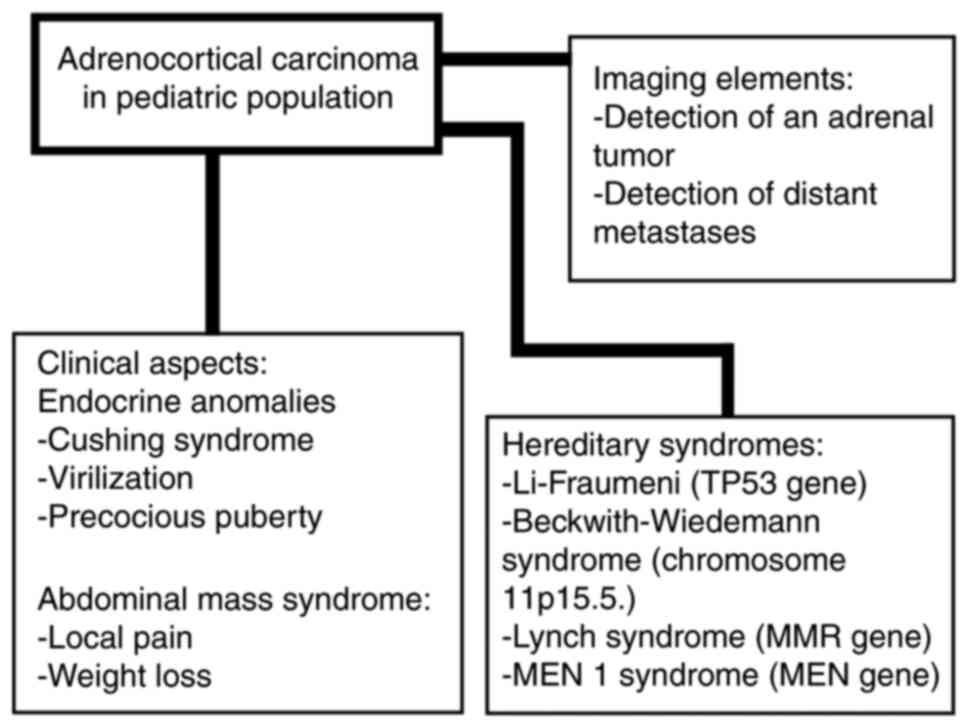
Adrenocortical carcinoma (adrenal cortex-derived
cancer), an orphan malignancy, is a very aggressive disease that
affects both adults and children with an annual incidence of 1-2
cases/million for adults vs. 0.2-0.38 cases/million in children,
except for markedly higher rates in Southern Brazil due to high
prevalence of tumor protein (TP53) p. R337H (R337H) mutation
(1-3).
It has a female predominance based on most studies (1-3).
A strong genetic background for adrenocortical carcinoma in the
pediatric population has been described (1,2). A
total of 50-80% of cases involve anomalies of genes such as TP53
(in half of the cases), formerly known as p53, insulin-like growth
factor 2 (IGF2) overexpression, IGF1R, ZNRF3, CTNNB1, multiple
endocrine neoplasia type 1 (MEN1), PRKAR1A, 11p15.5 (chromosome 11
anomalies) (3,4). Recently, mutations in epidermal growth
factor receptor (EGFR) have been reported in teenagers and young
children diagnosed with adrenocortical carcinoma, indicating the
potential future use of tyrosine kinase inhibitors in addition to
traditional medication such as mitotane and chemotherapy regimens
which are introduced at different stages during management, pre and
post-tumor removal if surgery is feasible (1,5).
Adrenalectomy is the first line therapy offering the best
prognosis, while the adjuvant therapies are less standardized in
children; genetic testing is recommended before surgery and
lifelong follow-up is required (6,7). The
tumor rapidly spreads at the local level and to distant sites
(6,8). In general, the pattern of this
condition renders it a rather distinct entity than the same tumor
identified in adults, despite the fact than even in pediatric cases
the disease is extremely severe (for instance, in children the
5-year survival rate is 46-55%) (9). In addition, the prognostic factors
described in adults are not necessarily similar in children
(10). Further markers of abnormal
pathways in adrenal cancer such as IGF or mechanistic target of
rapamycin (mTOR) are required for an improved therapeutic
intervention (11). The aim of the
present review was to focus on adrenocortical carcinoma based on a
multi-disciplinary approach, particularly in the pediatric
population. The present review represents an assessment of
literature using PubMed database. Researched key words in different
combinations included ‘adrenocortical carcinoma’, ‘adrenal cancer’,
‘adrenal tumor’, ‘children’ and ‘pediatric’. A total of 70
references are cited (between 2009 and 2021) based on their
clinical relevance. The level of statistical evidence varies from
case reports, cases series to different types of studies. The
majority of them are retrospective cohort/observational studies,
population-based studies, certain are National Registry-based
analyses, and also a meta-analysis that was published in 2021 was
included.
