Introduction
Non-alcoholic fatty liver disease (NAFLD) is a
disease that involves the ectopic accumulation of fat in
hepatocytes, which is usually caused by simple steatosis (simple
fatty liver), insulin resistance, type 2 diabetes mellitus (T2DM)
and dyslipidemia (1-3).
Non-alcoholic steatohepatitis (NASH), which is fatty liver disease
with inflammatory damage and/or fibrosis of hepatocytes, is a more
aggressive and irreversible disease that is associated with an
increased risk of end-stage liver disease [including cirrhosis and
hepatocellular carcinoma (HCC)] (4). In recent years, the prevalence of
NAFLD and NASH has been increasing. Estimates of the global
prevalence of NAFLD ranged from 24 to 30% in 2018, and patients
tend to be younger (5). As a
chronic multisystem disease, NASH can also lead to numerous
complications in other organs, such as T2DM, cardiovascular,
chronic kidney and heart disease. However, there is no effective
treatment for NASH. The only effective way to prevent NASH is
moderate exercise and lifestyle improvement (6,7).
Therefore, it is very urgent to find a new and effective
therapeutic regimen.
At present, ‘multiple-hit model theory’ is widely
regarded as the pathogenesis of NASH (8). The ‘first hit’ usually involves
excessive accumulation of fat in the liver and insulin resistance.
Subsequently, ‘subsequent multiple hits’ occur, showing an
interaction of oxidative stress, endoplasmic reticulum (ER) stress,
inflammatory cytokines and numerous other factors. Pro-inflammatory
cytokines play a key role in the development of NASH. For example,
TNF-α overexpression results in upregulation of sterol regulatory
element binding protein-1, a key nuclear transcription factor in
lipid metabolism (9). In
pathogenesis, this cytokine also leads to lipid metabolism
disorder. ER stress leads to the activation of three unfolded
proteins, blocking the protein-response signaling pathways for
novel protein synthesis, ER chaperone production and misfolded
protein degradation (10,11). Under certain conditions, these
events can lead to inflammation and even cell death, suggesting
that ER stress is closely associated with inflammation and lipid
metabolism disorders (10,11).
The active natural products extracted from herbs are
one of the vital sources for anti-metabolic disease drug
development. Ganoderma lucidum (G. lucidum) is one of
the momentous Asian fungi known as Ling Zhi in China and Korea, and
Reishi mushroom in Japan (12).
G. lucidum is not only used in traditional medicine to
improve health and promote longevity, but also potentially treats a
variety of diseases, such as tumors, HIV, hypoglycemia, sedation
and myocardial ischemia; it also participates in the regulation of
blood lipids and liver protection (13,14).
It was also identified that the powdered mycelium of G.
lucidum caused plasma cholesterol to decrease in rats (15). Several oxygenated lanostane-type
triterpenoids isolated from G. lucidum have been shown to
inhibit β-hydroxy β-methylglutaryl-CoA (HMG-CoA) reductase activity
in vitro (15-17).
For instance, ganoderiol F and ganodermic acid Q demonstrated
inhibitory activity against HMG Co-A reductase and against acyl-CoA
acyltransferase (18,19). Although ganoderic acid A (GAA) has
been revealed to improve metabolic syndromes such as obesity,
hyperlipidemia and insulin resistance induced by high-fat diet, its
anti-NASH effect has not been reported to date. In addition, its
anti-NASH mechanism is not clear. Therefore, further elucidation of
the anti-NASH effect of GAA and its underlying molecular mechanisms
is highly warranted. Consequently, the present study was conducted
to investigate whether GAA is able to alleviate the development of
NASH in high fat-high cholesterol (HFHC)-fed mice with
experimentally induced NASH and which are the underlying molecular
mechanisms of the effects of GAA in the aforementioned model.
Materials and methods
Animals and treatment
All experiments and animal care were conducted in
accordance with the Provision and General Recommendation of Chinese
Experimental Animals Administration Legislation and were approved
by the Ethics Committee of Science and Technology Department of
Jiangsu [approval no. SYXK (SU) 2016-0011]. A total of 30 Male
C57BL/6 mice (6-8 weeks old; weight: 18-22 g) were purchased from
Nanjing Biomedical Research Institute and were randomly maintained
on a basal diet [the normal diet (ND group); 360 kcal/100 g;
comprising 13.3 g/100 g fat, 26.2 g/100 g protein and 60.5 g/100 g
carbohydrate] or a HFHC diet (506.8 kcal/100 g; comprising 10 g/100
g lard, 2 g/100 g cholesterol, 5 g/100 g egg yolk power, 10 g/100 g
sucrose, 2 g/100 g propylthiouracil and basal diet, 72.8 g/100 g).
Animals were housed at 24˚C and 55% relative humidity in a barrier
facility under a 12 h light/dark cycle with free access to food and
water. The basal diet and HFHC diet were provided by the Jiangsu
Xietong Medical and Biological Corporation. Subsequently, mice were
randomly divided into the following three groups (n=6-8 per group):
Vehicle-treated chow group, vehicle-treated HFHC group and
GAA-treated HFHC group (25 or 50 mg/kg/day). The administration
concentration was determined to have therapeutic effect in a
previous study (20). HFHC-fed
mice were gavaged with GAA, which was purchased from Shanghai Yousi
Biotechnology Co., Ltd. and was dissolved in 0.5% sodium
carboxymethyl cellulose (Sigma-Aldrich; Merck KGaA) for 12 weeks.
All mice were sacrificed by cervical dislocation after overnight
fasting at the termination of the experiment. Subsequently, mouse
liver tissue was obtained. Before euthanasia, 300 µl blood was
collected in the 3% isoflurane-anaesthetized (ForeneTM; Abbott
Laboratories SA; oxygen flow rate of 4 l/min) mice by retroorbital
venous plexus method. All animal experiments were performed in
accordance with the approved guidelines.
Serum biochemical analysis
Serum was collected after sacrifice instantaneously
by centrifugation at 1,200 x g for 15 min at room temperature. The
serum lipids, including total cholesterol (TC; cat. no. A111-1-1),
total triglycerides (TG; cat. no. A110-1-1), low-density
lipoprotein cholesterol (LDL-c; cat. no. A113-1-1) and high-density
lipoprotein cholesterol (HDL-c; cat. no. A112-1-1) were detected.
Aspartate transaminase (AST; cat. no. C010-2-1) and alanine
transaminase (ALT; cat. no. C009-2-1) levels were evaluated to
assess the hepatic injury. All serum lipids were evaluated by
commercial kits (Nanjing Jiancheng Bioengineering Institute). ELISA
measurements of serum IL-1β (cat. no. EMC001b.96), TNF-α (cat. no.
EMC102a.96) and IL-6 (cat. no. EMC004.96) were performed following
the manufacturer's protocol (Neobioscience Technology Co.,
Ltd.).
Histopathological analysis
Histopathological analysis was performed with
standardized specimens from specified portions of liver. Liver
tissues were fixed in 4% paraformaldehyde for 4 h at room
temperature, dehydrated in a series of ethanol and embedded in
paraffin wax. Sections (4-mm-thick) were stained with
hematoxylin-eosin (H&E) at room temperature for 15 min before
being analyzed under a light microscope (BX53; Olympus
Corporation). The NAFLD activity score (NAS) of each group was
calculated as previously described (21). Lipid droplets of the fresh liver
samples were stained by Oil Red O (cat. no. O8010; Beijing Solarbio
Science & Technology Co., Ltd.) for 15 min at room temperature
and analyzed to quantify lipid content by cell imaging under an
Olympus-BX53 light microscope. Sirius Red (SR; cat. no. ab150681;
Abcam) staining, anti-CD68 (cat. no. ab31630; Abcam; 1:200) and
anti-F4/80 antibodies (cat. no. ab16911; Abcam; 1:200) were used
for histological analysis under a light microscope (BX53; Olympus
Corporation). Areas of stained droplets (Sirius Red staining for 1
h at room temperature) or SR and the intensity of
immunohistochemical staining were determined using ImageJ 1.8.0
(National Institutes of Health).
