
LightCycler® 480. The results were analyzed by the
2-ΔΔCq method (6).
Primer sequences used are shown as follows: Sestrin2 forward,
5'-GACCATGGCTA CTCGCTGAT-3' and reverse,
5'-CCAAAGACGCAGTGGATGTA-3'; β-actin forward, 5'-AGGGAAATCGTGCGTGA
CAT-3' and reverse, 5'-GAACCGCTCATTGCCGATAG-3'.
Histological and immunofluorescence
analysis
Myocardial tissues were gathered and fixed in 4%
phosphate-buffered paraformaldehyde overnight at room temperature.
Paraffin-embedded tissues were sectioned at a thickness of 5 µm for
staining with hematoxylin and eosin (H&E) for 3 min or Masson's
trichrome for 5 min at room temperature. The histological changes
of the myocardium and collagen deposition were imaged using a light
microscope (Olympus; magnification, x400).
TUNEL assay
Myocardial apoptosis was detected using TUNEL assay.
The heart tissue samples were fixed in 4% paraformaldehyde at room
temperature overnight, embedded in paraffin and sectioned into 5-µm
slices. Paraffin sections were dehydrated, dewaxed, and digested at
room temperature for 8 min. An in situ cell detection kit
(Roche Applied Science) was used to identify the apoptotic cells.
The TUNEL-positive cells were observed and counted under a
fluorescence microscope (Olympus). Left ventricle samples were also
stained with primary antibodies against Sestrin2 (catalog no.
8487S; 1:200; Cell Signaling Technology, Inc.) at 4˚C overnight,
followed by the secondary antibody (catalog no. GB23303; 1:3,000;
Wuhan Servicebio Technology Co., Ltd.). The sections were then
stained with 3,3'-diaminobenzidine and a fluorescence microscope
(Olympus) was used to obtain images of the tissues.
Cell apoptosis assay
H9c2 cells were inoculated into a 6-well plate at a
concentration of 1x106 cells/well, and cultured in a
humidified environment containing 5% CO2 at 37˚C for 24
h. Next, a ReadiDrop™ dual propidium iodide (PI)/fluorescein
isothiocynate-conjugated (FITC) Annexin V commercial staining kit
(Bio-Rad) was used to measure the cell apoptosis rate, according to
the manufacturer's instructions. The change in apoptosis was
analyzed with a flow cytometer (FACSCanto; BD Biosciences) and
FLOWJO (version 10; BD Biosciences) after staining.
Measurement of intracellular ROS
generation
For the detection of intracellular ROS generation,
the indicated cells were incubated with
2',7'-dichlorodihydrofluorescein diacetate (DCFH-DA; Sigma-Aldrich)
for 15 min at 37˚C. To determine the ROS production, the
fluorescence intensity was measured by flow cytometry (FACSCanto)
and FLOWJO (version 10).
Measurement of antioxidant and oxidant
levels
The Cell Mitochondria Isolation Kit (Beijing
Solarbio Science & Technology) was used to isolate the
mitochondria of H9c2 cells. The activities of superoxide dismutase
(SOD), mitochondrial SOD, and malondialdehyde (MDA) were determined
with the corresponding detection kit according to the
manufacturer's instructions (Nanjing Jiancheng Biotechnology
Institute).
Measurement of mitochondrial membrane
potential
JC-1 was broadly used for observing mitochondrial
membrane potential and identifying fluorescence characteristic
change from green (530 nm) to red (590 nm) depending on the
mitochondrial membrane potential. A red/green fluorescence
intensity ratio of JC-1 indicated a decrease in depolarized
mitochondria due to disruption of red fluorescent J-aggregates
(Beyotime Biotechnology). H9c2 cells were collected, JC-1 working
solution was added, and the cells were cultured at 37˚C for 30 min.
Imaging Buffer solution was added, and the cells were analyzed by
flow cytometry.
Intracellular Ca2+
measurement
The cells were collected and loaded with Fluo 3-AM
(2 M) in the HEPES-buffered salt solution (HBSS) (NaCl 137 mM, KCl
2.7 mM, NaH2PO4 0.4 mM, CaCl2 0.9
mM, 0.5 mM MgCl2, 10 mM HEPES, and 5.5 mM glucose, pH
7.4) for 30 min at 37˚C. The cells were resuspended, and then
incubated with the Fluo-3/AM (3 mM) for 30 min at 37˚C.
Subsequently, protein blocking was completed with 0.2% BSA which
was flushed with Ca2+-free HBSS three times. Finally,
flow cytometry was used to detect intracellular calcium levels at
an excitation wavelength of 488 nm and an emission wavelength of
525 nm.
Measurement of MPTP opening
The mitochondrial permeability transition pore
(MPTP) opening of H9c2 cells was measured by the MPTP Assay Kit
(Beyotime Biotechnology). After 0, 24, and 36 h compression, the
cells were collected. Subsequently, 500 µl preheated cleaning
solution (Reagent A) and isopyknic working solution containing
neutralization and staining solution (Reagent B) were added into
the cell suspension. The cell suspension was then mixed gently and
fully and incubated for 20 min at 37˚C in the dark. Subsequently,
the samples were resuspended in Reagent A and analyzed by flow
cytometry.