Adrenocortical carcinoma in children, representing
0.2% of all cancers in children, is considered rather a standalone
condition when compared with adults with the same diagnosis
(12,13). A total of 80-90% of cases have an
adrenal-related endocrine dysfunction (indicating that 10-20%, or
even 43% of the tumors, depending on the study, are non-functional)
such as Cushing syndrome, virilization (single or associated with
Cushing syndrome), or pubertal anomalies which are due to androgen
with or without glucocorticoid excess (Fig. 1) (12-14).
The incidentaloma scenario of detection is rarer than in adults
(15). Precocious puberty (PP) of
iso- or hetero-sexual pattern is independent of
gonadotropin-releasing hormone (GnRH) with high testosterone or
estrogens (with consecutive low FSH and LH) but post-operative
activation of GnRH may be expected with central (hypothalamic) PP
(16-18).
PP is accompanied by accelerated growth while Cushing syndrome by
arrest growth (19,20). Breast development may appear in both
males and females (19,21). Abnormally high levels of
dehydroepiandrosterone (DHEAS) have also been reported (22). Pure androgen-secreting tumors
underlying an adrenocortical carcinoma are exceptionally observed
(23). Steroidogenic machinery may
be provided by fetal adrenal tissue or even by Leydig cells as they
have been revealed in isolated cases (24). As expected, hypercortisolemia of
adrenal cause needs to be confirmed by specific tests (25). Another type of presentation is not
related to endocrine features, but with the presence of a very
large abdominal mass causing local pain and weight loss or
associated metastases of the lung or liver (26,27).
Genetic anomalies are more important in pediatric
than adult cases of adrenocortical carcinoma and they ideally need
to be evaluated before surgery (28). A potential involvement in adrenal
cancer as well as certain types of renal cancer and meningioma have
been described in association with ubiquitin-proteasome system
(UPS) as a regulator of the Armadillo repeat containing (ARMC5)
gene system (29). ARMC5 anomalies
have also been described in bilateral adrenal hyperplasia of
macronodular type associated with endocrine dysfunctions such as
glucocorticoid with or without mineralocorticoid excess (30,31).
Adrenocortical carcinoma is part of a more complex tumorigenesis
process in patients with Li-Fraumeni syndrome also introducing
sarcomas of both bone and soft tissues, breast cancer at a young
age, gastrointestinal cancers particularly of the colon and
stomach, leukemia, and brain cancer (32,33).
The underlying mutation is TP53(32). This is most frequently observed in
the pediatric population confirmed with adrenocortical cancer
(34). A retrospective
single-centric study involving 23 patients admitted between 1977
and 2017 revealed a 50% prevalence of the mutation (10 out of 20
tested patients) on a cohort with a female/male ratio of 3.6:1 and
a bimodal incidence (one peak incidence earlier in life between the
age of 0 and 6 years and the other one after the age of 12 years)
(34). TP53 R337H mutation is very
frequent in Brazil where large population studies have been
developed with a reported incidence of adrenocortical tumors of 4.8
in adults, and 6.4/million/year (depending on geographic area) in
children younger than 10 years old (35). Another study including data from 19
population-based registries of Brazil revealed an incidence of
adrenocortical cancer of 4 cases/million/year in individuals
younger than 10 years old (36). In
one study on TP53 R337H carriers, a higher count of cytotoxic T
lymphocytes (CD8-CTL) was correlated with the diagnosis of
adrenocortical cancer at very young age and stage I of the disease
while the Ki-67 value and Weiss score were not survival predictors
(37). Adrenocortical carcinoma has
an extremely low prevalence of 0.47-3.2% in subjects with Lynch
syndrome. However, in other cancers, such as colorectal cancer and
endometrial cancer, Lynch sundrome has a very high prevalence
underlying the mutation of MMR (DNA mismatch repair) gene (38,39).
Due to poor prognosis and the importance of early recognition that
offers an improved prognosis via adrenalectomy, screening of
adrenocortical carcinoma in patients diagnosed with Lynch syndrome
is recommended (38,39).
In addition, alternative lengthening of telomeres
(ALT) represents a potential dysfunctional mechanism in cancerous
cells of adrenal cancer (40).