Hepatic lipid and hepatocellular
oxidative stress analysis of the liver
Lipids extracted from murine liver tissue were
dissolved in isopropanol, after which hepatic TG and TC contents
were detected as aforementioned. Hepatic malondialdehyde (MDA) was
measured using a MDA assay kit (TBA method; cat. no. A003-1-2;
Nanjing Jiancheng Bioengineering Institute) in accordance with the
manufacturer's protocols. Briefly, 0.1 ml sample was mixed with
1,1,3,3-tetramethoxypropane, 0.75 ml TBA working solution (0.37%)
and perchloric acid. The resulting solution was incubated at 95˚C
for 45 min. After cooling (10 min in ice water bath), the
flocculent precipitate was removed by centrifugation (4,000 x g 10
min at room temperature). The supernatant was analyzed at 532 nm
using a multi-scan spectrum microplate spectrophotometer at room
temperature. Hepatic superoxide dismutase (SOD) levels was measured
using a SOD assay kit (WST-1 method; cat. no. A001-3-2; Nanjing
Jiancheng Bioengineering Institute). Briefly, the tissue were lysed
with No-nidet P-40 lysis buffer (1% NP-40, 50 mmol/l Tris-HCl [pH
7.5], 0.05 mmol/l ethylenediamine tetra-acetate) for 20 min at 4˚C.
The lysates were centrifuged at 300 g for 10 min, and 20 µl of this
sample solution was used for determination of SOD enzyme activity
according to the manufacturer's instructions. The value for each
treatment group was converted to the percentage of control.
Reverse transcription-quantitative
(RT-q) PCR
Total RNA extraction was performed using
TRIzol® (Vazyme Biotech Co., Ltd.) according to the
manufacturer's protocol. The purity and concentration of RNA was
determined by spectrophotometric analysis via absorbance at 260/280
and 260/230 nm. RNA concentrations were equalized and converted to
cDNA using Hiscript II reverse transcriptase (Vazyme Biotech Co.,
Ltd.) according to the manufacturer's protocol. Gene expression was
measured using a LightCycler 96 Real-Time PCR System (Roche
Diagnostics) using SYBR-green dyes (Roche Diagnostics). The
thermocycling conditions were as follows: Initial denaturisation at
95˚C for 10 min followed by 40 cycles for 10 sec at 95˚C, 10 sec at
55˚C and 30 sec at 72˚C, with a final elongation step at 72˚C for 7
min. All data were analyzed using GAPDH gene expression as an
internal standard. Relative gene expression levels were analyzed
using the 2-ΔΔCq method (22,23).
The sequences of the murine PCR primers were as follows: TNF-α
forward, 5'-AAGGGAGAGTGGTCAGGTTG-3' and reverse,
5'-TCTGTGAGGAAGGCTGTGC-3'; IL-1β forward,
5'-AACCTGCTGGTGTGTGACGTTC-3' and reverse,
5'-CAGCACGAGGCTTTTTTGTTGT-3'; IL-6 forward,
5'-CGGAGAGGAGACTTCACAGAG-3' and reverse,
5'-CATTTCCACGATTTCCCAGA-3'; α-smooth muscle actin (α-SMA) forward,
5'-CCCTGAAGTATCCGATAGAACA-3' and reverse,
5'-TGCCTGGGTACATGGTAGTG-3'; TGF-β forward,
5'-CTTTGTACAACAGCACCCGC-3' and reverse, 5'-TAGATTGCGTTGTTGCGGTC-3';
MMP-13 forward, 5'-GTGACTCTTGCGGGAATCCT-3' and reverse,
5'-CAGGCACTCCACATCTTGGT-3'; and GAPDH forward,
5'-AACAGCAACTCCCACTCTTC-3' and reverse,
5'-CCTGTTGCTGTAGCCGTATT-3'.
Western blot analysis
Liver tissues were homogenized at 4˚C with lysis
buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Nonidet P-40, 0.5%
sodium deoxycholate, 0.1% SDS, 1 mM EDTA and protease inhibitors).
After sonication, the samples were centrifuged for 10 min at room
temperature at 13,000 x g. The protein concentration were then
detected by performing a BCA assay (Beyotime Institute of
Biotechnology). 10% SDS-PAGE was conducted by loading equal amounts
of protein per lane (40 µg). Gels were then transferred to PVDF
membranes (EMD Millipore) and blocked with 5% non-fat milk in TBST
buffer for 1 h at room temperature. The membranes were then
incubated with the indicated primary antibodies overnight at 4˚C,
and washed three times with TBST (containing 0.1% Tween 20 in TBS)
for a total of 10 min. Thereafter, the membranes were incubated
with horseradish peroxidase-conjugated secondary antibody (1:2,000;
cat. no. A0208; Beyotime Institute of Biotechnology) for 1 h at
room temperature and washed three times with TBST. The blots were
visualized with the Amersham ECL Plus (Amersham; Cytiva) western
blotting detection reagents according to the manufacturer's
protocol. The primary antibodies (all dilutions were 1:500)
including glucose-regulated protein 78 (GRp78; cat. no. 3177),
phosphorylated (p)-eukaryotic initiation factor-2α (eIF-2α; cat.
no. 3398), eIF-2α (cat. no. 5324), p-JNK (cat. no. 4668), JNK (cat.
no. 9252), ERp57 (cat. no. 2887S), p-AKT (cat. no. 4060), AKT (cat.
no. 9272) p-MAPK (cat. no. 4370), MAPK (cat. no. 9102) and GAPDH
(cat. no. 8884) were obtained from Cell Signaling Technology, Inc.
Immunoreactive signals were detected with Chemi-Lumi One Ultra
(Tanon Science and Technology Co., Ltd.). The densitometric
analysis of the blots was performed by Image Pro Plus 6.0 software
(Media Cybernetics, Inc.).
Statistical analysis
Statistical analysis was performed using Prism
version 7.0 statistical software (GraphPad Software Inc.). All
quantitative values are presented as the mean ± SEM. Statistical
data were analyzed using one-way ANOVA with Dunnett's post hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
GAA improves the metabolic profiles in
HFHC-fed mice
The effect of GAA on the weight gain and metabolic
features of serum lipids in C57BL/6J mice is demonstrated in
Fig. 1. Mice fed with a HFHC diet
had significantly increased body weights, without significant
difference in food intake, compared with the ND-fed mice (Fig. 1A and B). Weight gain was also accompanied by
serum lipid disorder, including higher TG, TC, LDL-cholesterol
(LDL-c) and HDL-cholesterol (HDL-c) in the HFHC group compared with
the ND-fed group (Fig. 1C-F). In
addition, serum ALT and AST levels of mice were significantly
increased in the HFHC group compared with the ND group, which
revealed hepatic injury in model group (Fig. 1G and H). The chemical structure of GAA is
presented in Fig. 1I.
