Introduction
Pneumonia is a respiratory disease characterized by
lung inflammation that is mainly caused by pathogens (1). The main features of pneumonia include
fever, cough, dyspnea, somnolence and chest pain, and is a common
disease in children, accounting for 24.5-65.2% of pediatric
inpatients (2-4).
Lipopolysaccharide (LPS), the major bioactive component of the cell
wall, serves a pivotal role in inducing an inflammatory response in
patients with pneumonia (5).
Despite substantial advances in the therapeutic approaches to
pneumonia, further investigation is required (6). Thus, current research should focus on
the mechanisms underlying the inflammatory response and aim to
identify novel treatment options for pneumonia. Emerging evidence
has indicated that inflammation-related proteins, including TNF-α,
IL-1β and IL-6, as well as apoptosis-related proteins Bcl-2 and
Bax, were associated with LPS-induced acute pneumonia (7-10).
Glaucocalyxin A (GLA), an ent-kauranoid diterpene
derived from Rabdosia japonica var. glaucocalyx,
possesses a number of antibacterial, anti-oxidative and
anti-neuroinflammatory properties (11,12).
The results of a previous study revealed the therapeutic potential
of GLA in the treatment of LPS-mediated septic shock and
inflammation, in which GLA suppressed the activation of the NACHT,
LRR and PYD domains-containing protein 3 inflammasome (13). Furthermore, GLA attenuated
myocardial ischemia/reperfusion injury by alleviating microvascular
thrombosis in a mouse model (14).
HY1702, a novel small molecule diterpene acquired from the
modification of GLA, reduced the injury of acute respiratory
distress syndrome in mice induced by LPS (15). GLA also alleviated pulmonary
fibrosis in mice, which improved the survival rates of mice with
this disease (16). The results of
previous studies demonstrated that GLA possessed anti-inflammatory
and anti-oxidative stress effects (17,18).
Furthermore, GLA inhibited the activation of signal transducer and
activator of transcription 3 (STAT3) in osteosarcoma cells, and
STAT3 inhibition alleviated LPS-induced acute lung injury (19-21).
In addition, a previous study demonstrated that STAT3 binds to the
promoter site of activating transcription factor 6 to increase its
expression, thereby inducing endoplasmic reticulum stress (ERS)
(22). Therefore, the present
study aimed to investigate the role of GLA in ERS inhibition
regulated by ATF6 and the effects on STAT3 activity. In addition,
the role of GLA in LPS-induced inflammation and apoptosis in human
pulmonary microvascular endothelial cells (hPMVECs) and endothelial
cell permeability injury was also analyzed.
Materials and methods
Cell lines and treatment
GLA was purchased from MedChemExpress. Immortalized
hPMVECs were obtained from The Cell Bank of Type Culture Collection
of The Chinese Academy of Sciences (https://www.biomart.cn/infosupply/33021544.htm?from=search_1)
and cultured in DMEM, supplemented with 10% FBS and 1%
penicillin/streptomycin (all, Invitrogen; Thermo Fisher Scientific,
Inc.), at 37˚C in an incubator with 5% CO2. LPS
(Sigma-Aldrich; Merk KGaA; resuspended in DMEM) was added to the
cells for cell model establishment, at a concentration of 100 ng/ml
for 24 h. In subsequent experiments, 1.25, 2.5, 5 and 10 µM GLA
(MedChemExpress; resuspended in 100 mg/m DMSO) and 0.5 µM colivelin
(MedChemExpress; resuspended in DMEM), which is an activator of
STAT3, were used for the treatment of the cells (18,23,24).
Cell Counting Kit (CCK)-8 assay
Briefly, hPMVECs were seeded in 96-well plates, at a
density of 3x105 cells/well. After the cells were
treated with GLA, 10 µl CCK-8 reagent (Dojindo Molecular
Technologies, Inc.) was added to the cells and incubated for 4 h,
according to the manufacturer's protocol. Cell viability was
determined by measuring the optical density at 450 nm using a
microplate reader (BioTek Instruments, Inc.).
Reverse transcription-quantitative
(RT-q) PCR analysis
Following LPS and GLA treatment, total RNA was
extracted from the hPMVECs using TRIzol® (Invitrogen;
Thermo Fisher Scientific, Inc.). Single-stranded cDNA was
subsequently synthesized from total RNA using the
PrimeScript™ RT Reagent kit, with gDNA Eraser (cat. no.
RR047A; Takara Biotechnology Co., Ltd.) according to the
manufacturer's protocol. SYBR Green qPCR Master Mix
(MedChemExpress) was used for qPCR. The following thermocycling
conditions were used for qPCR: Initial denaturation for 2 min at
95˚C; followed by 40 cycles at 94˚C for 30 sec, 60˚C for 30 sec and
72˚C for 2 min; and a final extension at 72˚C for 5 min. mRNA
expression levels were determined using the 2-∆∆Cq
method (25) and GAPDH was used as
the internal control for quantitative normalization of mRNA. The
following primers were used: TNF-α forward,
5'-CCTCTCTCTAATCAGCCCTCTG-3' and reverse,
5'-GAGGACCTGGGAGTAGATGAG-3'; IL-1β forward,
5'-ATGATGGCTTATTACAGTGGCAA-3' and reverse,
5'-GTCGGAGATTCGTAGCTGGA-3', IL-6, forward,
5'-ACTCACCTCTTCAGAACGAATTG-3' and reverse,
5'-CCATCTTTGGAAGGTTCAGGTTG-3'; GAPDH forward,
5'-GGAGCGAGATCCCTCCAAAAT-3' and reverse,
5'-GGCTGTTGTCATACTTCTCATGG-3'.
TUNEL assay
A TUNEL assay was used to detect the apoptosis of
cultured hPMVECs. After the cells were fixed with 4%
paraformaldehyde at room temperature for 30 min, a TUNEL assay kit
(Roche Diagnostics GmbH) was used to detect the apoptotic cells
according to the manufacturer's protocol. Apoptotic cells of each
section were stained using 50 µl TUNEL at 37˚C for 60 min.
Following nuclear staining with DAPI (0.1 µg/ml) at room
temperature for 5 min in the dark, Antifade mounting medium was
used to mount the sections (Beyotime Institute of Biotechnology).
Fluorescent images taken at four random fields of view were
captured using an inverted microscope and subsequently analyzed
using ImageJ (v1.38; National Institutes of Health).