Murine experiments revealed that besides the p53/Rb pathway, the
Wnt/β-catenin signaling pathway may also be involved (41). Clinical studies on
β-catenin/N-cadherin indicated the potential role of this pathway
in tumorigenesis underlying adrenal cortex-derived carcinoma
(41,42). β-Catenin activity in association
with CTNNB1 genes has been reported in relation to sclerosing bone
dysplasia with adrenal tumors (43). A retrospective cohort study based on
the German Childhood Cancer Registry, published in 2020, involving
321 subjects diagnosed with Beckwith-Wiedemann syndrome (which is
associated with anomalies of chromosome 11p15.5.) revealed a
33-fold higher risk for different cancers such as hepatoblastoma,
nephroblastoma but also, more rarely, adrenocortical carcinoma
(44). In addition, the association
of 11p15 or TP53 genes with abnormal activity of phosphodiesterases
has been reported (45).
Genome-wide association studies (GWAS) based on single nucleotide
polymorphism (SNP) assays identified retinoic acid pathway
anomalies in adrenocortical carcinoma (46). MEN type 1 syndrome caused by MEN1
gene mutation, which is associated with pituitary tumors or
gastrointestinal and pancreatic neuroendocrine neoplasia has been
reported to be present in adrenal tumors (47-49).
However, pediatric adrenocortical carcinoma is rarer than adult
adrenal cancer in MEN1 syndrome (50).
Weiss score, rather than I-IV staging according to
the European Network for the Study of Adrenal Tumors (ENSAT) is
essential for prognosis (51). A
retrospective case series published in 2021 on 8 patients
(male/female ratio of 2:6; median age of 6 years) revealed that
survival for the next 2 years after diagnosis was 100% for stage II
and 75% for stage IV (without achieving statistical significance),
but in patients with a Weiss score <6 the 2-year survival rate
was 100% which was statistically significantly higher when compared
with the subjects with a Weiss score at least 6 or more who had a
2-year survival rate of 35% (P=0.043); Weiss score being considered
the most important prognostic factor (51). The combination of mitotane,
etoposide, Adriamycin and cisplatin was revealed to be most useful
in improving the prognosis (51). A
comparative study from 2021, evaluated adults (n=146) vs. children
<15 years old (n=44), and the results revealed: Female
prevalence (more evident in adults: 84 vs. 61%); adrenal Cushing
syndrome more often observed in adults while virilization was more
frequently identified in children (both clinical features with
statistically significant differences between the two studied
populations); and a Ki-67 index of proliferation based on the
immunohistochemistry report which was higher in children compared
with adult correspondents, emerging as a useful prognostic marker,
as well as the Weiss score (52). A
Ki-67 value of least 15% represents a powerful predictor of poor
outcome in the pediatric population (52). Ki-67 is an independent prognostic
factor in adults as well (53). An
extensive German study suggested the following Ki-67 cut-offs:
<10%, 10-19%, and ≥20% (54).
Another study revealed that a Ki-67 cut-off value of 10% was useful
for prediction (55). Variations
between manual/digital assessments of Ki-67 have been reported
(with digital assessment superior to manual analysis which displays
less reproducible values) (56).
Surgical removal of the tumor, if feasible, remains the first line
of therapy; procedures of approach vary, but successful tumor
removal represents a major predictor of favorable outcome (57,58).
Metastases removal may be useful in addition to mitotane, although
chemotherapy regimens in children are less clear when compared to
adults (59). A retrospective study
from 2021 on 24 children with adrenal tumors (median age at
diagnosis, 78 months; confirmed adrenocortical carcinoma in 14
cases) reported the use of a transabdominal approach (in
combination with chemotherapy in certain cases), but also an
additional thoraco-abdominal approach which was applied if the
tumor had spread to the lungs or the primary adrenal tumor was
extensive (57). A complex
meta-analysis published in 2021 based on 42 studies between 1994
and 2020 (including a total of 1,006 children aged <18 years)
highlighted the following factors of favorable outcome with
statistical significance: Age at diagnosis <4 years old,
complete tumor surgical removal, decreased tumor volume, size, and
weight, adrenal cancer without hormonal burden (the presence of
adrenal Cushing syndrome was an indicator of poor prognosis) and
stage I disease (60). A previous
study revealed that virilization was a favorable prognostic factor
(12). In addition, the control of
tumor-related Cushing syndrome may be achieved not only through
mitotane, but also by using other adrenal blockers that are mostly
used in adults, and only exceptionally used in children to date
(61). In 2020, a case of a
2-month-old female treated with etomidate (0.03 mg/hg/h) to control
high cortisol levels was published, and further implementation of
the mentioned drug in complex regimes is required (61). Mitotane, an adrenolytic
chemotherapeutic, causes adrenal insufficiency which requires