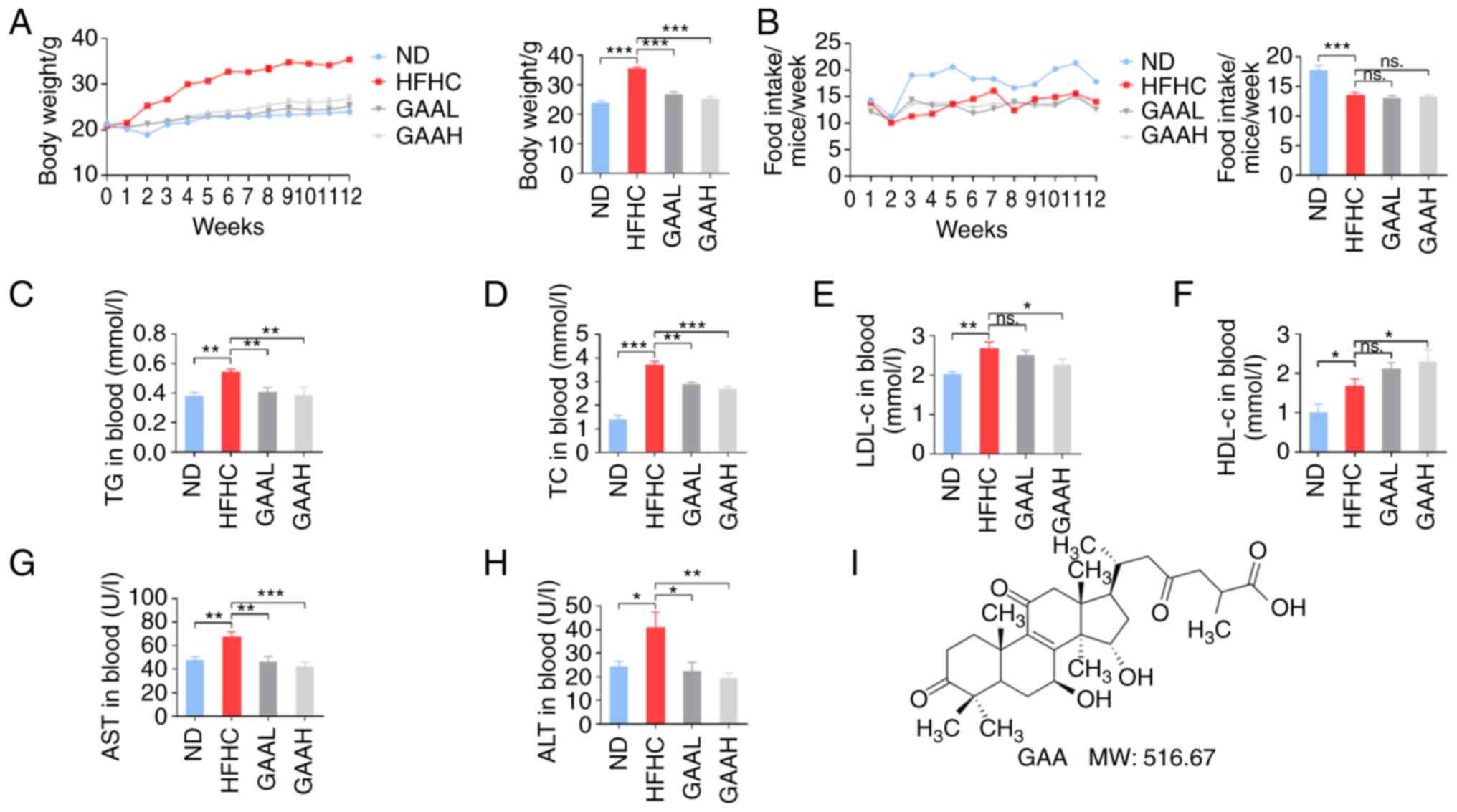 | Figure 1GAA improves the metabolic profiles
of HFHC-fed mice. (A) Body weight, (B) food intake, (C) TG, (D) TC,
(E) LDL-c, (F) HDL-c, (G) AST and (H) ALT levels were determined in
murine blood. Mice were fed an ND or HFHC diet with or without
indicated doses of GAA treatment (n=6-8). (I) The chemical
structure and molecular weight of GAA. Assays were repeated three
times. Data are expressed as the mean ± SEM. *P<0.05,
**P<0.01 and ***P<0.001 vs. the HFHC
group. ALT, alanine aminotransferase; AST, aspartate
aminotransferase; GAA, ganoderic acid A; GAAH, GAA 50 mg/kg/day;
GAAL, GAA 25 mg/kg/day; HDL-c, high density
lipoprotein-cholesterol; HFHC, high-fat high-cholesterol; LDL-c,
low density lipoprotein-cholesterol; MW, molecular weight; ns, not
significant; ND, normal diet; TC, total cholesterol; TG,
triglyceride. |
Compared with the model group, the body weight of
HFHC-fed mice treated with GAA was significantly reduced (Fig. 1A). Serum lipid disorders (increased
levels of TG, TC, LDL-c) were also reversed by GAA treatment in
HFHC-fed mice (Fig. 1C-F).
Additionally, the serum ALT and AST levels were significantly lower
in both GAA groups compared with the HFHC group, demonstrating a
hepatoprotective role of GAA against liver injury (Fig. 1G and H). These results suggested that GAA
improved the metabolic profiles of HFHC-fed mice.
GAA alleviates hepatic steatosis in
HFHC-fed mice
Hepatic lipid accumulation was evaluated by Oil Red
O staining and biochemical parameter assays. As revealed in
Fig. 2A and B, compared with the ND group, the Oil Red
O staining positive area in HFHC group mice was increased
significantly. By contrast, the positive area was reduced by GAA in
a dose-dependent manner compared with the model group (Fig. 2A and B). Similar results were observed
following liver biochemical parameter assays. The ratio of liver
weight to body weight and the levels of hepatic TG and TC were
significantly increased in the HFHC group compared with the ND
group. Conversely, oral administration of 25 or 50 mg/kg of GAA
reduced the HW/BW, TG and TC levels (Fig. 2C-E). These results indicated that
GAA alleviated hepatic steatosis in HFHC-fed mice.