Western blot analysis
Total protein was extracted from the hPMVECs using
RIPA Lysis Buffer (Beyotime Institute of Biotechnology) according
to the manufacturer's protocol. Total protein was quantified using
a BCA kit (Beyotime Institute of Biotechnology) and the proteins
(30 µg per lane) were separated using 12% SDS-PAGE. The separated
proteins were subsequently transferred onto PVDF membranes and
blocked with 5% (w/v) skimmed milk for 2 h at room temperature. The
membranes were incubated overnight at 4˚C with the following
primary antibodies at 1:1,000 dilution: Anti-Bcl-2 (cat. no.
ab32124), anti-Bax (cat. no. ab32503), anti-vascular endothelial
(VE)-cadherin (cat. no. ab232880), anti-zona occludens protein-1
(ZO-1; cat. no. ab216880), anti-claudin 5 (cat. no. ab131259),
anti-phosphorylated (p)-STAT3 (cat. no. ab76315), anti-STAT3 (cat.
no. ab68153), anti-ATF6 (a cat. no. b122897), anti-C/EBP-homologous
protein (CHOP; cat. no. ab11419), anti-glucose regulated protein 78
(GRP78; cat. no. ab21685) and anti-GAPDH (cat. no. ab9485) (all
from Abcam). The membranes were subsequently incubated with
horseradish peroxidase-conjugated secondary antibodies (cat. no.
sc-2357; 1:10,000; Santa Cruz Biotechnology, Inc.) for 1 h at room
temperature. GAPDH was used as an internal control. Protein bands
were visualized using ECL (Thermo Fisher Scientific, Inc.).
FITC-dextran assay
The FITC-dextran assay was performed as previously
described (26). Briefly,
monolayers were cultured using the treated hPMVECs with LPS
combined with GLA or GLA and colivelin at 2x105
cells/cm2 at 37°C. Subsequently, 500 µg/ml
FITC-dextran was added to the apical compartment of the transwell
chamber (Corning, Inc.), and 200 µl samples from serum-free DMEM
(lower chamber) were collected 2 h later at 37°C,
following the addition of FITC-dextran. The absorbance was measured
using a microplate reader with excitation at 485 nm and emission at
535 nm.
Statistical analysis
The data are presented as the mean ± SD. The
comparisons between more than two groups were assessed using
one-way ANOVA followed by Tukey's post hoc test, using GraphPad
Prism 5 (GraphPad Software Inc.). Each experiment was repeated at
least three times. P<0.05 was considered to indicate a
statistically significant difference.
Results
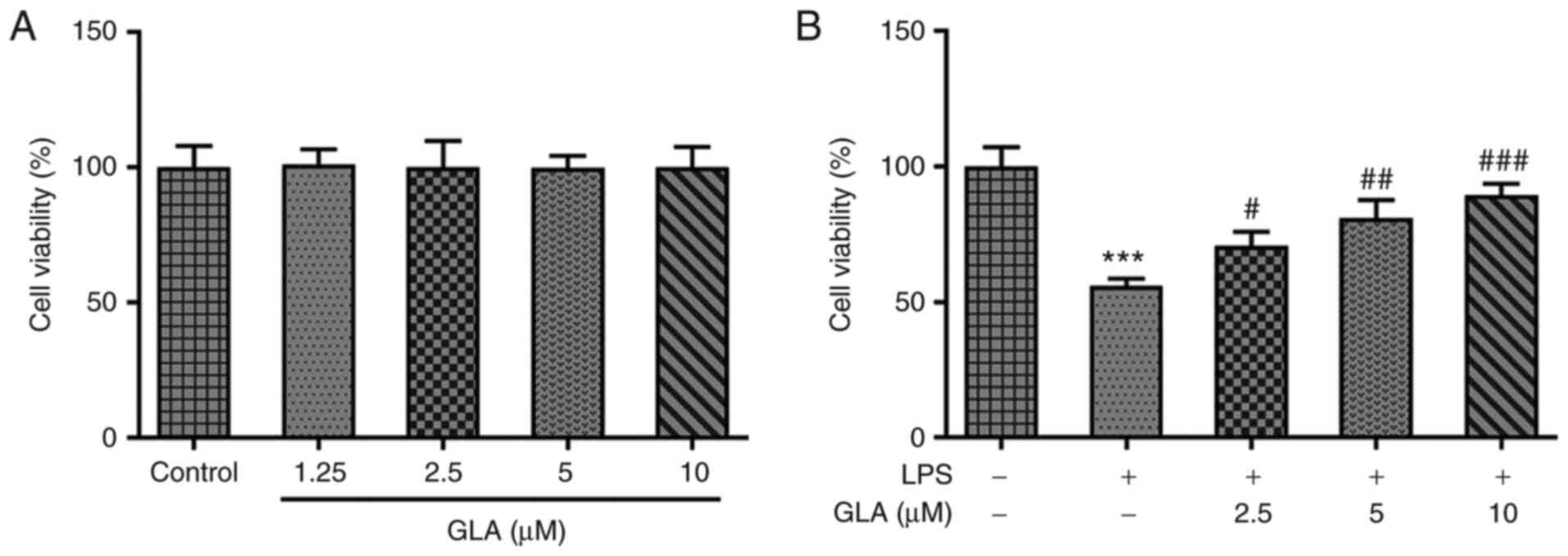
GLA ameliorates LPS-induced hPMVEC
injury
The dysfunction of endothelial cells has been
associated with the pathophysiological consequences of acute lung
injury (27). To investigate the
role of GLA in acute pneumonia, hPMVECs were treated with different
concentrations of GLA to observe the corresponding cytotoxicity. As
exhibited in Fig. 1A, low, medium
and high doses of GLA did not impose cytotoxic effects on hPMVECs.
Thus, 2.5, 5 and 10 µM GLA concentrations were used for subsequent
experiments. In addition, following LPS induction, the viability of
the hPMVECs was reduced to almost half of that in the control
group, whereas this trend was reversed by increasing the dose of
GLA (Fig. 1B).