glucocorticoid and mineralocorticoid substitution, increased liver
enzymes, high cholesterol, gynecomastia, and primary hypothyroidism
in adults and children (62). An
estrogen-like effect was suggested as a possible cause of PP
(62). An analysis from Mayo Clinic
between 1950 and 2017 included 41 subjects with adrenal cortex
carcinoma having an onset before the age of 21 years (63). The study revealed: A median age at
diagnosis of 15.7 years; 54% of children had at least one hormonal
dysfunction; metastases were more often identified in children
older than 12 years (particularly at the hepatic and pulmonary
level); the 2-year survival rate was 61%, while the 5-year survival
rate was 46% (63). Metastases
represent an independent factor of poor outcome (63). The European Cooperative Study Group
for Pediatric Rare Tumors (EXPeRT) revealed in a
multi-centric-based European study that included individuals
younger than 18 years diagnosed with adrenocortical carcinoma
(n=82) that the 3-year progression-free survival rate was 51%,
while the overall survival rate was 55% (64). Poor prognostic factors were:
Vascular invasion and incomplete surgery, co-presence of
metastases; large tumor volume if the disease was localized
(64). The ‘Helsinki score’, a
model of prediction for metastases in adrenocortical carcinomas,
combines pathological data and immunohistochemistry data (Ki-67)
(65). A previous study on 255
adrenal cortex carcinomas confirmed a good equivalence of the
Helsinki score with the Weiss score and a good predictive value
(65). A retrospective,
observational study based on the Brazilian National Institute of
Cancer, between 1997 and 2015 included 27 pediatric (median age,
3.6 years) adrenocortical carcinomas with a median carcinoma
diameter of 8.2 cm (66). The most
frequent medical drug that was used as adjuvant medication was
mitotane (66). Poor prognostic
factors included increased tumor size, a high Weiss score,
incomplete surgery, and histological data revealing capsular
invasion (66). The National Cancer
Data Base (NCDB)-related study on 111 subjects (18 years or
younger) confirmed with adrenocortical cancer (between 1998 and
2011) revealed a median age at diagnosis of 4 years (48% of the
patients were younger than 3 years old); female sex predominance
(69%); a median diameter of 9.5 cm; and 1- and 3-year survival
rates of 70 and 64%, respectively (67). Survival depended on the initial
tumor size, associated metastases, local spreading of the disease
and the success of the surgical procedure as reflected by the
margin status (67). Another
pediatric study on 27 cases revealed an increased expression of
endoglin MVD (intra-tumoral microvessel density) and a low
expression of CD34 MVD as poor prognostic factors while vascular
endothelial growth factor (VEGF) was not relevant (68). The routine use of growth factors as
prognostic factors is currently under evaluation.
Multiple aspects of adrenocortical carcinoma remain
poorly understood. Another clinical association, probably less
relevant in every day practice, is the co-presence of pulmonary
arteriovenous malformation (based on a case report with statistical
evidence), however, the current level of statistical evidence is
low (69). Uncommon predictors of
poor outcome such as Fetal and Adult Testis-Expressed 1 (FATE1)
expression have been revealed in adults, not in children and
further studies are necessary (70).
At present, pediatric adrenocortical carcinoma still
represents a severe condition that requires prompt intervention and
a multidisciplinary team. Further development of molecular markers
is required for a deeper understanding of the disease thus
improving the protocols of approach and the prognosis.
Not applicable.
Funding: Not applicable.
Not applicable.
FS drafted the manuscript and critically revised the
final version. RCP researched the literature. MC drafted the
manuscript, researched the bibliography, and created the figure. AP
approved the final version and refined the references. MCD wrote
the manuscript and is the corresponding author and AG critically
revised the final version. All authors read and approved the final
manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Akhavanfard S, Yehia L, Padmanabhan R,
Reynolds JP, Ni Y and Eng C: Germline EGFR variants are
over-represented in adolescents and young adults (AYA) with
adrenocortical carcinoma. Hum Mol Genet. 29:3679–3690.
2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Jarzembowski JA: New prognostic indicators
in pediatric adrenal tumors: Neuroblastoma and adrenal cortical
tumors, can we predict when these will behave badly? Surg Pathol
Clin. 13:625–641. 2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Kamilaris CDC, Hannah-Shmouni F and
Stratakis CA: Adrenocortical tumorigenesis: Lessons from genetics.
Best Pract Res Clin Endocrinol Metab. 34(101428)2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ribeiro TC, Jorge AA, Almeida MQ, Mariani
BM, Nishi MY, Mendonca BB, Fragoso MC and Latronico AC:
Amplification of the insulin-like growth factor 1 receptor gene is
a rare event in adrenocortical adenocarcinomas: Searching for
potential mechanisms of overexpression. Biomed Res Int.