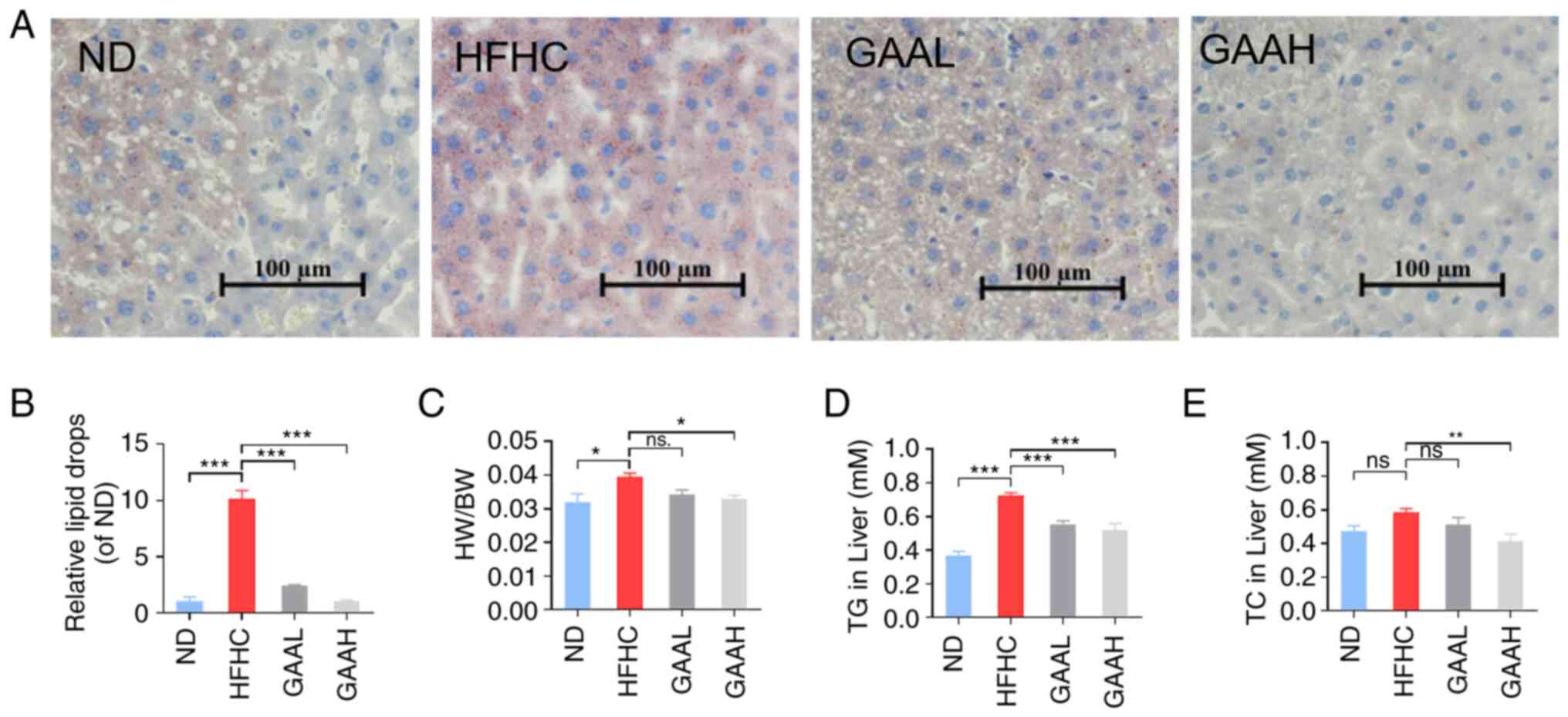 | Figure 2GAA alleviates hepatic steatosis in
HFHC-fed mice. (A) Livers were stained with Oil red O. (B) The area
of Oil red O was determined using ImageJ 1.8.0 software. (C) The
ratio of HW/BW was determined. (D and E) Hepatic TC and TG levels
were evaluated. Mice were fed an ND or HFHC diet with or without
indicated doses of GAA treatment (n=6-8). Assays were repeated
three times. Data are expressed as the mean ± SEM.
*P<0.05, **P<0.01 and
***P<0.001 vs. the HFHC group. GAA, ganoderic acid A;
GAAH, GAA 50 mg/kg/day; GAAL, GAA 25 mg/kg/day; HFHC, high-fat
high-cholesterol; HW/BW, liver weight to body weight; ND, normal
diet; ns, not significant; TC, total cholesterol; TG,
triglyceride. |
GAA reduces the hepatic inflammation
response in HFHC-fed mice
To evaluate the effect of GAA on the hepatic injury
process from NAFLD to NASH, liver histopathology was detected by
typical H&E staining (Fig.
3A). Compared with mice in the ND group, the typical
pathological phenomenon of NASH, including NAS, hepatocellular
ballooning, lobular inflammation and inflammatory cell infiltration
were observed in the livers of HFHC-fed mice (Fig. 3A). Furthermore, the number of CD68
positive marking Kupffer cells and F4/80 positive marking
macrophage infiltration were significantly enhanced in the livers
of HFHC-fed mice, compared with the ND group (Fig. 3B-E), which suggested that hepatic
inflammation had occurred in the model group. Circulating
inflammation factors, such as IL-1β, TNF-α and IL-6, were also
significantly increased in the model group (Fig. 3F-H). In addition, the mRNA
expression of inflammation factors, IL-1β, TNF-α and IL-6, was
significantly increased, compared with the corresponding control
group (Fig. 3I-K).
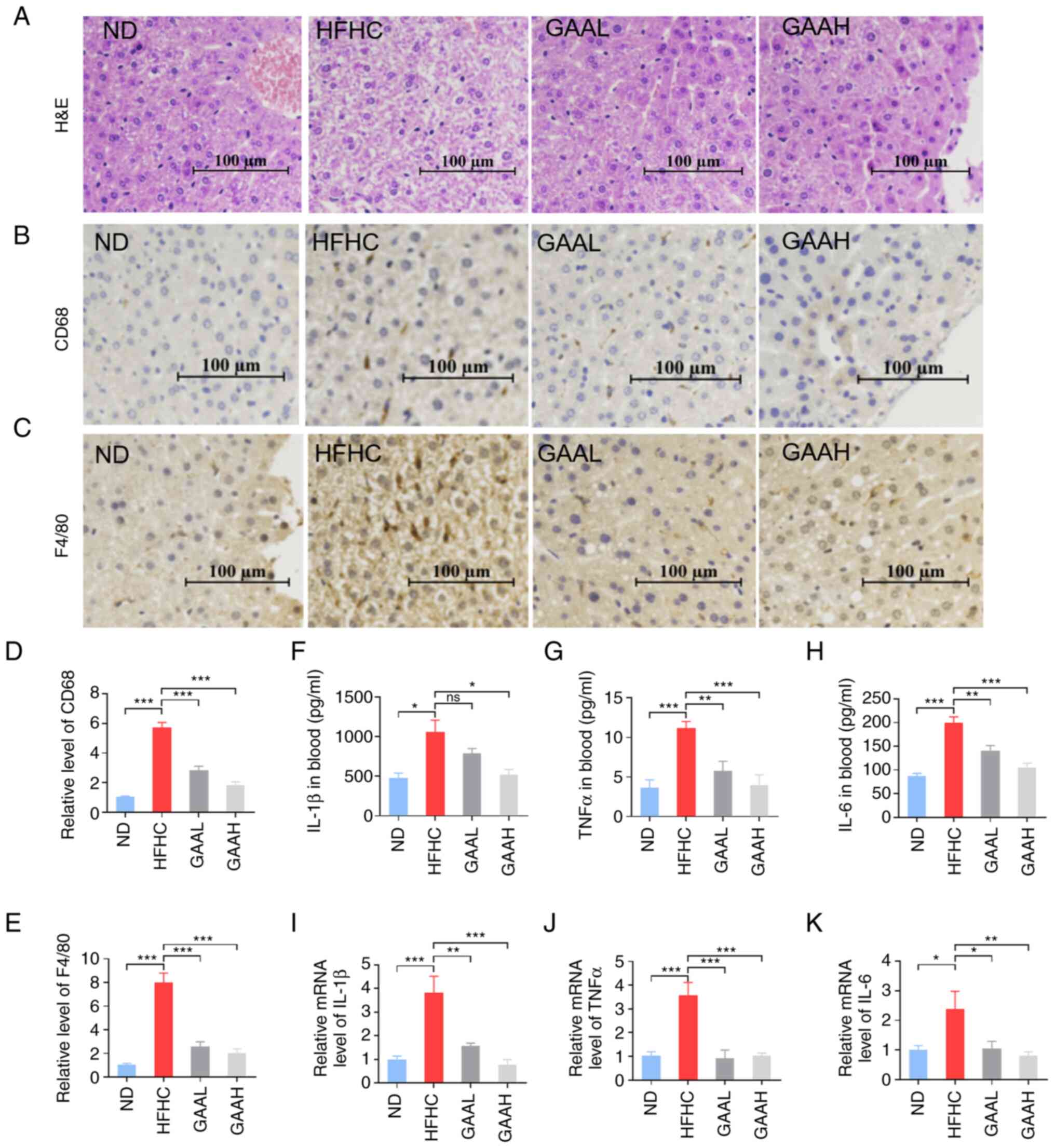 | Figure 3GAA reduces the hepatic inflammation
response in HFHC-fed mice. (A) H&E staining of liver tissue. (B
and C) CD68 and F4/80 levels in liver were detected by
immunohistochemistry. (D and E) Relative levels of CD68 and F4/80
were measured. (F) IL-1β, (G) TNFα and (H) IL-6 levels in murine
blood. mRNA levels of (I) IL-1β, (J) TNFα and (K) IL-6 levels in
liver tissue. Mice were fed an ND or HFHC diet with or without
indicated doses of GAA treatment (n=6-8). Assays were repeated
three times. Data are expressed as the mean ± SEM.