Measurement of ATP levels
The level of ATP in H9c2 cells was measured by an
ATP assay kit (Beyotime Biotechnology). H9c2 cells were rinsed with
PBS and lysed with lysis buffer on ice to collect the lysate and
centrifuged for 4 min at 12,000 x g at 4˚C. Sample supernatant was
then added containing 100 µl ATP detection solution and mixed
vibration. A multifunctional microplate reader (VICTOR X5;
PerkinElmer, Inc.) was used to determine the ATP content of the
sample.
Statistical analysis
Data were analyzed using SPSS 16.0 (SPSS, Inc.)
statistical software and presented as the mean ± standard deviation
(SD). One-way analysis of variance (ANOVA) test and post hoc
analysis with a Bonferroni test were used for multi-group
comparisons. Unpaired Student's t-test was used to compare two
groups. Two-sided P<0.05 was considered to indicate a
statistically significant difference.
Results
Sestrin2 is significantly upregulated
in DCM conditions
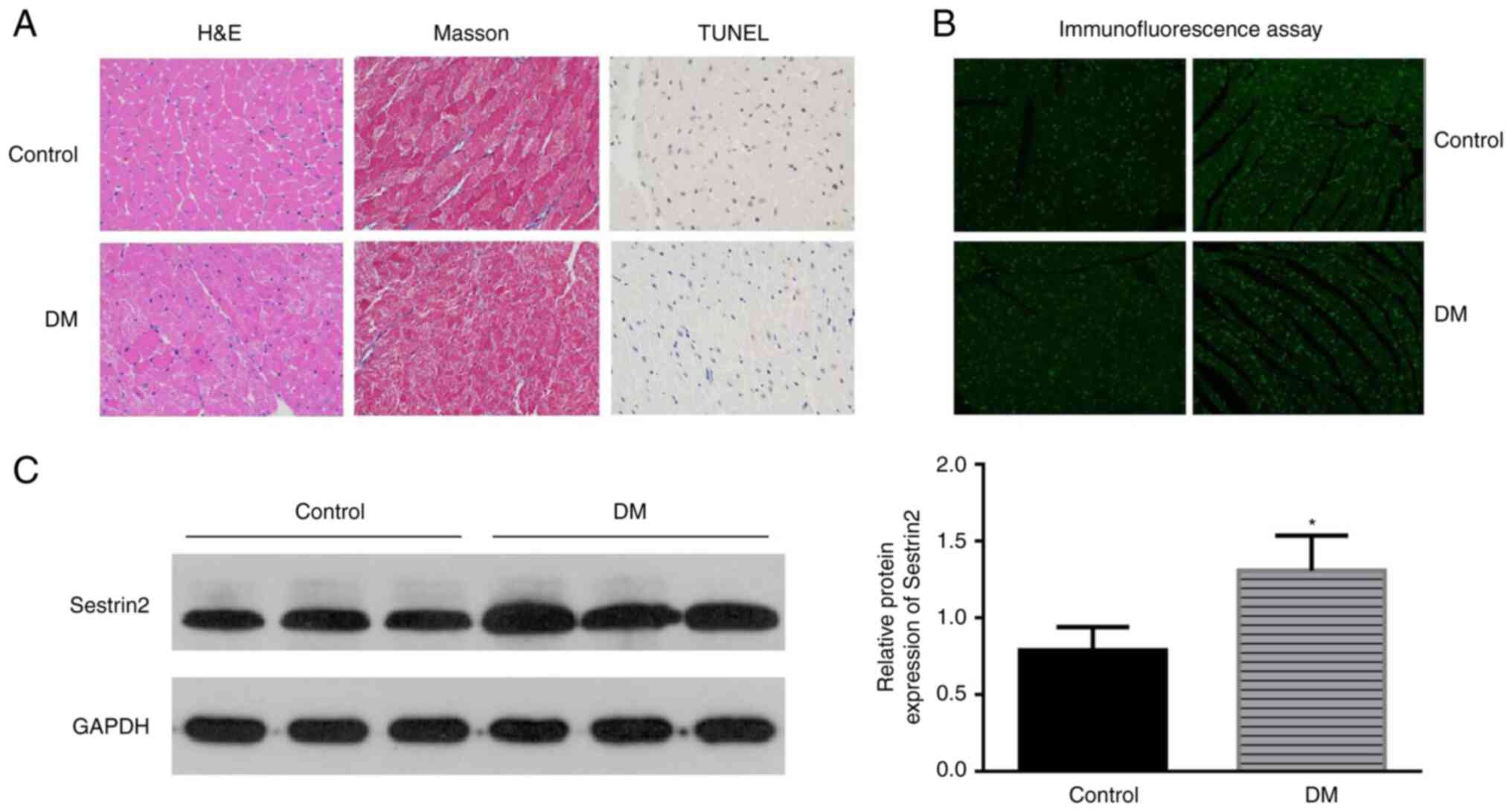
The characteristics of DCM were found in the left
ventricular sections of DM mice by H&E and Masson staining. The
results showed that the myocardial fibers were arranged disorderly,
the nucleus sizes became different and more vacuoles and collapse
of myofibers were identified in mice in the DM group. Apoptotic
cells were identified by the TUNEL assay. Brown-stained nuclei
indicated apoptotic cells, and blue-green or tan shades indicated
non-apoptotic cells in diabetic mouse hearts (Fig. 1A). Few apoptotic cells were
observed in the control group, while increased apoptotic
cardiomyoblasts were observed in DM mice (Fig. 1C). To assess the potential effect
of Sestrin2 in DCM, Sestrin2 expressions in DCM models were
measured by immunofluorescence assay (Fig. 1B), western blotting and RT-qPCR
(Fig. 1C). The results revealed
that the expression of Sestrin2 was significantly increased in
myocardial tissues of DM mice (P<0.001, Fig. 1) and H9c2 cardiomyocytes in HG
conditions (P<0.01, Fig.