2014(936031)2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Traynor MD Jr, Sada A, Thompson GB, Moir
CR, Bancos I, Farley DR, Dy BM, Lyden ML, Habermann EB and McKenzie
TJ: Adrenalectomy for non-neuroblastic pathology in children.
Pediatr Surg Int. 36:129–135. 2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Brondani VB and Fragoso MCBV: Pediatric
adrenocortical tumor-review and management update. Curr Opin
Endocrinol Diabetes Obes. 27:177–186. 2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zekri W, Hammad M, Rashed WM, Ahmed G,
Elshafie M, Adly MH, Elborai Y, Abdalla B, Taha H, Elkinaae N, et
al: The outcome of childhood adrenocortical carcinoma in Egypt: A
model from developing countries. Pediatr Hematol Oncol. 37:198–210.
2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Longui CA: Adrenal cortical carcinoma in
infancy. Rev Paul Pediatr. 37:2–3. 2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Breidbart E, Cameo T, Garvin JH, Hibshoosh
H and Oberfield SE: Pubertal outcome in a female with virilizing
adrenocortical carcinoma. J Pediatr Endocrinol Metab. 29:503–509.
2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Erickson LA: Challenges in surgical
pathology of adrenocortical tumours. Histopathology. 72:82–96.
2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
De Martino MC, van Koetsveld PM, Feelders
RA, de Herder WW, Dogan F, Janssen JAMJL, Hofste Op Bruinink D,
Pivonello C, Waaijers AM, Colao A, et al: IGF and mTOR pathway
expression and in vitro effects of linsitinib and mTOR inhibitors
in adrenocortical cancer. Endocrine. 64:673–684. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Pinto EM, Zambetti GP and
Rodriguez-Galindo C: Pediatric adrenocortical tumours. Best Pract
Res Clin Endocrinol Metab. 34(101448)2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Kastenberg ZJ and Scaife ER:
Adrenocortical tumors in children. Semin Pediatr Surg.
29(150927)2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Sipayya V, Yadav YK, Arora R, Sharma U and
Gupta K: Virilizing adrenocortical carcinoma in a child: A rare
enigma. Indian J Endocrinol Metab. 16:621–623. 2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Khan MS, Ali A, Tariq I, Khan MA, Bakar MA
and Anwar AW: A clinical study and treatment results of
adrenocortical carcinoma patients presented in Shaukat Khanum
Memorial cancer hospital and research center, Lahore. J Pak Med
Assoc. 69:717–719. 2019.PubMed/NCBI
|
|
16
|
Goyal A, Malhotra R and Khadgawat R:
Precocious pseudopuberty due to virilising adrenocortical carcinoma
progressing to central precocious puberty after surgery. BMJ Case
Rep. 12(e229476)2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Shimelis D, Tadesse A, Schneider J and
Tilahun B: Adrenocortical carcinoma in a 3% years old girl with
hetro-sexual precocity. Ethiop Med J. 52:91–94. 2014.PubMed/NCBI
|
|
18
|
Miyoshi Y, Oue T, Oowari M, Soh H,
Tachibana M, Kimura S, Kiyohara Y, Yamada H, Bessyo K, Mushiake S,
et al: A case of pediatric virilizing adrenocortical tumor
resulting in hypothalamic-pituitary activation and central
precocious puberty following surgical removal. Endocr J.
56:975–982. 2009.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Takeuchi T, Yoto Y, Ishii A, Tsugawa T,
Yamamoto M, Hori T, Kamasaki H, Nogami K, Oda T, Nui A, et al:
Adrenocortical carcinoma characterized by gynecomastia: A case
report. Clin Pediatr Endocrinol. 27:9–18. 2018.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Ghazizadeh F, Ebadi M, Alavi S, Arzanian
M, Shamsian B and Jadali F: Adrenocortical carcinoma presenting
with heterosexual pseudoprecocious puberty shortly after birth:
Case report and review. Ecancermedicalscience.
7(289)2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wendt S, Shelso J, Wright K and Furman W:
Neoplastic causes of abnormal puberty. Pediatr Blood Cancer.
61:664–671. 2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Idkowiak J, Elhassan YS, Mannion P, Smith
K, Webster R, Saraff V, Barrett TG, Shaw NJ, Krone N, Dias RP, et
al: Causes, patterns and severity of androgen excess in 487
consecutively recruited pre- and post-pubertal children. Eur J
Endocrinol. 180:213–221. 2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Tong A, Jiang J, Wang F, Li C, Zhang Y and
Wu X: Pure androgen-producing adrenal tumor: Clinical features and
pathogenesis. Endocr Pract. 23:399–407. 2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Fujisawa Y, Sakaguchi K, Ono H, Yamaguchi
R, Kato F, Kagami M, Fukami M and Ogata T: Combined steroidogenic
characters of fetal adrenal and Leydig cells in childhood
adrenocortical carcinoma. J Steroid Biochem Mol Biol. 159:86–93.