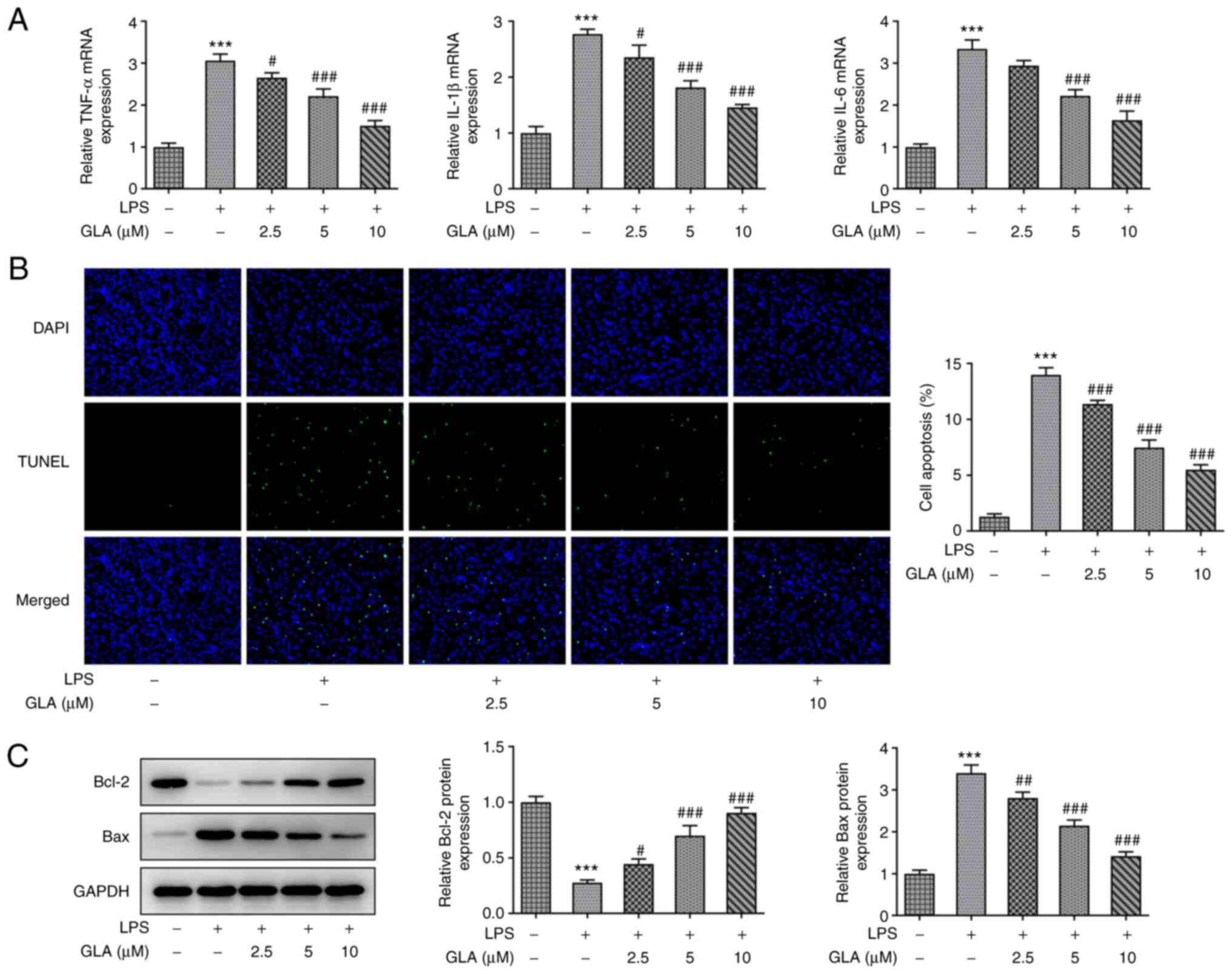
GLA ameliorates inflammation,
apoptosis and permeability injury in LPS-induced hPMVECs
The effect of GLA on inflammation and apoptosis in
LPS-induced hPMVECs was determined. The results of RT-qPCR analysis
revealed that LPS increased the mRNA expression levels of the
inflammatory factors, IL-6, IL-1α and TNF-α, while various doses of
GLA caused a reduction in these expression levels in a
dose-dependent manner (Fig. 2A).
The apoptosis of hPMVECs was significantly increased following
treatment with LPS, and the level of apoptosis was decreased
following GLA treatment (Fig. 2B).
Apoptosis-related Bcl-2 protein was markedly decreased compared
with control, while Bax levels were increased after LPS
stimulation. These effects were considerably reversed by GLA
treatment (Fig. 2C).
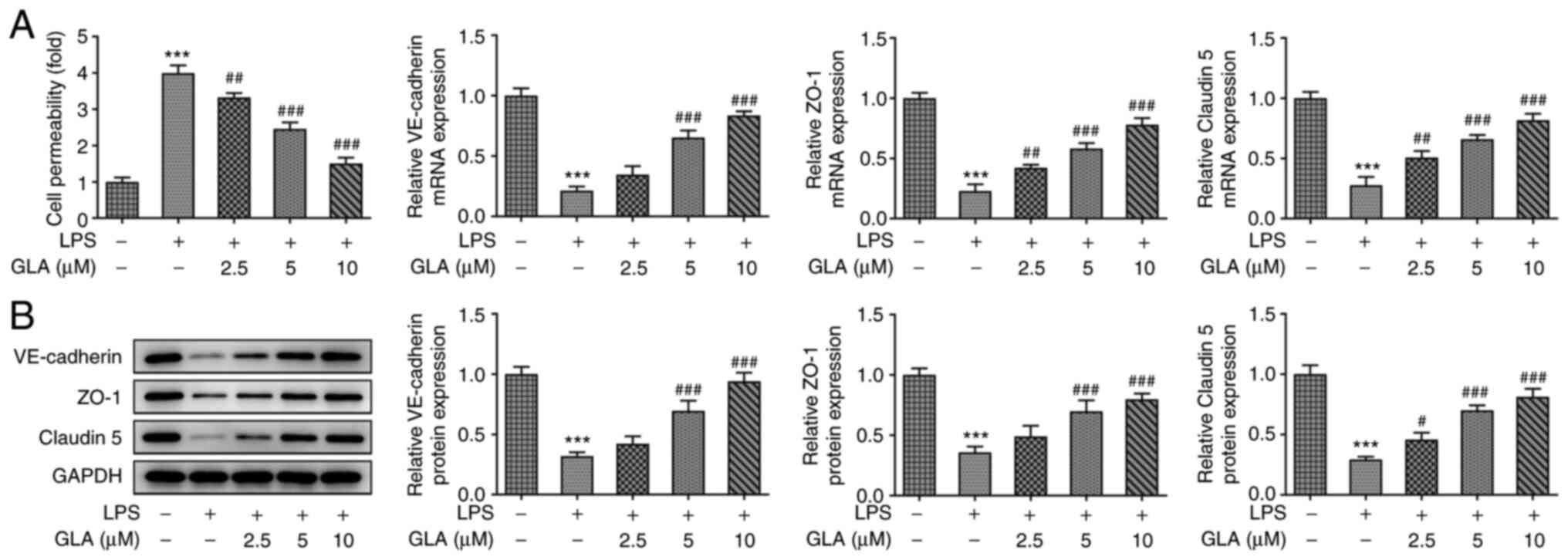
FITC-dextran analysis demonstrated that LPS
increased the permeability of hPMVECs and reduced the mRNA and
protein expression levels of VE-cadherin, ZO-1 and claudin 5. These
effects were reversed following treatment with GLA (Fig. 3A and B).
Collectively, the present results demonstrated that
GLA ameliorated inflammation, apoptosis and dysfunctional
permeability of LPS-induced hPMVECs.