*P<0.05, **P<0.01 and
***P<0.001 vs. the HFHC group. GAA, ganoderic acid A;
GAAH, GAA 50 mg/kg/day; GAAL, GAA 25 mg/kg/day; H&E,
hematoxylin and eosin; HFHC, high-fat high-cholesterol; ND, normal
diet; ns, not significant. |
The typical pathology of NASH, including NAS,
hepatocellular ballooning, lobular inflammation and inflammatory
cell infiltration, was significantly decreased in the liver of
GAA-treated (25 or 50 mg/kg) HFHC-fed mice (Fig. 3A). As demonstrated in Fig. 3B-E, GAA (25 or 50 mg/kg) treatment
markedly reduced the number of Kupffer cells and macrophage
infiltration in the livers of HFHC-fed mice. Additionally, the
upregulated serum inflammation factors in HFHC-fed mice were
significantly reduced by GAA treatment for 12 weeks (Fig. 3F-H). In consistency with these
results, the mRNA expression of inflammation factors, such as
IL-1β, TNF-α and IL-6, was significantly decreased in HFHC-fed mice
treated with GAA (Fig. 3I-K).
These results suggested that GAA reduced the hepatic inflammation
response in HFHC-fed mice.
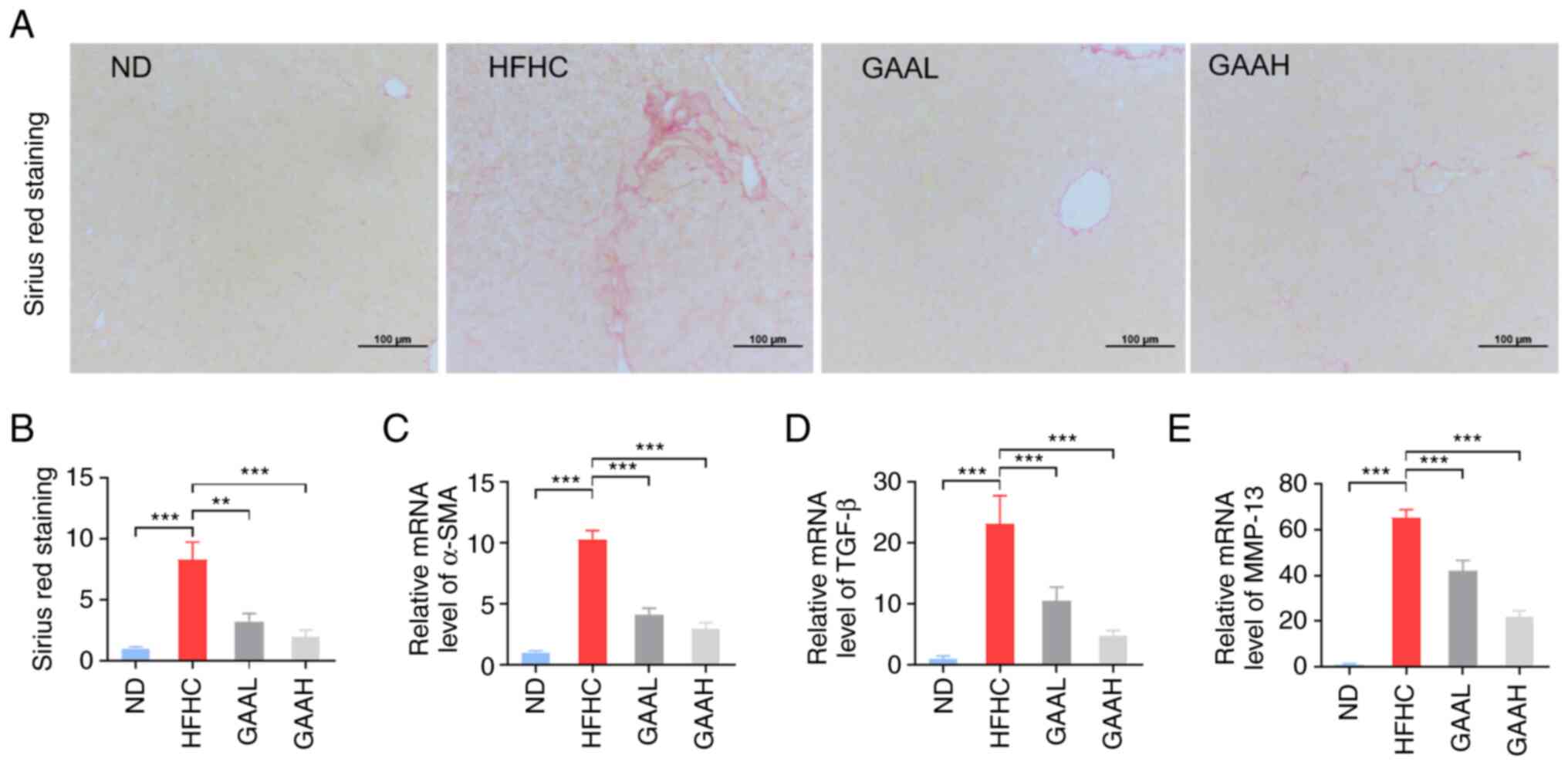
GAA decreases the hepatic fibrosis in
HFHC-fed mice
To certify the pathological progression from NASH to
hepatic fibrosis, SR staining was used to indirectly reflect
activated stellate cells in the livers of HFHC-fed mice. As
presented in Fig. 4A and B, the hepatic collagen formation of model
mice was increased, compared with the ND group. Additionally, the
mRNA levels of α-SMA, TGF-β and MMP-13 were also significantly
upregulated in HFHC-fed mice (Fig.
4C-E).
Administration of GAA (25 or 50 mg/kg) reduced the
number of activated stellate cells in the livers of HFHC-fed mice,
as determined by SR staining (Fig.
4A and B). The transcription
of fibrosis-related genes, including α-SMA, TGF-β and MMP-13, was
also decreased by GAA treatment (Fig.
4C-E). These results indicated that GAA could decrease the
hepatic fibrosis in HFHC-fed mice.
GAA hepatoprotection is associated
with the hepatic oxidative stress and ER stress response
It has been reported that oxidative stress plays a
key role in the occurrence and development of NASH (24). To explore the underlying mechanisms
of GAA, the contents of MDA and SOD in the livers of mice fed with
an HFHC diet were evaluated. As revealed in Fig. 5A and B, the hepatic level of MDA was
significantly increased in HFHC-fed mice compared with the ND
group, whereas it was decreased in HFHC-fed mice treated with GAA
when compared with the HFHC group. Conversely, the hepatic level of
SOD was significantly decreased in HFHC-fed mice compared with the
ND group, whereas it was increased in HFHC-fed mice treated with
GAA.