2).
Downregulation of Sestrin2 increases
cell viability and decreases cell apoptosis induced by HG
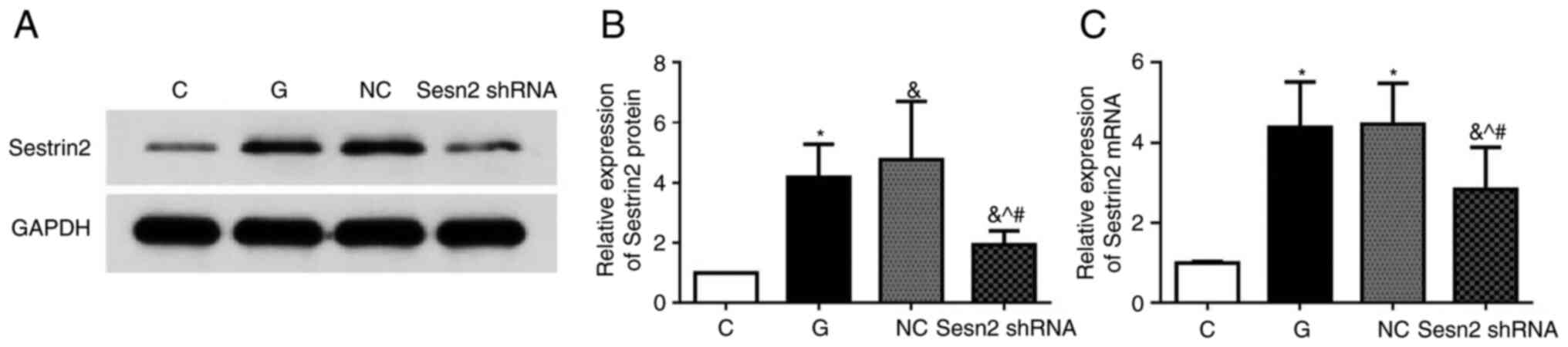
Sestrin2 shRNA lentivirus was constructed for
Sestrin2 silencing. The efficiency of siRNA-mediated inhibition of
Sestrin2 was evaluated by detecting Sestrin2 protein by western
blot analysis and mRNA levels by RT-qPCR. Compared with the NC
group, levels of protein expression and mRNA of Sestrin2 were
significantly decreased in the Sesn2 shRNA group (P<0.05)
(Fig. 2).