GLA reduces the release of ERS-related
proteins, the inflammation and the apoptosis of LPS-induced hPMVECs
by inhibiting STAT3 signaling
To examine whether the function of STAT3 was
associated with the underlying mechanisms by which GLA exerted
protective effects on LPS-induced hPMVECs, western blot analysis
was conducted. Notably, treatment with LPS increased the protein
expression levels of p-STAT3 and ERS-related proteins (ATF6, CHOP
and GRP78), which were decreased following GLA treatment (Fig. 4A and B).
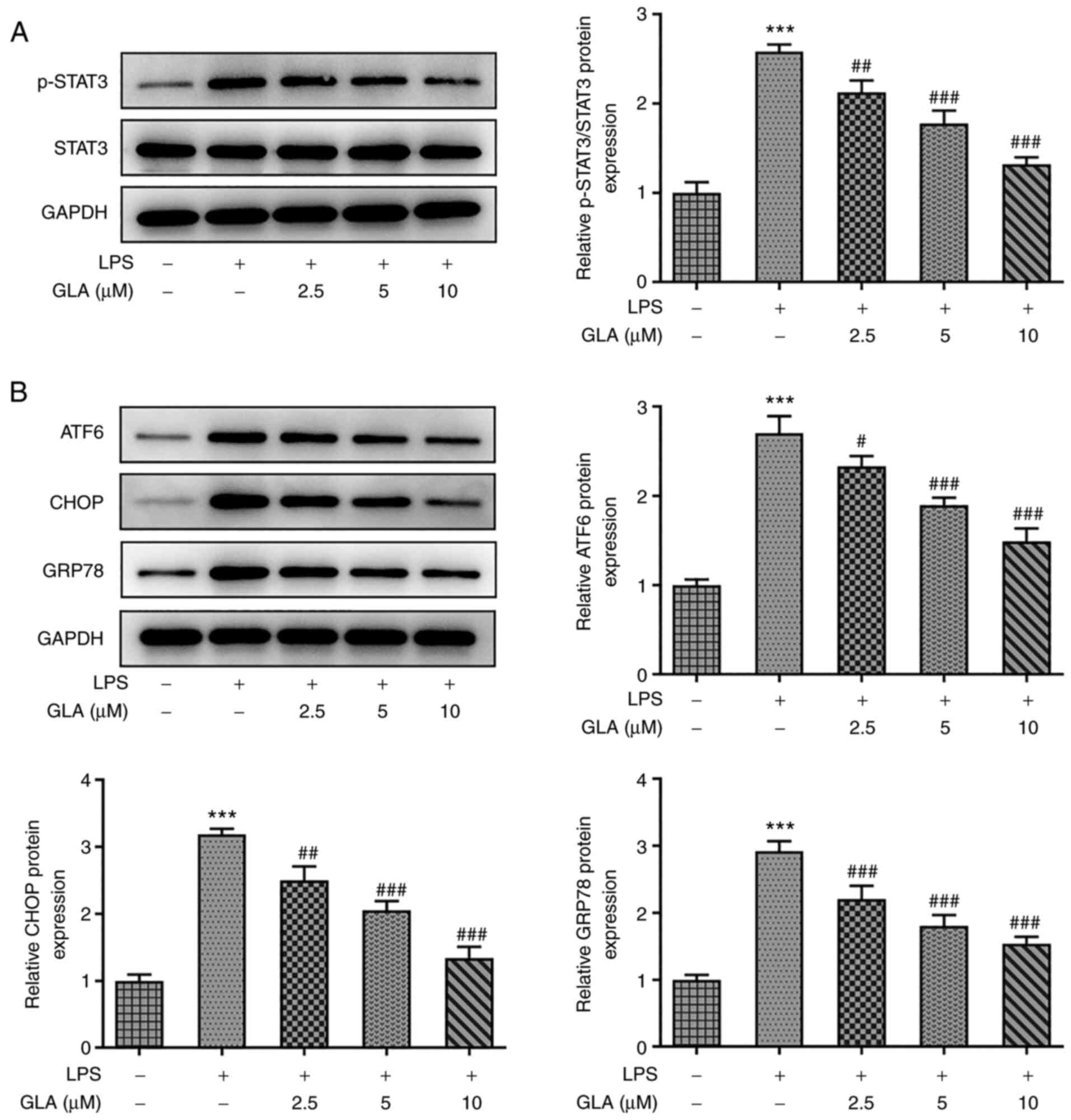 | Figure 4GLA reduces the STAT3-mediated
expression of ERS-related proteins in LPS-induced hPMVECs treated
with GLA. Protein expression level of (A) p-STAT3 and STAT3 and (B)
ERS-related proteins in LPS-induced hPMVECs treated with GLA was
analyzed using western blot analysis. ***P<0.001 vs.
LPS (-) + GLA (µM) (-); #P<0.05,
##P<0.01, ###P<0.00 vs. LPS (+) + GLA
(µM) (-). hPMVECs, human pulmonary microvascular endothelial cells;
GLA, glaucocalyxin A; LPS, lipopolysaccharide; p, phosphorylated,
ERS, endoplasmic reticulum stress; GRP78, glucose regulated protein
78; ATF6, activating transcription factor 6; CHOP, C/EBP-homologous
protein; STAT3, signal transducer and activator of transcription
3. |
However, following treatment with 0.5 µM colivelin
in LPS-induced hPMVECs treated with GLA, the protein expression
levels of p-STAT3, ATF6, CHOP and GRP78 were increased, whereas no
significant changes were observed in the expression levels of total
STAT3 (Fig. 5). The impacts of
colivelin in STAT2 are similar to those reported in a previous
study (28).