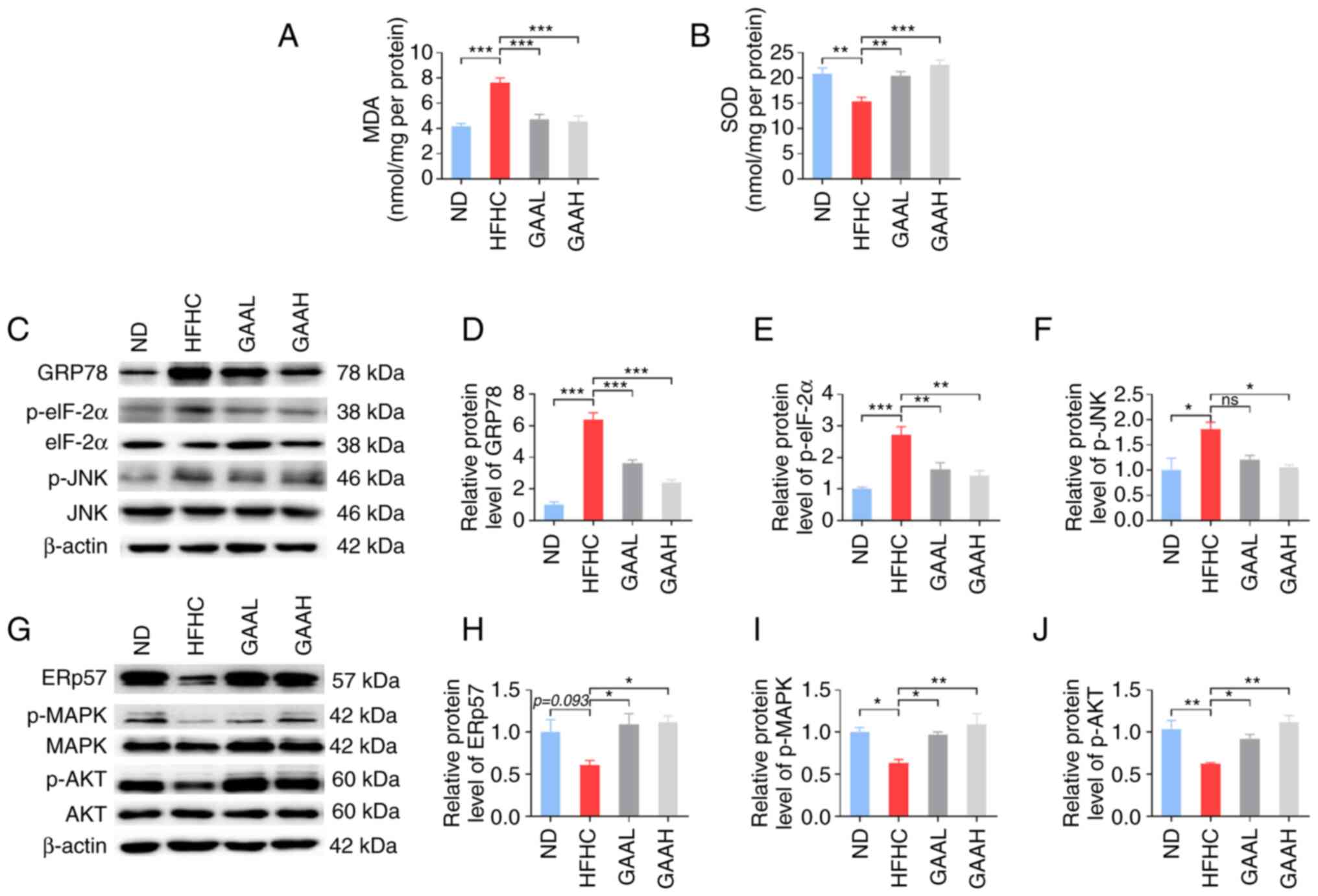 | Figure 5GAA hepatoprotection is associated
with hepatic oxidative stress and the ER stress response. (A) MDA
and (B) SOD levels were detected in murine livers. (C and G)
Western blotting was performed to determine the relative protein
levels of (D) GRp78, (E) p-eIF-2α, (F) p-JNK, (H) ERp57, (I) p-MAPK
and (J) p-AKT. Mice were fed an ND or HFHC diet with or without
indicated doses of GAA treatment (n=6-8). Assays were repeated
three times. Data are expressed as the mean ± SEM.
*P<0.05, **P<0.01 and
***P<0.001 vs. the HFHC group. eIF-2α, eukaryotic
initiation factor-2α GAA, ganoderic acid A; GAAH, GAA 50 mg/kg/day;
GAAL, GAA 25 mg/kg/day; GRp78, glucose-regulated protein 78; HFHC,
high-fat high-cholesterol; MDA, malondialdehyde; ND, normal diet;
ns, not significant; p, phosphorylated; SOD, superoxide
dismutase. |
Subsequently, the expression levels of essential ER
stress-related genes were detected. As demonstrated in Fig. 5C-F, administration of the HFHC diet
to mice resulted in an increase in GRp78, p-eIF-2α and p-JNK
protein expression, which was significantly reversed by GAA
treatment (25 or 50 mg/kg). Furthermore, compared with the ND
group, the levels of ERp57, p-Akt, and p-MAPK were decreased in
HFHC-fed mice, whereas GAA treatment increased their levels
(Fig. 5G-J). These results
suggested that the reduction of hepatic oxidative stress and the ER
stress response by GAA may be related to the prevention of NASH
progression in mice.
Discussion
As an important herbal medicine, G. lucidum
is widely used in the treatment of multiple diseases due to its
anti-inflammatory and antioxidant activities, including
inflammation-associated diseases, cancer, cardiovascular and
cerebrovascular diseases (25).
The main category of biologically active compounds produced in
G. lucidum, are the triterpenoids, which are known as
ganoderic acids (26). GAA, which
is derived from G. lucidum mushrooms, is considered to be a
potential therapeutic candidate for the treatment of a variety of
diseases, such as obesity, NAFLD, cancer and hepatic toxicity, but
is focused on alternative or complementary therapies for these
diseases (27-30).
A previous study has revealed that ganoderic acids have
hepatoprotective and hypocholesterolemic effects. In addition, a
previous study revealed that GAA improved lipid accumulation and
insulin resistance by inhibiting the sterol regulatory
element-binding protein signaling pathway (20). Liu et al (28) also found that GAA attenuated
high-fat-diet-induced liver injury in rats by regulating lipid
oxidation and liver inflammation. However, in the present study,
HFHC-induced mice were used to construct NAFLD and NASH models and
for the first time, to the best of our knowledge. The results
revealed that GAA significantly reduced liver lipid accumulation
and markedly improved liver inflammation and fibrosis. In terms of
the underlying mechanism of action, the results of the present
study preliminarily demonstrated that GAA improved NASH by
inhibiting hepatic oxidative and ER stress. The in-depth mechanism
of GAA in the improvement of NASH should be investigated in
subsequent studies.
The first step in the progression of NASH is the
accumulation of excessive TG in the liver. Therefore, suppression
of hepatic TG accumulation may be a potential therapeutic approach
for NASH (31). In the present
study, the role of GAA was investigated using a HFHC diet-induced
NASH model that simulates the development of human NASH. The
results demonstrated that oral administration of GAA effectively
inhibited lipid accumulation in the liver, markedly reduced the oil
red O staining area and largely reduced liver TG.
The HFHC diet is high in fat (~41%) and cholesterol
(~0.21%). A deficiency of choline and methionine, two essential
nutrients, leads to a reduction in the production of very-LDL
particles, which leads to fat accumulation in the liver. Therefore,
unlike the high-fat diet model, the HFHC diet model does not lead
to excessive fat absorption in the gut, but induces fat
accumulation via the lack of choline and methionine, thus blocking
the TG transfer pathway to the liver (32). In a previous study, it has been
demonstrated that GAA does not affect intestinal fat absorption in
mice with high-fat diet-induced fatty liver (20). Therefore, the beneficial effects of
GAA are not due to changes in intestinal fat absorption.