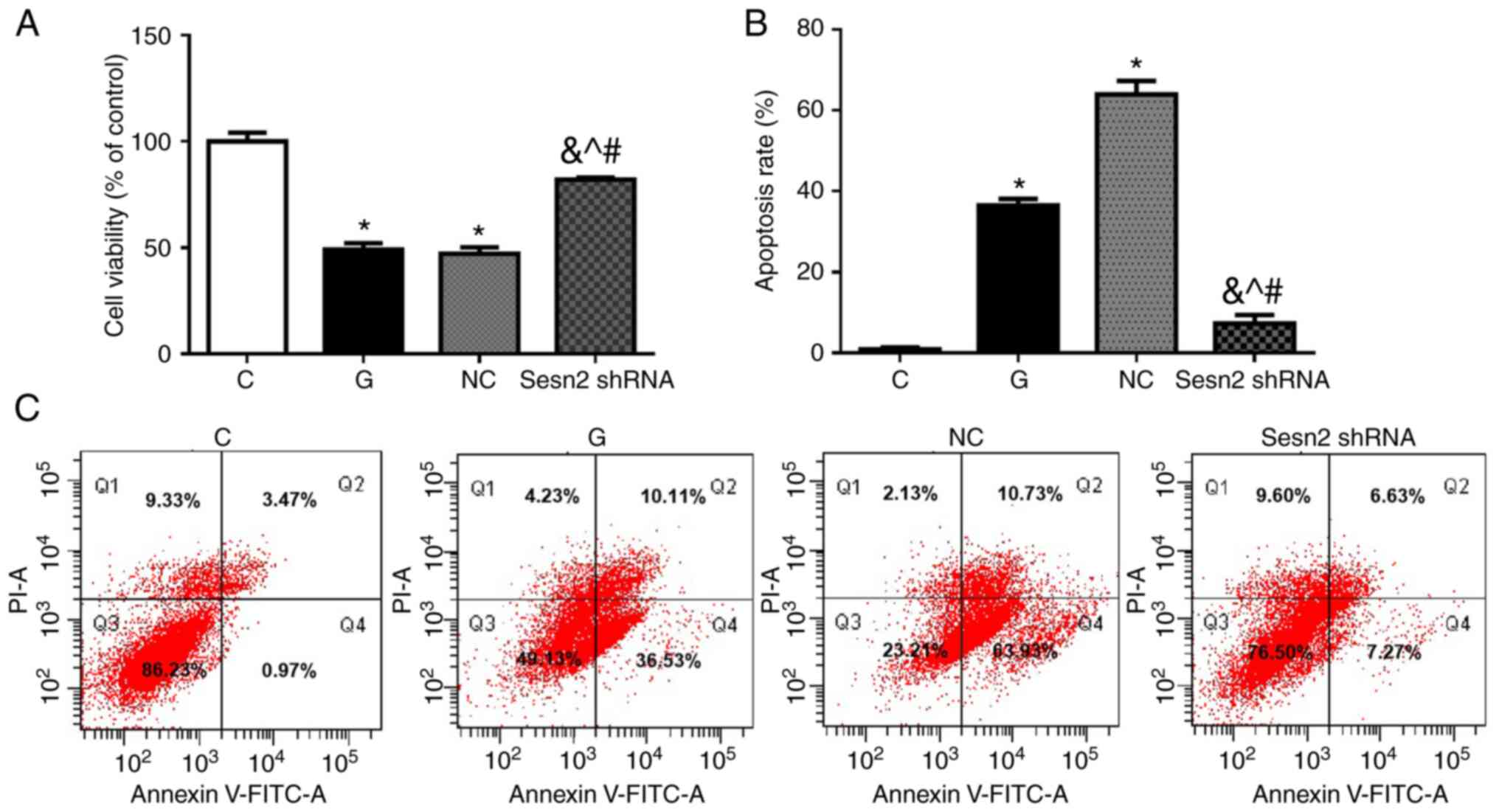
The cell viabilities were measured by MTS analysis.
As shown in Fig. 3A, cell
viabilities were decreased greatly in G group compared to C group
(P<0.001). The downregulation of Sestrin2 significantly
increased cell viability of H9c2 cells, compared with the G and NC
groups (P<0.001). To evaluate the possible effects of
downregulation of Sestrin2 on HG-treated H9c2 cell apoptosis, the
apoptotic rates in the four groups were determined by flow
cytometry using V-FITC/PI staining. As shown in Fig. 3B and C, despite the high PI cells (about 10% of
the total) in Q1 in flow cytometry, the apoptotic rate was very low
in C group, while it increased significantly in G group
(P<0.001). The apoptotic rate was significantly decreased after
Sestrin2 shRNA transfection, compared with the G and NC groups
(P<0.001). These results indicated that the downregulation of
Sestrin2 could provide cardioprotective effects in hyperglycemic
conditions by attenuating cardiomyocyte injury and apoptosis.
Specifically, significant populations of high PI cells in Q1 in
flow cytometry were identified in repetitive experiments performed
by different members of the research group, without observing
obvious cell necrosis during cell culture. Since a common cause for
this situation is a mechanical lesion, the HG concentration in the
culture medium may lead to a higher permeability of PI through the
cell membrane.