2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Chen QL, Su Z, Li YH, Ma HM, Chen HS and
Du ML: Clinical characteristics of adrenocortical tumors in
children. J Pediatr Endocrinol Metab. 24:535–541. 2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Jaruratanasirikul S, Patarapinyokul S and
Mitranun W: Androgen-producing adrenocortical carcinoma: Report of
3 cases with different clinical presentations. J Med Assoc Thai.
95:816–20. 2012.PubMed/NCBI
|
|
27
|
Mittal A, Aggarwal M and Debata P: Large
adrenocortical carcinoma presenting as an adenoma with precocious
puberty. Indian J Pediatr. 79:820–821. 2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Pinto EM, Chen X, Easton J, Finkelstein D,
Liu Z, Pounds S, Rodriguez-Galindo C, Lund TC, Mardis ER, Wilson
RK, et al: Genomic landscape of paediatric adrenocortical tumours.
Nat Commun. 6(6302)2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Yan G, Liu N, Tian J, Fu Y, Wei W, Zou J,
Li S, Wang Q, Li K and Wang J: Deubiquitylation and stabilization
of ARMC5 by ubiquitin-specific processing protease 7 (USP7) are
critical for RCC proliferation. J Cell Mol Med. 25:3149–3159.
2021.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Espiard S and Bertherat J: The genetics of
adrenocortical tumors. Endocrinol Metab Clin North Am. 44:311–334.
2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Jouinot A, Armignacco R and Assié G:
Genomics of benign adrenocortical tumors. J Steroid Biochem Mol
Biol. 193(105414)2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Daly MB, Pal T, Berry MP, Buys SS, Dickson
P, Domchek SM, Elkhanany A, Friedman S, Goggins M, Hutton ML, et
al: Genetic/familial high-risk assessment: Breast, ovarian, and
pancreatic, version 2.2021, NCCN clinical practice guidelines in
oncology. J Natl Compr Canc Netw. 19:77–102. 2021.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ferreira AM, Brondani VB, Helena VP,
Charchar HLS, Zerbini MCN, Leite LAS, Hoff AO, Latronico AC,
Mendonca BB, Diz MDPE, et al: Clinical spectrum of Li-Fraumeni
syndrome/Li-Fraumeni-like syndrome in Brazilian individuals with
the TP53 p.R337H mutation. J Steroid Biochem Mol Biol. 190:250–255.
2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Brenna CTA, Michaeli O, Wasserman JD and
Malkin D: Clinical outcomes of children with adrenocortical
carcinoma in the context of germline TP53 status. J Pediatr Hematol
Oncol. 43:e635–e641. 2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Costa TEJ, Gerber VKQ, Ibañez HC, Melanda
VS, Parise IZS, Watanabe FM, Pianovski MAD, Fiori CMCM, Fabro ALMR,
Silva DBD, et al: Penetrance of the TP53 R337H mutation and
pediatric adrenocortical carcinoma incidence associated with
environmental influences in a 12-year observational cohort in
Southern Brazil. Cancers (Basel). 11(1804)2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Kostiainen I, Hakaste L, Kejo P,
Parviainen H, Laine T, Löyttyniemi E, Pennanen M, Arola J, Haglund
C, Heiskanen I and Schalin-Jäntti C: Adrenocortical carcinoma:
Presentation and outcome of a contemporary patient series.
Endocrine. 65:166–174. 2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Parise IZS, Parise GA, Noronha L, Surakhy
M, Woiski TD, Silva DB, Costa TEB, Del-Valle MHCP, Komechen H,
Rosati R, et al: The prognostic role of CD8+ T
lymphocytes in childhood adrenocortical carcinomas compared to
Ki-67, PD-1, PD-L1, and the weiss score. Cancers (Basel).
11(1730)2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Raymond VM, Everett JN, Furtado LV,
Gustafson SL, Jungbluth CR, Gruber SB, Hammer GD, Stoffel EM,
Greenson JK, Giordano TJ and Else T: Adrenocortical carcinoma is a
lynch syndrome-associated cancer. J Clin Oncol. 31:3012–3018.
2013.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Domènech M, Grau E, Solanes A, Izquierdo
A, Del Valle J, Carrato C, Pineda M, Dueñas N, Pujol M, Lázaro C,
et al: characteristics of adrenocortical carcinoma associated with
lynch syndrome. J Clin Endocrinol Metab. 106:318–325.