During the development of NASH, lipid disturbances
can increase inflammation. TNF-α is a key factor in the progression
of NASH, as it induces key molecules in hepatic lipid metabolism
associated with inflammation and fibrosis (31). GAA has an effective
anti-inflammatory effect (33). In
the present study, the data revealed that GAA treatment
significantly suppressed the HFHC diet-induced upregulated
expression of IL-6 and TNFα in the serum and liver of NASH mice. At
present, evidence supports the view that inhibition of IL-6
improves the symptoms of HFHC-induced NASH. For example, it has
been reported that IL-6 inhibition suppresses NASH induced
inflammation and liver damage (34). Consistent with other current
pharmacological studies, resveratrol, a natural product, reduces
liver steatosis and inflammation in NASH mice, indicating a
reduction in serum IL-6 levels (35-37).
The transition from steatosis to steatohepatitis represents an
important step in liver damage progression, which eventually
culminates in hepatic fibrosis and cirrhosis (38). Therefore, the effect of GAA on
liver fibrosis was also investigated. The results demonstrated that
GAA also inhibited the expression of α-SMA, TGF-β and MMP-13
induced by HFHC.
Pathological analysis further confirmed the
anti-inflammatory effect of GAA, reflecting the aggregation of
inflammatory cells; however, the anti-inflammatory mechanism of GAA
is not clear. Recent studies demonstrated that activation of the ER
stress pathway can trigger or aggravate fat accumulation and the
inflammatory response, eventually leading to hepatocyte damage or
even cell death in certain conditions. These are important factors
in the pathogenesis of NASH (39,40).
In the present study, it was found that GAA effectively reduced ER
stress responses induced by the HFHC diet in NASH mice. Meanwhile,
the decrease of GRp78 also decreased the expression of p-JNK,
thereby inhibiting the expression of IL-6 and TNF-α to improve the
inflammatory response, and increasing p-AKT to avoid cell damage.
Evidence has demonstrated the role of GRp78 in promoting cell
survival by activating MAPK (41).
In the present study, GAA largely upregulated p-MAPK levels,
suggesting that GAA may block hepatocyte death. Importantly, a
previous study has shown that ERp57 is a protective factor against
ER stress (42). GAA markedly
upregulated ERp57 levels in the liver tissue of NASH mice, serving
a protective role and indicating that GAA may be associated with
ERp57 and ER stress.
Oxidative stress occurs as a result of the imbalance
between antioxidant response and the pro-oxidation reaction. The
accumulation of excessive free radicals in the body lead to DNA
oxidative damage and the abnormal expression of various cytotoxic
related proteins, which is closely associated the occurrence of
NAFLD (43-45).
In the present study, the effects of GAA on hepatic oxidative
stress were investigated from the two aspects of scavenging and
producing reactive oxygen species. The hepatic level of MDA was
significantly upregulated in HFHC-fed mice and this effect was
reversed in HFHC-fed mice treated with GAA. Conversely, the hepatic
level of SOD was significantly downregulated in HFHC-fed mice and
this effect was reversed in HFHC-fed mice treated with GAA. The
results demonstrated that GAA improved NASH-related oxidative
stress injury by increasing hepatic anti-oxidase activity and
suppressing the activities of free radical generating enzymes.
The present study had certain limitations. For
example, there is no specific drug for the treatment of NASH on the
market. It was therefore difficult to locate a recognized positive
drug as a reference when pharmacodynamic evaluation was conducted.
Therefore, no positive control was used in the experimental design
of the present study. Furthermore, it was identified that GAA
improved HFHC-induced NASH; however, this experimental data was
produced using a mouse model, meaning that it is difficult to
hypothesize whether GAA will exert similar effects in patients with
NASH. The promotion of the clinical pharmacodynamic validation of
GAA requires at least the conduction of pharmacodynamic,
pharmacokinetic and pharmaco-toxicological experiments in a variety
of different animal experiments to ensure the safety and
effectiveness of GAA.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Medical Health
Science and Technology Project of Zhejiang Province (grant no.
2019RC069), the Program of Zhejiang University of traditional
Chinese Medicine (grant no. 2018ZY24) and the Science and
Technology Development Program of Nanjing Medical University (grant
no. 2017NJMU086). The authors alone are responsible for the content
and writing of the paper.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JZ and JJ conceived and designed the current study.
JZ and JD acquired the data. JZ and SL analyzed and interpreted the
data. JZ and JJ confirmed the authenticity of all the raw data. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
All experiments and animal care were conducted in
accordance with the Provision and General Recommendation of Chinese
Experimental Animals Administration Legislation and approved [SYXK
(SU) 2016-0011] by the Science and Technology Department of Jiangsu
Province.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kitade H, Chen G, Ni Y and Ota T:
Nonalcoholic fatty liver disease and insulin resistance: New
insights and potential new treatments. Nutrients.
9(387)2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Milic S, Lulic D and Stimac D:
Non-alcoholic fatty liver disease and obesity: Biochemical,
metabolic and clinical presentations. World J Gastroenterol.
20:9330–9337. 2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Sheka AC, Adeyi O, Thompson J, Hameed B,
Crawford PA and Ikramuddin S: Nonalcoholic steatohepatitis: A
review. JAMA. 323:1175–1183. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Anstee QM, Reeves HL, Kotsiliti E, Govaere
O and Heikenwalder M: From NASH to HCC: Current concepts and future
challenges. Nat Rev Gastroenterol Hepatol. 16:411–428.
2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Younossi Z, Anstee QM, Marietti M, Hardy
T, Henry L, Eslam M, George J and Bugianesi E: Global burden of
NAFLD and NASH: Trends, predictions, risk factors and prevention.
Nat Rev Gastroenterol Hepatol. 15:11–20. 2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Bonora E and Targher G: Increased risk of
cardiovascular disease and chronic kidney disease in NAFLD. Nat Rev
Gastroenterol Hepatol. 9:372–381. 2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Ferguson D and Finck BN: Emerging
therapeutic approaches for the treatment of NAFLD and type 2
diabetes mellitus. Nat Rev Endocrinol. 17:484–495. 2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Buzzetti E, Pinzani M and Tsochatzis EA:
The multiple-hit pathogenesis of non-alcoholic fatty liver disease
(NAFLD). Metabolism. 65:1038–1048. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Horton JD, Goldstein JL and Brown MS:
SREBPs: Activators of the complete program of cholesterol and fatty
acid synthesis in the liver. J Clin Invest. 109:1125–1131.
2002.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Han J and Kaufman RJ: The role of ER
stress in lipid metabolism and lipotoxicity. J Lipid Res.
57:1329–1338. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Yan J and Horng T: Lipid metabolism in
regulation of macrophage functions. Trends Cell Biol. 30:979–989.
2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wachtel-Galor S, Yuen J, Buswell JA and
Benzie IFF: Ganoderma lucidum (Lingzhi or Reishi): A
Medicinal Mushroom. In: Herbal Medicine: Biomolecular and Clinical
Aspects. Benzie IFF and Wachtel-Galor S (eds). 2nd edition. CRC
Press/Taylor & Francis, Boca Raton, FL, 2011.
|
|
13
|
Paterson RR: Ganoderma-a therapeutic
fungal biofactory. Phytochemistry. 67:1985–2001. 2006.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Sliva D: Cellular and physiological
effects of Ganoderma lucidum (Reishi). Mini Rev Med Chem.