Downregulation of Sestrin2 attenuates
oxidative stress in H9c2 cells exposed to HG
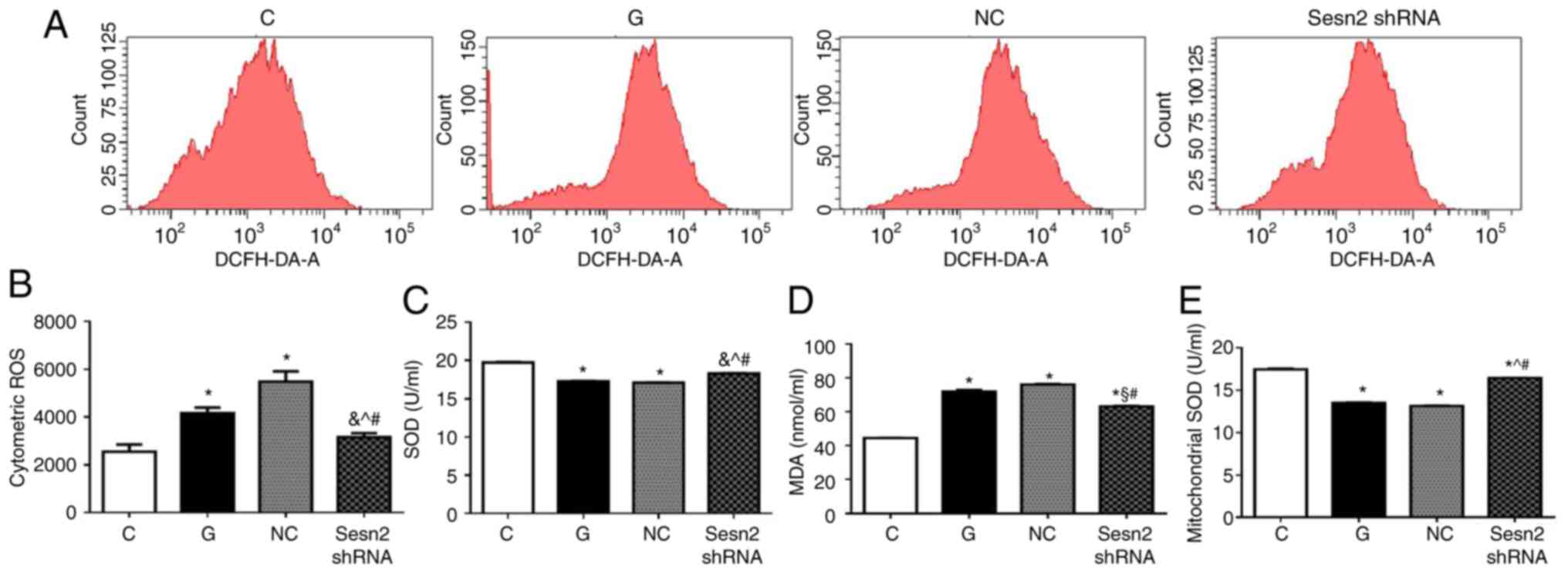
Overwhelming oxidative stress is the key procedure
of DCM. Excessive ROS production can induce mitochondrial
dysfunction of cardiomyocytes, thereby causing apoptosis (7). DCFH-DA was used to detect cellular
oxidative stress. As shown in Fig.
4A, intracellular ROS production was markedly increased in H9c2
cells under HG conditions. However, the downregulation of Sestrin2
significantly reduced ROS level in H9c2 cells, compared with G and
NC groups (P<0.001). In addition, oxidant (MDA) and antioxidant
levels (SOD and mitochondrial SOD) were determined. As shown in
Fig. 4B, MDA content in G group
increased in comparison with that in C group (P<0.001), which
was markedly decreased by Sestrin2 inhibition. The antioxidative
enzymes SOD and mitochondrial SOD were reduced in G group
(P<0.001), suggesting that antioxidant capacity was compromised
under HG conditions. However, subsequent to Sestrin2 silencing,
antioxidant levels increased significantly (P<0.001).
Collectively, the results indicated that downregulation of Sestrin2
protects H9c2 cells against HG-induced injuries through alleviation
of oxidative stress.
Downregulation of Sestrin2 alleviates
HG-induced mitochondrial injury in H9c2 cells
To investigate the effect of Sestrin2 on
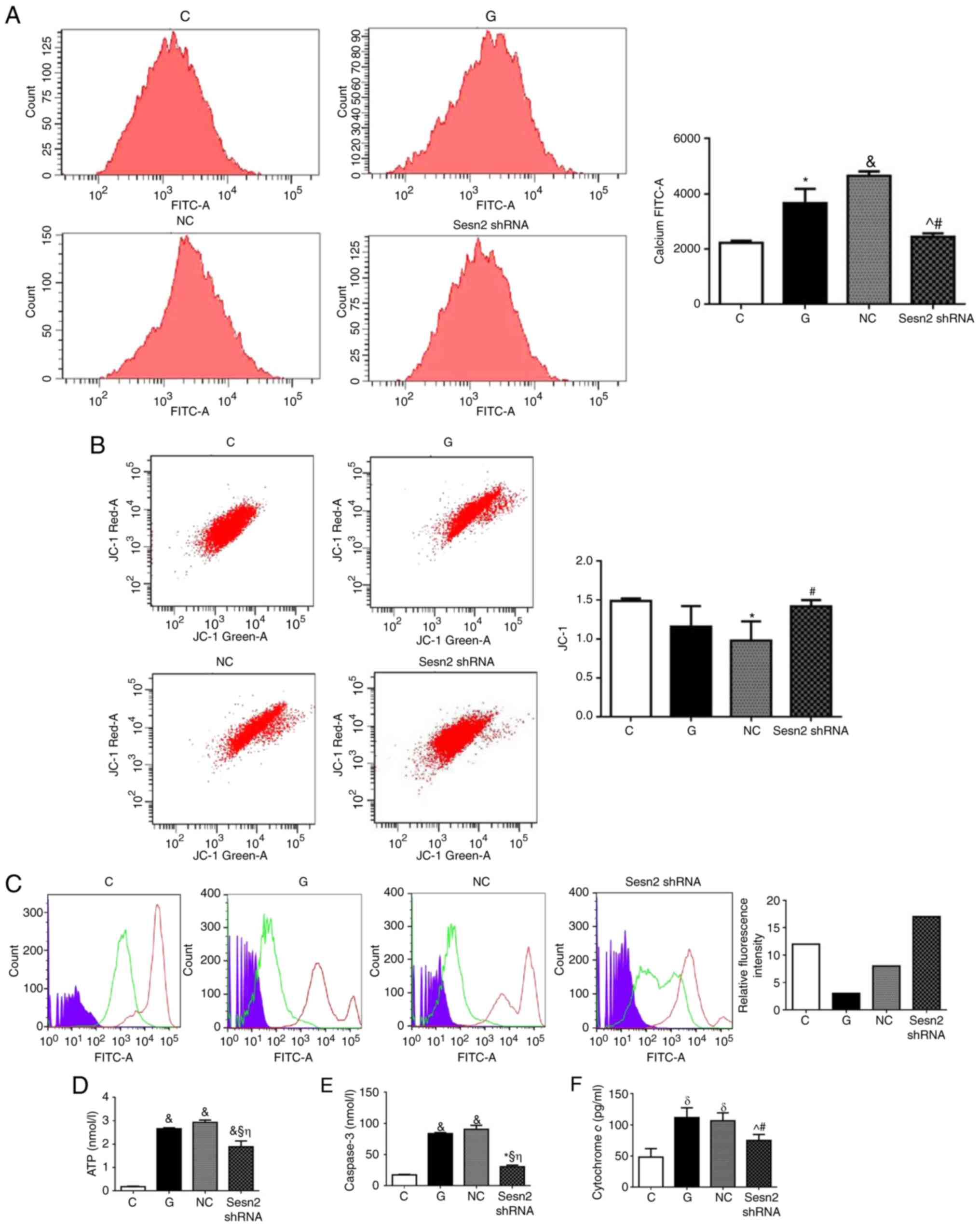
mitochondrial function, intracellular calcium (Fig. 5A) and mitochondrial membrane
potential in H9c2 cells was measured (Fig. 5B) by flow cytometry. Results
demonstrated that the intracellular calcium concentration of G
group was increased compared with C group (P<0.05; Fig. 5), while the inhibition of Sestrin2
decreased the intracellular calcium level compared with G and NC
groups (P<0.05). The mitochondrial membrane potential was
decreased under HG conditions, while the inhibition of Sestrin2
significantly increased mitochondrial membrane potential compared
with NC group (P<0.05). The MPTP opening was increased in HG
status while the inhibition of Sestrin2 decreased MPTP opening.
Furthermore, ATP, caspase-3, and cytochrome c generations
were increased in G group, while Sestrin2 downregulation decreased
at all their levels. Taken together, these results demonstrated
that the inhibition of Sestrin2 ameliorated mitochondrial function