2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Sung JY, Lim HW, Joung JG and Park WY:
Pan-cancer analysis of alternative lengthening of telomere
activity. Cancers (Basel). 12(2207)2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Borges KS, Pignatti E, Leng S, Kariyawasam
D, Ruiz-Babot G, Ramalho FS, Taketo MM, Carlone DL and Breault DT:
Wnt/β-catenin activation cooperates with loss of p53 to cause
adrenocortical carcinoma in mice. Oncogene. 39:5282–5291.
2020.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Rubin B, Regazzo D, Redaelli M, Mucignat
C, Citton M, Iacobone M, Scaroni C, Betterle C, Mantero F, Fassina
A, et al: Investigation of N-cadherin/β-catenin expression in
adrenocortical tumors. Tumour Biol. 37:13545–13555. 2016.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Peng H, Jenkins ZA, White R, Connors S,
Hunter MF, Ronan A, Zankl A, Markie DM, Daniel PB and Robertson SP:
An activating variant in CTNNB1 is associated with a sclerosing
bone dysplasia and adrenocortical neoplasia. J Clin Endocrinol
Metab. 105(dgaa034)2020.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Cöktü S, Spix C, Kaiser M, Beygo J,
Kleinle S, Bachmann N, Kohlschmidt N, Prawitt D, Beckmann A, Klaes
R, et al: Cancer incidence and spectrum among children with
genetically confirmed Beckwith-Wiedemann spectrum in Germany: A
retrospective cohort study. Br J Cancer. 123:619–623.
2020.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Pinto EM, Faucz FR, Paza LZ, Wu G,
Fernandes ES, Bertherat J, Stratakis CA, Lalli E, Ribeiro RC,
Rodriguez-Galindo C, et al: Germline variants in phosphodiesterase
genes and genetic predisposition to pediatric adrenocortical
tumors. Cancers (Basel). 12(506)2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Surakhy M, Wallace M, Bond E, Grochola LF,
Perez H, Di Giovannantonio M, Zhang P, Malkin D, Carter H, Parise
IZS, et al: A common polymorphism in the retinoic acid pathway
modifies adrenocortical carcinoma age-dependent incidence. Br J
Cancer. 122:1231–1241. 2020.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Harada K, Yasuda M, Hasegawa K, Yamazaki
Y, Sasano H and Otsuka F: A novel case of myxoid variant of
adrenocortical carcinoma in a patient with multiple endocrine
neoplasia type 1. Endocr J. 66:739–744. 2019.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Wang W, Han R, Ye L, Xie J, Tao B, Sun F,
Zhuo R, Chen X, Deng X, Ye C, et al: Adrenocortical carcinoma in
patients with MEN1: A kindred report and review of the literature.
Endocr Connect. 8:230–238. 2019.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Al-Salameh A, Cadiot G, Calender A, Goudet
P and Chanson P: Clinical aspects of multiple endocrine neoplasia
type 1. Nat Rev Endocrinol. 17:207–224. 2021.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Else T and Rodriguez-Galindo C: 5th
International ACC Symposium: Hereditary predisposition to childhood
ACC and the associated molecular phenotype: 5th International ACC
symposium session: Not just for kids! Horm Cancer. 7:36–39.
2016.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Li J, Zhang W, Hu H, Zhang Y, Wen Y and
Huang D: Adrenocortical carcinoma in eight children: A report and
literature review. Cancer Manag Res. 13:1307–1314. 2021.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Martins-Filho SN, Almeida MQ, Soares I,
Wakamatsu A, Alves VAF, Fragoso MCBV and Zerbini MCN: Clinical
impact of pathological features including the Ki-67 labeling index
on diagnosis and prognosis of adult and pediatric adrenocortical
tumors. Endocr Pathol. 32:288–300. 2021.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Zhang F, Zhang F, Liu Z, Wu K, Zhu Y and
Lu Y: Prognostic role of Ki-67 in adrenocortical carcinoma after
primary resection: A retrospective mono-institutional study. Adv
Ther. 36:2756–2768. 2019.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Beuschlein F, Weigel J, Saeger W, Kroiss
M, Wild V, Daffara F, Libé R, Ardito A, Al Ghuzlan A, Quinkler M,
et al: Major prognostic role of Ki67 in localized adrenocortical
carcinoma after complete resection. J Clin Endocrinol Metab.
100:841–849. 2015.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Yamazaki Y, Nakamura Y, Shibahara Y,
Konosu-Fukaya S, Sato N, Kubota-Nakayama F, Oki Y, Baba S,
Midorikawa S, Morimoto R, et al: Comparison of the methods for
measuring the Ki-67 labeling index in adrenocortical carcinoma:
Manual versus digital image analysis. Hum Pathol. 53:41–50.