4:873–879. 2004.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Tong CC, Choong YK, Mohamed S, Mustapha NM
and Umar NA: Nutrition & Food Science. Efficacy of Ganoderma
lucidum on plasma lipids and lipoproteins in rats fed with high
cholesterol diet. Nutrition Food Sci. 38:229–238. 2008.
|
|
16
|
Wasser SP: Reishi or ling zhi
(Ganoderma lucidum). Encyclopedia Dietary Suppl. 1:603–622.
2005.
|
|
17
|
Kabir Y, Kimura S and Tamura T: Dietary
effect of Ganoderma lucidum mushroom on blood pressure and
lipid levels in spontaneously hypertensive rats (SHR). J Nutr Sci
Vitaminol. 34:433–438. 1988.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Li C, Li Y and Sun HH: New ganoderic
acids, bioactive triterpenoid metabolites from the mushroom
Ganoderma lucidum. Nat Prod Res. 20:985–991. 2006.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Xu JW, Zhao W and Zhong JJ:
Biotechnological production and application of ganoderic acids.
Appl Microbiol Biotechnol. 87:457–466. 2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhu J, Jin J, Ding J, Li S, Cen P, Wang K,
Wang H and Xia J: Ganoderic Acid A improves high fat diet-induced
obesity, lipid accumulation and insulin sensitivity through
regulating SREBP pathway. Chem Biol Interact. 290:77–87.
2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Lang S, Demir M, Martin A, Jiang L, Zhang
X, Duan Y, Gao B, Wisplinghoff H, Kasper P, Roderburg C, et al:
Intestinal virome signature associated with severity of
nonalcoholic fatty liver disease. Gastroenterology. 159:1839–1852.
2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Sun R, Liang H, Guo H, Wang Z and Deng Q:
PMCA4 gene expression is regulated by the androgen receptor in the
mouse testis during spermatogenesis. Mol Med Rep.
23(152)2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ucar F, Sezer S, Erdogan S, Akyol S,
Armutcu F and Akyol OJ: The relationship between oxidative stress
and nonalcoholic fatty liver disease: Its effects on the
development of nonalcoholic steatohepatitis. Redox Rep. 18:127–133.
2013.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Pan K, Jiang Q, Liu G, Miao X and Zhong D:
Optimization extraction of Ganoderma lucidum polysaccharides
and its immunity and antioxidant activities. Int J Biol Macromol.
55:301–306. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Liang C, Tian D, Liu Y, Li H, Zhu J, Li M,
Xin M and Xia J: Review of the molecular mechanisms of Ganoderma
lucidum triterpenoids: Ganoderic acids A, C2, D, F, DM, X and
Y. Eur J Med Chem. 174:130–141. 2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Boh B, Berovic M, Zhang J and Zhi-Bin L:
Ganoderma lucidum and its pharmaceutically active compounds.
Biotechnol Annu Rev. 13:265–301. 2007.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Liu F, Shi K, Dong J, Jin Z, Wu Y, Cai Y,
Lin T, Cai Q, Liu L and Zhang Y: Ganoderic acid A attenuates
high-fat-diet-induced liver injury in rats by regulating the lipid
oxidation and liver inflammation. Arch Pharm Res. 43:744–754.
2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wu GS, Lu JJ, Guo JJ, Li YB, Tan W, Dang
YY, Zhong ZF, Xu ZT, Chen XP and Wang YT: Ganoderic acid DM, a
natural triterpenoid, induces DNA damage, G1 cell cycle arrest and
apoptosis in human breast cancer cells. Fitoterapia. 83:408–414.
2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Lixin X, Lijun Y and Songping H: Ganoderic
acid A against cyclophosphamide-induced hepatic toxicity in mice. J
Biochem Mol Toxicol. 33(e22271)2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Bessone F, Razori MV and Roma MG:
Molecular pathways of nonalcoholic fatty liver disease development
and progression. Cell Mol Life Sci. 76:99–128. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Bala S, Ganz M, Babuta M, Zhuang Y, Csak
T, Calenda CD and Szabo G: Steatosis, inflammasome upregulation,
and fibrosis are attenuated in miR-155 deficient mice in a high
fat-cholesterol-sugar diet-induced model of NASH. Lab Invest.
101:1540–1549. 2021.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ma JQ, Zhang YJ and Tian ZK: Anti-oxidant,
anti-inflammatory and anti-fibrosis effects of ganoderic acid A on
carbon tetrachloride induced nephrotoxicity by regulating the
Trx/TrxR and JAK/ROCK pathway. Chem Biol Interact.
344(109529)2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Ganz M and Szabo G: Immune and
inflammatory pathways in NASH. Hepatol Int. 7 (Suppl 2):S771–S781.
2013.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Heebøll S, Thomsen KL, Clouston A,
Sundelin EI, Radko Y, Christensen LP, Ramezani-Moghadam M,
Kreutzfeldt M, Pedersen SB, Jessen N, et al: Effect of resveratrol
on experimental non-alcoholic steatohepatitis. Pharmacol Res.
95-96:34–41. 2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Ji G, Wang Y, Deng Y, Li X and Jiang Z:
Resveratrol ameliorates hepatic steatosis and inflammation in
methionine/choline-deficient diet-induced steatohepatitis through
regulating autophagy. Lipids Health Dis. 14(134)2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Kessoku T, Imajo K, Honda Y, Kato T, Ogawa
Y, Tomeno W, Kato S, Mawatari H, Fujita K, Yoneda M, et al:
Resveratrol ameliorates fibrosis and inflammation in a mouse model
of nonalcoholic steatohepatitis. Sci Rep. 6(22251)2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Trautwein C, Friedman SL, Schuppan D and
Pinzani M: Hepatic fibrosis: Concept to treatment. J Hepatol. 62
(Suppl 1):S15–S24. 2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Arroyave-Ospina JC, Wu Z, Geng Y and
Moshage H: Role of oxidative stress in the pathogenesis of
non-alcoholic fatty liver disease: Implications for prevention and
therapy. Antioxidants (Basel). 10(174)2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Chen Z, Tian R, She Z, Cai J and Li H:
Role of oxidative stress in the pathogenesis of nonalcoholic fatty
liver disease. Free Radic Biol Med. 152:116–141. 2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Darling NJ and Cook SJ: The role of MAPK
signalling pathways in the response to endoplasmic reticulum
stress. Biochim Biophys Acta. 1843:2150–2163. 2014.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Parakh S, Jagaraj CJ, Vidal M, Ragagnin
AMG, Perri ER, Konopka A, Toth RP, Galper J, Blair IP, Thomas CJ,
et al: ERp57 is protective against mutant SOD1-induced cellular
pathology in amyotrophic lateral sclerosis. Hum Mol Genet.
27:1311–1331. 2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Koruk M, Taysi S, Savas MC, Yilmaz O,
Akcay F and Karakok M: Oxidative stress and enzymatic antioxidant
status in patients with nonalcoholic steatohepatitis. Ann Clin Lab
Sci. 34:57–62. 2004.PubMed/NCBI
|
|
44
|
Sutti S, Jindal A, Locatelli I, Vacchiano
M, Gigliotti L, Bozzola C and Albano E: Adaptive immune responses
triggered by oxidative stress contribute to hepatic inflammation in
NASH. Hepatology. 59:886–897. 2014.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Gabbia D, Cannella L and De Martin S: The
Role of Oxidative Stress in NAFLD-NASH-HCC Transition-Focus on
NADPH Oxidases. Biomedicines. 9(687)2021.PubMed/NCBI View Article : Google Scholar
|