under HG status. The maintenance of mitochondrial function is
mainly involved in the protective mechanism.
Discussion
It has been well documented that mitochondrial
dysfunction plays a critical role in the development of DCM.
Several aspects of common mitochondrial functions have been
described as being altered in DCM, including impaired energy
metabolism, compromised mitochondrial dynamics, deficiencies in
Ca2+ handling, increases in reactive oxygen species
production, and a higher propensity for MPTP opening (8). Enhanced oxidative stress, decreased
cell viability and impaired mitochondrial function in an in
vitro DCM model have been previously observed (7). Increased apoptosis and necrosis of
cardiomyocytes were observed in clinical and animal studies in DCM,
which was closely related to mitochondrial dysfunction (9). The present study demonstrated
increased cellular apoptosis, enhanced oxidative stress, and
impaired mitochondrial integrity in DCM, which was in accordance
with results of previous studies (8).
Sestrin2 is an important oxidative stress response
protein. It has been indicated that activation of Sestrin2 plays
important roles in reducing ROS accumulation, maintaining energy
balance, enhancing autophagy, reducing protein synthesis,
modulating cell growth, and retaining the progression of metabolic
diseases (10). Sestrin2 was
reported to be involved in various diseases. Previously, there were
some researches which investigated the role of Sestrin2 in
cardiovascular diseases, but mainly in ischemic and age-related
heart diseases. Sestrin2 alleviated oxidative stress and ER stress,
and activated autophagy in these experimental models (11). In aged hearts, Sestrin2 expression
was reduced (12). Additionally,
Sestrin2 knockout enhanced pressure overload-induced cardiac
hypertrophy and Sestrin2 rescue partially alleviated cardiac
hypertrophy in aged mice. In research concerning diabetes, Sestrin2
was considered to be involved in maintaining insulin sensitivity.
Sestrin2 deficiency increased obesity-induced insulin resistance
and accelerated the progression of diabetes (13). The expression of Sestrin2 was found
to be decreased in podocytes and monocytes in HG conditions. The
activation of Sestrin2 ameliorated mitochondrial dysfunction and
was involved in the regulation of HG-mediated atherosclerosis
(14,15).