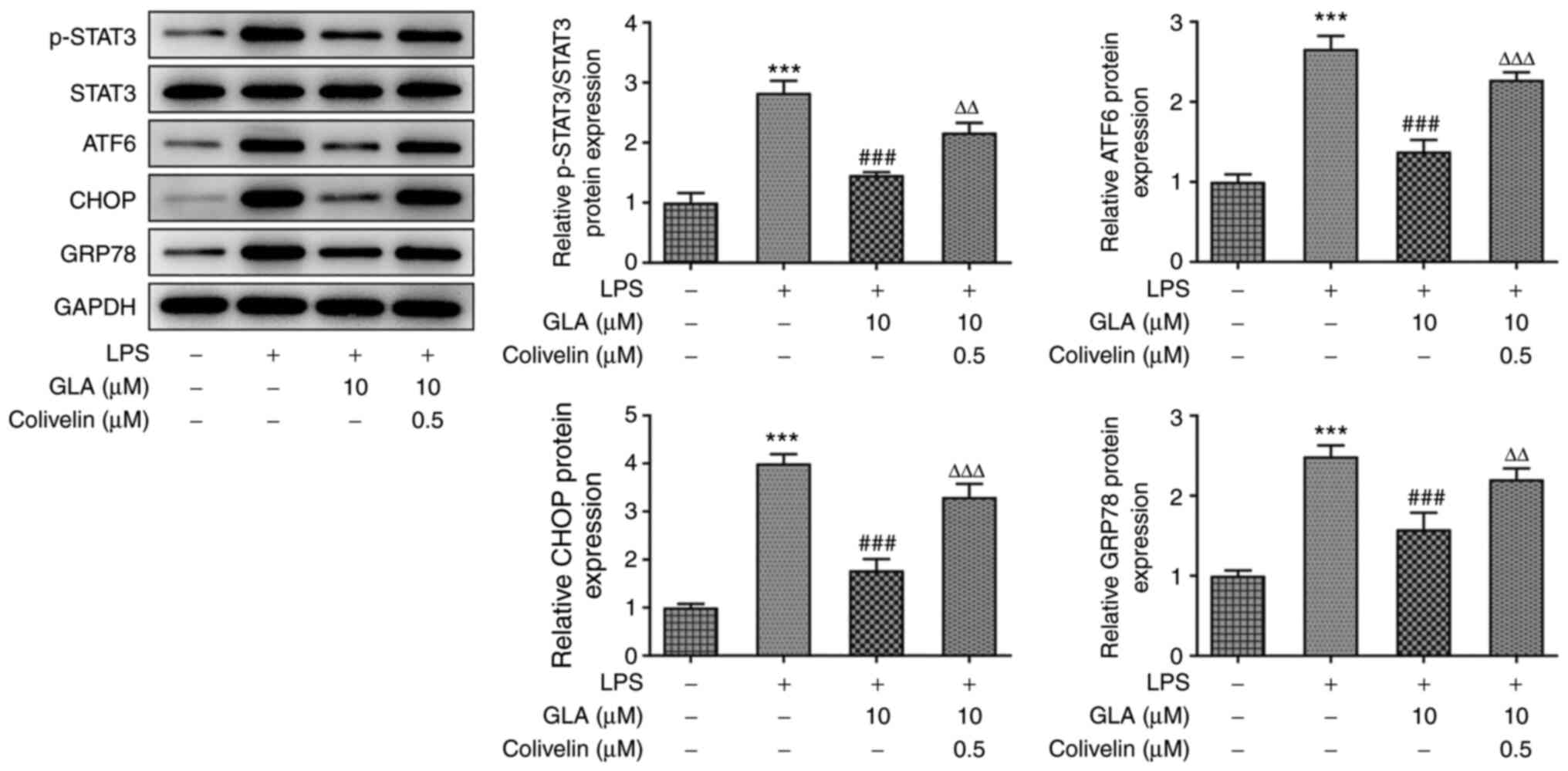 | Figure 5GLA reduces the release of ERS in
LPS-induced hPMVECs by inhibiting STAT3 signal. The protein
expression level of p-STAT3, STAT3 and ERS-related protein
expression in LPS-induced hPMVECs co-treated with GLA and colivelin
was analyzed using western blot analysis and subsequently
quantified using densitometry. ***P<0.001 vs. LPS (-)
+ GLA (µM) (-); ###P<0.001 vs. LPS (+) + GLA (µM) (-)
+ Colivelin (µM) (-); ∆∆P<0.01,
∆∆∆P<0.001 vs. LPS (+) + GLA (µM) (10) + Colivelin (µM) (-). hPMVECs, human
pulmonary microvascular endothelial cells; GLA, glaucocalyxin A;
LPS, lipopolysaccharide; p, phosphorylated, ERS, endoplasmic
reticulum stress; ATF6, activating transcription factor 6; CHOP,
C/EBP-homologous protein; STAT3, signal transducer and activator of
transcription 3; GRP78, glucose regulated protein 78. |
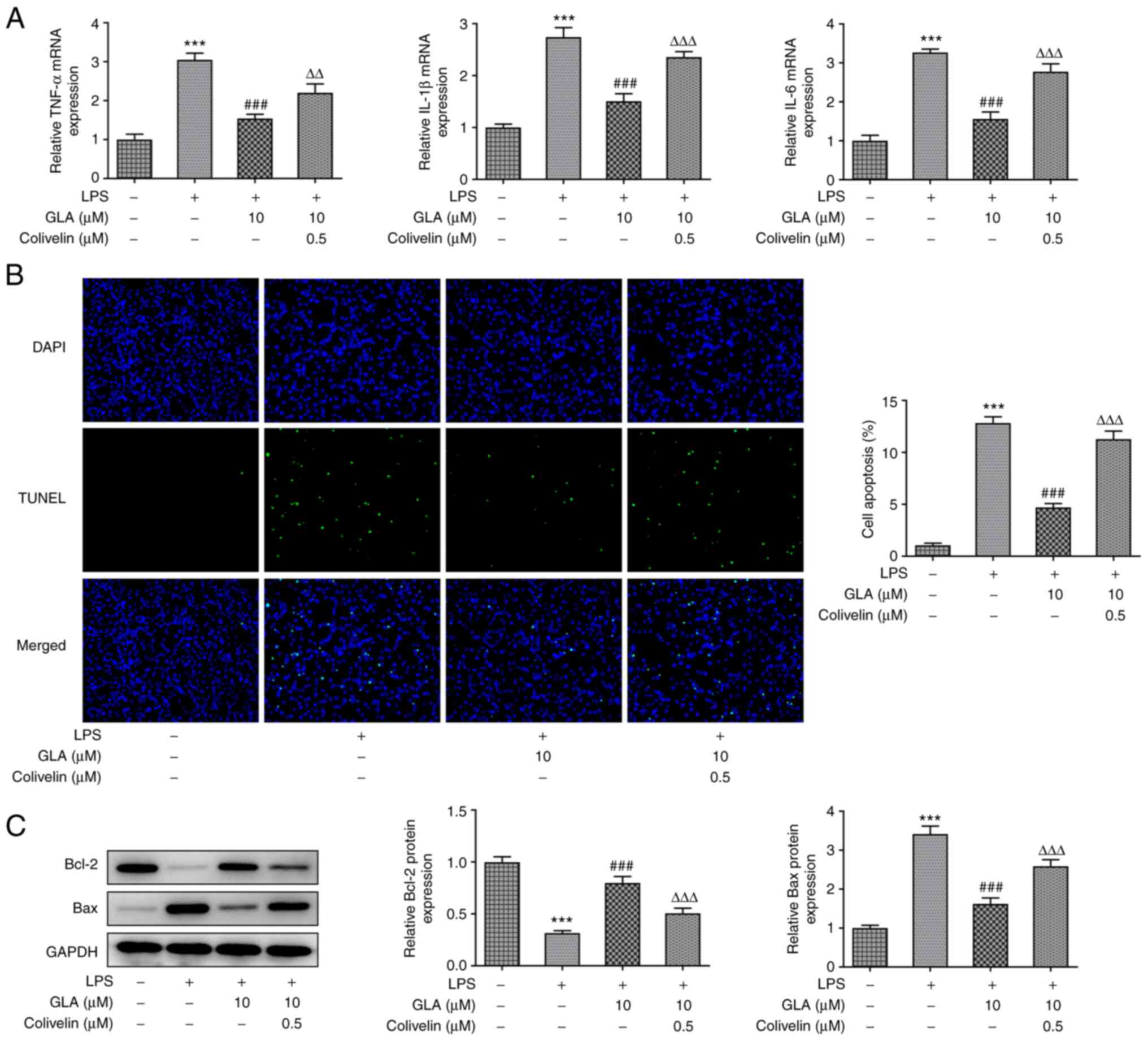
Following colivelin treatment, the mRNA expression
level of inflammatory cytokines (TNF-α, IL-1β and IL-6) were
analyzed by RT-qPCR, while apoptosis was detected by performing a
TUNEL assay. The results revealed that the mRNA expression of
inflammatory cytokines and apoptosis were increased, indicating
that colivelin treatment reversed the suppressive effects of GLA on
inflammation and cell apoptosis (Fig.