2016.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Papathomas TG, Pucci E, Giordano TJ, Lu H,
Duregon E, Volante M, Papotti M, Lloyd RV, Tischler AS, van
Nederveen FH, et al: An international Ki67 reproducibility study in
adrenal cortical carcinoma. Am J Surg Pathol. 40:569–576.
2016.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Ardicli B, User IR, Ciftci AO, Akyuz C,
Kutluk MT, Yalcin B, Gonc N, Ozon ZA, Alikasifoglu A, Oguz B, et
al: Adrenocortical tumours in children: A review of surgical
management at a tertiary care centre. ANZ J Surg. 91:992–999.
2021.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Orbach L, Nachmany I, Goykhman Y, Lahat G,
Yossepowitch O, Beri A, Ben-Gal Y, Klausner JM and Lubezky N:
Surgical approach to abdominal tumors involving the inferior vena
cava. Isr Med Assoc J. 22:364–368. 2020.PubMed/NCBI
|
|
59
|
Mirsharifi A, Vasei M, Sadeghian E,
Ghorbani-Abdehgah A and Naybandi Atashi S: Extra-adrenal,
non-functional adrenocortical carcinoma presenting with acute
abdomen: A case report. J Med Case Rep. 14(107)2020.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Zambaiti E, Duci M, De Corti F, Gamba P,
Dall'Igna P, Ghidini F and Virgone C: Clinical prognostic factors
in pediatric adrenocortical tumors: A meta-analysis. Pediatr Blood
Cancer. 68(e28836)2021.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Kwon A, Choi Y, Jung JW, Suh J and Kim HS:
Using etomidate in a 2-month-old infant with cushing syndrome due
to adrenocortical carcinoma. J Clin Res Pediatr Endocrinol 2020
(Epub ahead of print).
|
|
62
|
Oddie PD, Albert BB, Hofman PL, Jefferies
C, Laughton S and Carter PJ: Mitotane in the treatment of childhood
adrenocortical carcinoma: A potent endocrine disruptor. Endocrinol
Diabetes Metab Case Rep. 2018:18–0059. 2018.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Gupta N, Rivera M, Novotny P, Rodriguez V,
Bancos I and Lteif A: Adrenocortical carcinoma in children: A
clinicopathological analysis of 41 patients at the mayo clinic from
1950 to 2017. Horm Res Paediatr. 90:8–18. 2018.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Cecchetto G, Ganarin A, Bien E, Vorwerk P,
Bisogno G, Godzinski J, Dall'Igna P, Reguerre Y, Schneider D,
Brugières L, et al: Outcome and prognostic factors in high-risk
childhood adrenocortical carcinomas: A report from the European
Cooperative Study Group on Pediatric Rare Tumors (EXPeRT). Pediatr
Blood Cancer. 64:2017.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Duregon E, Cappellesso R, Maffeis V,
Zaggia B, Ventura L, Berruti A, Terzolo M, Fassina A, Volante M and
Papotti M: Validation of the prognostic role of the ‘Helsinki
Score’ in 225 cases of adrenocortical carcinoma. Hum Pathol.
62:1–7. 2017.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Bulzico D, de Faria PA, de Paula MP,
Bordallo MA, Pessoa CH, Corbo R, Ferman S, Vaisman M and Neto LV:
Recurrence and mortality prognostic factors in childhood
adrenocortical tumors: Analysis from the Brazilian National
Institute of Cancer experience. Pediatr Hematol Oncol. 33:248–258.
2016.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Gulack BC, Rialon KL, Englum BR, Kim J,
Talbot LJ, Adibe OO, Rice HE and Tracy ET: Factors associated with
survival in pediatric adrenocortical carcinoma: An analysis of the
National cancer data base (NCDB). J Pediatr Surg. 51:172–177.
2016.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Dias AI, Fachin CG, Avó LR, Frazão CV,
Caran EM, Schettini ST, Alves MT, Ribeiro RC and Abib Sde C:
Correlation between selected angiogenic markers and prognosis in
pediatric adrenocortical tumors: Angiogenic markers and prognosis
in pediatric ACTs. J Pediatr Surg. 50:1323–1328. 2015.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Rahat-Ul-Ain. Acquired pulmonary
arteriovenous malformation in a case of a pediatric adrenocortical
carcinoma. J Coll Physicians Surg Pak. 31:119–120. 2021.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Doghman-Bouguerra M, Finetti P, Durand N,
Parise IZS, Sbiera S, Cantini G, Canu L, Hescot S, Figueiredo MMO,
Komechen H, et al: Cancer-testis antigen fate1 expression in
adrenocortical tumors is associated with a pervasive autoimmune
response and is a marker of malignancy in adult, but not children,
ACC. Cancers (Basel). 12(689)2020.PubMed/NCBI View Article : Google Scholar
|