To the best of our knowledge, this is the first
research to investigate the role of Sestrin2 in DCM. An increased
expression of Sestrin2 was identified in DCM models. Considering
the previous researches, this finding led to speculation of
probable protection effect of Sestrin2 in DCM. However, beyond our
expectation, inhibition of Sestrin2 ameliorated cardiomyocyte
injury by alleviating mitochondrial dysfunction, which is in
contrast to previous studies in other inflammatory conditions
(11,12,16,17).
The levels of Sestrin2 were upregulated or
downregulated in different stress conditions. However, in most
in vitro and animal studies, the activation of Sestrin2 was
similarly reported to play protective roles while the
downregulation or knockdown of Sestrin2 was indicated to aggravate
these stress conditions. A study showed that the knockdown of
Sestrin2 increased lipopolysaccharide-induced oxidative stress and
apoptosis in H9c2 cells and heart tissues of mice (16). In rats, Sestrin2 knockdown was
reported to aggravate the cardiomyocyte hypertrophy induced by
phenylephrine, and Sestrin2 overexpression protected cardiomyocytes
from hypertrophy (17).
Previous findings have shown that the upregulation
of Sestrin2 was not suggested to be protective in some conditions.
Early researches in Sestrin2 have proven that overexpression of
Sestrin2 full-length cDNA was toxic for many types of cultured
cells (18). Clinical studies in
Sestrin2 are on the increase in recent years and contradictory
results were shown. For example, Sestrin2 concentrations were
increased in patients with chronic heart failure (CHF), and
correlated positively with the severity of CHF (19). Upregulated Sestrin2 significantly
increased the occurrence of major adverse cardiac events and
indicated poor outcome in patients with CHF (19). Similarly, plasma Sestrin2 level in
patients with coronary artery disease (CAD) was found to be high
and associated with the severity of CAD (20). In addition, serum Sestrin2
concentration showed an increasing trend in patients with metabolic
syndrome. Plasma sestrin2 concentrations were high in patients with
carotid plaque and were associated with plaque severity (21). In particular, concerning the
expression of Sestrin2 in diabetes, studies presented inconsistent
findings. A study in patients with newly diagnosed T2DM
demonstrated a trend for increased Sestrin2 level, which was
significantly related to insulin resistance and percentage body fat
(22). However, other studies in
patients with T2DM and in patients with diabetic nephropathy (DN)
reported significantly decreased Sestrin2 levels (23,24).
Thus, Sestrin2 seems to be involved in complex regulation
mechanisms and reacts differently in different stress conditions.
Based on findings of the present study and the aforementioned
studies, Sestrin2 levels may increase in a compensatory manner in
response to oxidative stress and exert detrimental effects in DCM.
The roles of Sestrin2 are more complicated than expected.
The present study has several limitations. First,
the findings were mainly derived from in vitro research.
Second, the molecular mechanisms that connect Sestrin2 and
mitochondrial function were not extensively explored. Further
investigations are required to clarify the roles and underlying
mechanisms of Sestrin2 in DCM.
In conclusion, the present study demonstrated that
inhibition of enhanced Sestrin2 expression ameliorates cardiac
injury in DCM, which may be largely attributed to restoration of
mitochondrial function.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81800726)
and the Natural Science Foundation of Guangdong, China (grant no.
2017A030310257).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XZ and WL contributed to the conception and design
of the study. XZ, XD and HY conducted the experiments. XZ and WL
confirm the authenticity of all the raw data. XZ, XD and ZC
analyzed the data and wrote the manuscript. XZ and WL reviewed and
edited the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal procedures were approved by the Ethics
Committee of the Second Affiliated Hospital of Guangzhou Medical
University (Guangzhou, China) (approval no.: 20200407).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cosentino F, Grant PJ, Aboyans V, Bailey
CJ, Ceriello A, Delgado V, Federici M, Filippatos G, Grobbee DE,
Hansen TB, et al: 2019 ESC Guidelines on diabetes, pre-diabetes,
and cardiovascular diseases developed in collaboration with the
EASD. Eur Heart J. 41:255–323. 2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Jia G, Whaley-Connell A and Sowers JR:
Diabetic cardiomyopathy: A hyperglycaemia and insulin
resistance-induced heart disease. Diabetologia. 61:21–28.
2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lee JH, Budanov AV, Park EJ, Birse R, Kim
TE, Perkins GA, Ocorr K, Rllisman MH, Bodmer R, Bier E and Karin M:
Sestrin as a feedback inhibitor of TOR that prevents age-related
pathologies. Science. 327:1223–1228. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Gao A, Li F, Zhou Q and Chen L: Sestrin2
as a potential therapeutic target for cardiovascular diseases.
Pharmacol Res. 159(104990)2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ding B, Parmigiani A, Divakaruni AS,
Archer K, Murphy AN and Budanov AV: Sestrin2 is induced by glucose
starvation via the unfolded protein response and protects cells
from non-canonical necroptotic cell death. Sci Rep.