6A and B). We also found that
the activator of STAT3 significantly blunted the impacts of GLA in
the expression of Bcl-2 and Bax in LPS-treated hPMVECs (Fig. 6C).
Collectively, the present results demonstrated that
GLA reduced the release of ERS-related proteins in LPS-induced
hPMVECs by inhibiting STAT3 signaling.
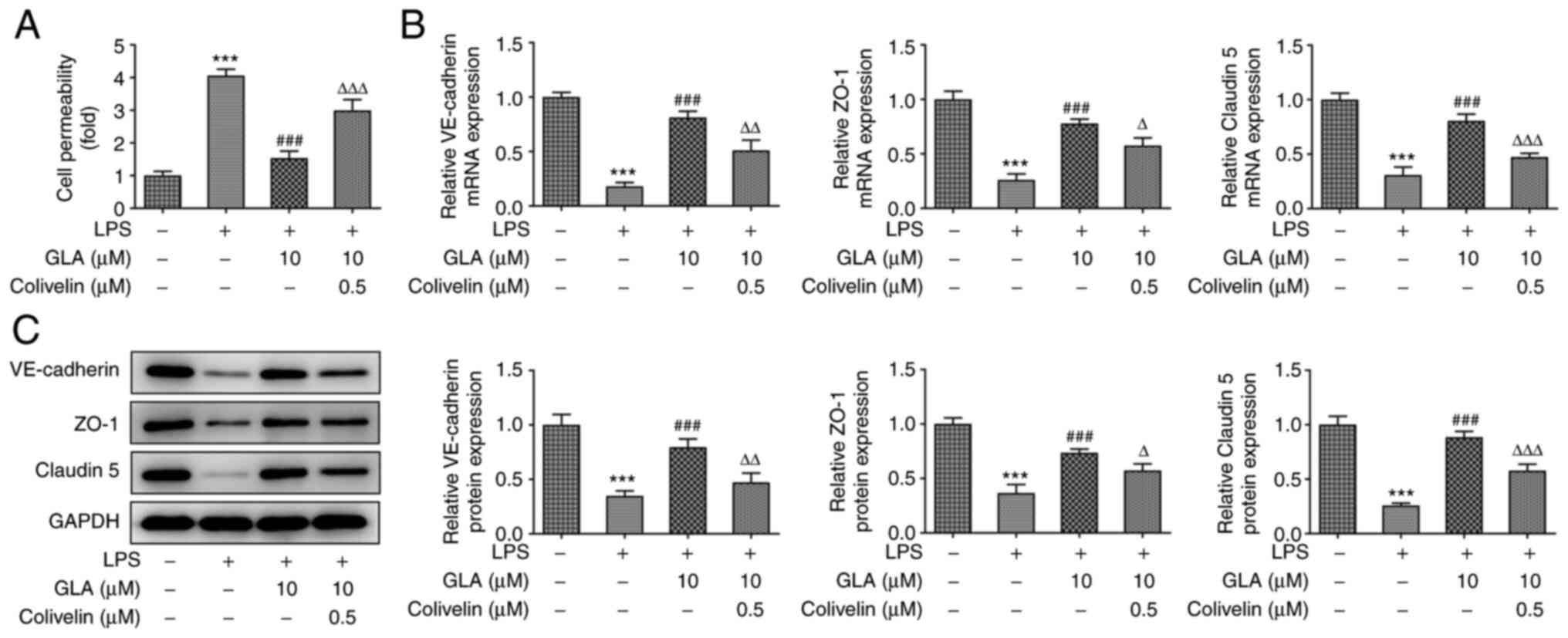
GLA ameliorates the permeability
injury of LPS-induced hPMVECs by inhibiting STAT3 signaling
In order to further investigate the functions of
GLA, FITC-dextran analysis was used to detect the effect of
colivelin on the permeability of LPS-induced hPMVECs treated with
GLA. The results demonstrated that the increased level of cell
permeability in LPS-induced hPMVECs was reduced following treatment
with GLA, whereas treatment with colivelin increased cell
permeability (Fig. 7A).
Furthermore, the mRNA expression levels of VE-cadherin, ZO-1 and
claudin 5 were decreased following treatment with LPS, while the
expression levels of these proteins were increased following GLA
treatment (Fig. 7B). The observed
increase in the mRNA expression levels was partially reversed
following treatment with colivelin. Similar results were found in
the protein expression levels of VE-cadherin, ZO-1 and claudin 5
(Fig. 7C). Collectively, the
present results indicated that GLA ameliorated the permeability
injury of LPS-induced hPMVECs by inhibiting STAT3 signaling.
Discussion
Pneumonia is one of the most infectious diseases
affecting both children and adults, with high prevalence and
morbidity rates (8). A number of
studies have described the pivotal role of inflammatory cytokines
and inflammation in the progression of pneumonia (29,30).
Furthermore, the aggregation of inflammatory cytokines in pneumonia
development induces cell apoptosis and microvascular dysfunction
(31). The results of the present
study initially demonstrated that GLA did not exert cytotoxicity in
hPMVECs. Furthermore, hPMVECs were treated with LPS to simulate
inflammation observed in the development of pneumonia. An increase
in the mRNA expression levels of TNF-α, IL-1βα and IL-6, and an
increase apoptosis were observed in hPMVECs following treatment
with LPS, which is consistent with previous studies (32,33).
Whereas treatment with GLA resulted in decreased inflammation and
apoptosis in LPS-induced hPMVECs, as indicated by the reduced mRNA
expression levels of inflammatory cytokines and a decrease in the
rate of apoptosis. Loss of endothelial barrier integrity is another
key characteristic of pneumonia (34), and the results from the present
study demonstrated that the enhanced endothelial permeability
stimulated by LPS was reversed following treatment with GLA.
STAT3 is a transcription factor that exerts
biological effects in immunity and inflammation (35). The results of a previous study
revealed the participation of STAT3 in bacterial Escherichia
coli pneumonia, as the protein levels of p-STAT3 were markedly
decreased in IL-6-deficient mice, highlighting STAT3 as a potential
therapeutic target for the treatment of this disease (36). Furthermore, a reduction in the
protein levels of STAT3 could aggregate lung injury during
pneumonia (37). These findings
associated STAT3 signaling with the severity and progression of
pneumonia. Another previous study also revealed that the
amelioration of ERS via caffeine treatment mitigated
hyperoxia-induced lung injury (38). In the present study, STAT3 protein
expression level was markedly elevated in response to LPS treatment
for 24 h in hPMVECs, which is consistent with the findings of a
previous study (20). Furthermore,
GLA treatment suppressed the phosphorylation of STAT3 and reduced
the protein expression levels of ERS-associated proteins in the
present study, which further suggests that GLA may alleviate ERS by
regulating STAT3 signaling in pneumonia. To verify this hypothesis,
hPMVECs were treated with colivelin to determine the changes in the
protein expression levels of p-STAT3 and ERS-associated proteins.
The suppressive effects of GLA on both the protein expression
levels of p-STAT3 and ERS-associated proteins were reversed
following treatment with colivelin. STAT3 was also suggested to be
essential for the inflammation and pathogenesis of acute lung
injury by regulating the expression of inflammation- and
immune-related proteins (39). The
present study indicated that the GLA-induced reduction in the mRNA
expression level of inflammatory cytokines was reversed following
treatment with colivelin in LPS-induced hPMVECs, indicating that
STAT3 may indirectly modulate the inflammatory response by
mediating associated cytokines to affect the progression of
pneumonia. In addition, further investigation also confirmed that
the GLA-induced levels of apoptosis and permeability injury in
LPS-induced hPMVECs were reversed following treatment with
colivelin, demonstrating that GLA inhibited apoptosis and
permeability injury in LPS-induced hPMVECs by inhibiting STAT3
signaling. It was reported that the mTOR/STAT3 pathway or the
JAK2/STAT3 signaling pathway was associated with lung injury, which
provides an insight on understanding how GLA regulates STAT3
(40,41), which still requires further
investigation.
In conclusion, GLA alleviated LPS-induced
inflammation, apoptosis and permeability injury of hPMVECs by
inhibiting STAT3 signaling, which revealed the therapeutic value of
GLA in the treatment of pneumonia. The functional effects of GLA in
LPS-induced hPMVECs were explored in vitro, and further
studies should aim to expand these investigations into in
vivo models.
Acknowledgements
No applicable.
Funding
Funding: The present study was supported by Zhongshan Medical
Scientific Research Item (grant no. 2018A020169).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JWC and KJZ made substantial contributions to the
conception and design of the current study. JWC, MLL, SFF and YYL
performed the experiments and collected data. JWC and KJZ
interpreted the data, and drafted and revised the manuscript for
important intellectual content. JWC and KJZ confirm the
authenticity of the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Li W, An X, Fu M and Li C: Emergency
treatment and nursing of children with severe pneumonia complicated
by heart failure and respiratory failure: 10 Case reports. Exp Ther
Med. 12:2145–2149. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Korppi M: Diagnosis and treatment of
community-acquired pneumonia in children. Acta Paediatr.
101:702–704. 2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Simonetti AF, Viasus D, Garcia-Vidal C and
Carratalà J: Management of community-acquired pneumonia in older
adults. Ther Adv Infect Dis. 2:3–16. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Mattila JT, Fine MJ, Limper AH, Murray PR,
Chen BB and Lin PL: Pneumonia. Treatment and diagnosis. Ann Am
Thorac Soc. 11 (Suppl 4):S189–S192. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Dreyfuss D and Ricard JD: Acute lung
injury and bacterial infection. Clin Chest Med. 26:105–112.
2005.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zhang J, Mao F, Zhao G, Wang H, Yan X and
Zhang Q: Long non-coding RNA SNHG16 promotes
lipopolysaccharides-induced acute pneumonia in A549 cells via
targeting miR-370-3p/IGF2 axis. Int Immunopharmacol.
78(106065)2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zeng M, Sang W, Chen S, Chen R, Zhang H,
Xue F, Li Z, Liu Y, Gong Y, Zhang H and Kong X: 4-PBA inhibits
LPS-induced inflammation through regulating ER stress and autophagy
in acute lung injury models. Toxicol Lett. 271:26–37.
2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zhang Y, Zhu Y, Gao G and Zhou Z:
Knockdown XIST alleviates LPS-induced WI-38 cell apoptosis and
inflammation injury via targeting miR-370-3p/TLR4 in acute
pneumonia. Cell Biochem Funct. 37:348–358. 2019.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Zhou Z, Zhu Y, Gao G and Zhang Y: Long
noncoding RNA SNHG16 targets miR-146a-5p/CCL5 to regulate
LPS-induced WI-38 cell apoptosis and inflammation in acute
pneumonia. Life Sci. 228:189–197. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Zang L, Song Y, Yu F and Liu X: Emodin
relieved lipopolysaccharide-evoked inflammatory damage in WI-38
cells by up-regulating taurine up-regulated gene 1. Biofactors.
46:860–868. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Dong Z, Gao Q and Guo H: Glaucocalyxin A
attenuates the activation of hepatic stellate cells through the
TGF-β1/smad signaling pathway. DNA Cell Biol. 37:227–232.
2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Xiang Z, Wu X, Liu X and Jin Y:
Glaucocalyxin A: A review. Nat Prod Res. 28:2221–2236.
2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hou X, Xu G, Wang Z, Zhan X, Li H, Li R,
Shi W, Wang C, Chen Y, Ai Y, et al: Glaucocalyxin A alleviates
LPS-mediated septic shock and inflammation via inhibiting NLRP3
inflammasome activation. Int Immunopharmacol.
81(106271)2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Liu X, Xu D, Wang Y, Chen T, Wang Q, Zhang
J, You T and Zhu L: Glaucocalyxin A ameliorates myocardial
ischemia-reperfusion injury in mice by suppression of microvascular
thrombosis. Med Sci Monit. 22:3595–3604. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wang M, Zhang T, Li L, Xie Q, Wang Y, Li Y
and Chen Z: Protective effects of HY1702 on
lipopolysaccharide-induced mild acute respiratory distress syndrome
in mice. Eur J Pharmacol. 887(173563)2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Yang F, Cao Y, Zhang J, You T and Zhu L:
Glaucocalyxin A improves survival in bleomycin-induced pulmonary
fibrosis in mice. Biochem Biophys Res Commun. 482:147–153.