6(2253)2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Livak KJ and Schmittgen TD: Analysis of
reactive gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zhang X, Zhong Z and Li W: Downregulation
of TRAP1 aggravates injury of H9c2 cardiomyocytes in a
hyperglycemic state. Exp Ther Med. 18:2681–2686. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Federico M, Fuente S, Palomeque J and Sheu
S: The role of mitochondria in metabolic disease: A special
emphasis on heart dysfunction. J Physiol. 599:3477–3493.
2021.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Kubli D and Gustafsson Å: Unbreak my
heart: Targeting mitochondrial autophagy in diabetic
cardiomyopathy. Antioxid Redox Signal. 22:1527–1544.
2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Budanov AV, Lee JH and Karin M: Stressin
Sestrins take an aging fight. EMBO Mol Med. 2:388–400.
2010.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Sun W, Wang Y, Zheng Y and Quan N: The
emerging role of Sestrin2 in cell metabolism, and cardiovascular
and age-related diseases. Aging Dis. 11:154–163. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Quan N, Li X, Zhang J, Han Y, Sun W, Ren
D, Tong Q and Li J: Substrate metabolism regulated by
Sestrin2-mTORC1 alleviates pressure overload-induced cardiac
hypertrophy in aged heart. Redox Biol. 36(101637)2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Lee JH, Budanov AV, Talukdar S, Park EJ,
Park HL, Park HW, Bandyopadhyay G, Li N, Aghajan M, Jang I, et al:
Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell
Metab. 16:311–321. 2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Lin Q, Ma Y, Chen Z, Hu J, Chen C, Fan Y,
Liang W and Ding G: Sestrin-2 regulates podocyte mitochondrial
dysfunction and apoptosis under high-glucose conditions via AMPK.
Int J Mol Med. 45:1361–1372. 2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Sundararajan S, Jayachandran I,
Balasubramanyam M, Mohan V, Venkatesan B and Manickam N: Sestrin2
regulates monocyte activation through AMPK-mTOR nexus under
high-glucose and dyslipidemic conditions. J Cell Biochem: Nov 18,
2018 (Epub ahead of print). doi: 10.1002/jcb.28102.
|
|
16
|
Hwang H, Kim JW, Chung HS, Seo JA, Kim SG,
Kim NH, Choi KM, Baik SH and Yoo HJ: Knockdown of Sestrin2
increases lipopolysaccharide-induced oxidative stress, apoptosis,
and fibrotic reactions in H9c2 cells and heart tissues of mice via
an AMPK-dependent mechanism. Mediators Inflamm.
2018(6209140)2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Dong B, Xue R, Sun Y, Dong Y and Liu C:
Sestrin 2 attenuates neonatal rat cardiomyocyte hypertrophy induced
by phenylephrine via inhibiting ERK1/2. Mol Cell Biochem.
433:113–123. 2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Budanov AV, Shoshani T, Faerman A, Zelin
E, Kamer I, Kalinski H, Gorodin S, Fishman A, Chajut A, Einat P, et
al: Identification of a novel stress-responsive gene Hi95 involved
in regulation of cell viability. Oncogene. 21:6017–6031.
2002.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Wang H, Li N, Shao X, Li J, Guo L, Yu X,
Sun Y, Hao J, Xiang J, Li X and Han X: Increased plasma sestrin2
concentrations in patients with chronic heart failure and predicted
the occurrence of major adverse cardiac events: A 36-month
follow-up cohort study. Clin Chim Acta. 495:338–344.
2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Kishimoto Y, Aoyama M, Saita E, Ikegami Y,
Ohmori R, Kondo K and Momiyama Y: Association between plasma
Sestrin2 Levels and the presence and severity of coronary artery
disease. Dis Markers. 2020(7439574)2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kishimoto Y, Saita E, Ohmori R, Kondo K
and Momiyama Y: Plasma sestrin2 concentrations and carotid
atherosclerosis. Clin Chim Acta. 504:56–59. 2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Chung HS, Hwang H, Hwang SY, Kim NH, Seo
JA, Kim SG, Kim NH, Sei Baik H, Choi KM and Yoo HJ: Association of
serum Sestrin2 level with metabolic risk factors in newly diagnosed
drug-naïve type 2 diabetes. Diabetes Res Clin Pract. 144:34–41.
2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Sundararajan S, Jayachandran I,
Subramanian SC, Anjana RM, Balasubramanyam M, Mohan V, Venkatesan B
and Manickam N: Decreased Sestrin levels in patients with type 2
diabetes and dyslipidemia and their association with the severity
of atherogenic index. J Endocrinol Invest. 44:1395–1405.
2021.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Mohany KM and Rugaie OA: Association of
serum sestrin 2 and betatrophin with serum neutrophil gelatinase
associated lipocalin levels in type 2 diabetic patients with
diabetic nephropathy. J Diabetes Metab Disord. 19:249–256.
2020.PubMed/NCBI View Article : Google Scholar
|