2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Peng Z, Zhang R, Pan L, Pei H, Niu Z, Wang
H, Lv J and Dang X: Glaucocalyxin A PRotects H9c2 cells against
hypoxia/reoxygenation-induced injury through the activation of
Akt/Nrf2/HO-1 pathway. Cell Transplant.
29(963689720967672)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Zhu S, Zhang J and Lv Y: Glaucocalyxin A
inhibits hydrogen peroxide-induced oxidative stress and
inflammatory response in coronary artery smooth muscle cells. Clin
Exp Pharmacol Physiol. 47:765–770. 2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Mao M, Zhang T, Wang Z, Wang H, Xu J, Yin
F, Wang G, Sun M, Wang Z, Hua Y and Cai Z: Glaucocalyxin A-induced
oxidative stress inhibits the activation of STAT3 signaling pathway
and suppresses osteosarcoma progression in vitro and in vivo.
Biochim Biophys Acta Mol Basis Dis. 1865:1214–1225. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhao J, Yu H, Liu Y, Gibson SA, Yan Z, Xu
X, Gaggar A, Li PK, Li C, Wei S, et al: Protective effect of
suppressing STAT3 activity in LPS-induced acute lung injury. Am J
Physiol Lung Cell Mol Physiol. 311:L868–L880. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wang L, Astone M, Alam SK, Zhu Z, Pei W,
Frank DA, Burgess SM and Hoeppner LH: Suppressing STAT3 activity
protects the endothelial barrier from VEGF-mediated vascular
permeability. bioRxiv: doi: 10.1101/2020.10.27.358374.
|
|
22
|
Meng J, Liu K, Shao Y, Feng X, Ji Z, Chang
B, Wang Y, Xu L and Yang G: ID1 confers cancer cell chemoresistance
through STAT3/ATF6-mediated induction of autophagy. Cell Death Dis.
11(137)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Jiang CQ, Ma LL, Lv ZD, Feng F, Chen Z and
Liu ZD: Polydatin induces apoptosis and autophagy via STAT3
signaling in human osteosarcoma MG-63 cells. J Nat Med. 74:533–544.
2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Qs L, K C, Ap L, F X, Qw H, Z L, Qh Y, YI
W, Zz Z and J Z: Roles of M3 receptor in the effect of
penehyclidine hydrochloride upregulated beta-arrestin-1 expression
in LPS-stimulated HPMVEC. J Recept Signal. 39:39–44.
2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Vallverdú J, Martínez García de la Torre
RA, Mannaerts I, Verhulst S, Smout A, Coll M, Ariño S, Rubio-Tomás
T, Aguilar-Bravo B, Martínez-Sánchez C, et al: Directed
differentiation of human induced pluripotent stem cells to hepatic
stellate cells. Nat Protoc. 16:2542–2563. 2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Zhou Y, Li P, Goodwin AJ, Cook JA,
Halushka PV, Chang E, Zingarelli B and Fan H: Exosomes from
endothelial progenitor cells improve outcomes of the
lipopolysaccharide-induced acute lung injury. Crit Care.
23(44)2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Fang YY and Zhang JH: MFG-E8 alleviates
oxygen-glucose deprivation-induced neuronal cell apoptosis by STAT3
regulating the selective polarization of microglia. Int J Neurosci.
131:15–24. 2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Mizgerd JP: Inflammation and pneumonia:
Why are some more susceptible than others? Clin Chest Med.
39:669–676. 2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Jae SY, Heffernan KS, Kurl S, Kunutsor SK,
Kim CH, Johnson BD, Franklin BA and Laukkanen JA: Cardiorespiratory
fitness, inflammation, and the incident risk of pneumonia. J
Cardiopulm Rehabil Prev. 41:199–201. 2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Redente EF, Keith RC, Janssen W, Henson
PM, Ortiz LA, Downey GP, Bratton DL and Riches DW: Tumor necrosis
factor-α accelerates the resolution of established pulmonary
fibrosis in mice by targeting profibrotic lung macrophages. Am J
Respir Cell Mol Biol. 50:825–837. 2014.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Jiang Z, Shen J, Ding J, Yuan Y, Gao L,
Yang Z and Zhao X: USP18 mitigates lipopolysaccharide-induced
oxidative stress and inflammation in human pulmonary microvascular
endothelial cells through the TLR4/NF-κB/ROS signaling. Toxicol In
Vitro. 75(105181)2021.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhang D, Zhou J, Ye LC, Li J, Wu Z, Li Y
and Li C: Autophagy maintains the integrity of endothelial barrier
in LPS-induced lung injury. J Cell Physiol. 233:688–698.
2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Wang T, Yegambaram M, Gross C, Sun X, Lu
Q, Wang H, Wu X, Kangath A, Tang H, Aggarwal S and Black SM: RAC1
nitration at Y32 IS involved in the endothelial barrier
disruption associated with lipopolysaccharide-mediated acute lung
injury. Redox Biol. 38(101794)2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Lee H, Jeong AJ and Ye SK: Highlighted
STAT3 as a potential drug target for cancer therapy. BMB Rep.
52:415–423. 2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Jones MR, Quinton LJ, Simms BT, Lupa MM,
Kogan MS and Mizgerd JP: Roles of interleukin-6 in activation of
STAT proteins and recruitment of neutrophils during Escherichia
coli pneumonia. J Infect Dis. 193:360–369. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
37
|
Quinton LJ, Jones MR, Robson BE, Simms BT,
Whitsett JA and Mizgerd JP: Alveolar epithelial STAT3, IL-6 family
cytokines, and host defense during Escherichia coli
pneumonia. Am J Respir Cell Mol Biol. 38:699–706. 2008.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Teng RJ, Jing X, Michalkiewicz T, Afolayan
AJ, Wu TJ and Konduri GG: Attenuation of endoplasmic reticulum
stress by caffeine ameliorates hyperoxia-induced lung injury. Am J
Physiol Lung Cell Mol Physiol. 312:L586–L598. 2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Severgnini M, Takahashi S, Rozo LM, Homer
RJ, Kuhn C, Jhung JW, Perides G, Steer M, Hassoun PM, Fanburg BL,
et al: Activation of the STAT pathway in acute lung injury. Am J
Physiol Lung Cell Mol Physiol. 286:L1282–L1292. 2004.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Meng SS, Guo FM, Zhang XW, Chang W, Peng
F, Qiu HB and Yang Y: mTOR/STAT-3 pathway mediates mesenchymal stem
cell-secreted hepatocyte growth factor protective effects against
lipopolysaccharide-induced vascular endothelial barrier dysfunction
and apoptosis. J Cell Biochem. 120:3637–3650. 2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhang L, Lu P, Guo X, Liu T, Luo X and Zhu
YT: Inhibition of JAK2/STAT3 signaling pathway protects mice from
the DDP-induced acute kidney injury in lung cancer. Inflamm Res.
68:751–760. 2019.PubMed/NCBI View Article : Google Scholar
|