Introduction
O'Donnel-Luria-Rodan (ODLURO) syndrome was first
reported in 2019 in 30 patients, with the majority of patients
being under the age of 10 years. To date, there have been 38
reported cases worldwide (1). It is
a neurodevelopmental disorder that leads to delayed development,
low intelligence quotient (IQ), speech delay, autism spectrum
disorders, anxiety and seizures. Other manifestations include
hypotonia, feeding difficulties and mild physical deformities
(2). The inheritance of ODLURO is
autosomal dominant, although the majority of cases appear de
novo (3). The clinical
manifestations develop at an early age and tend to have variable
expressivity that is primarily dependent on the biological sex of
the patient (4).
The lysine methyltransferase 2E (inactive) (KMT2E)
gene encodes a member of the lysine N-methyltransferase-2 family, a
group of enzymes that serve an important role in chromatin
remodeling through transcriptional regulation. ODLURO is caused by
mutations in the KMT2E gene that result in pathogenic enzyme
variants responsible for regulating histone 4 methylation on lysine
3 (H3K4). Lack of proper methylation leads to abnormalities in the
molecular activity of neurons. As methylation maintains the open
chromatin state, any dysregulation affects proper transcription
regulation. H3K4 methylation undergoes dynamic changes during the
neurodevelopmental phase, impacting both the environment of neural
and glial cells in the brain and the molecular activity of the
neurons (5,6).
O'Donnell-Luria identified 31 different mutations in
the heterozygous state for the KMT2E gene in a group of 34
individuals with ODLURO syndrome (OMIM 618512) (4). All patients received clinical and
molecular evaluation via exome or genome sequencing through a
collaboration of four research centers. The majority of the
identified mutations were consistent with haploinsufficiency due to
truncated proteins, except four patients with missense mutations
affecting highly conserved residues. As expected, the majority of
the mutations occurred de novo, dominant variant inheritance was
observed in three affected siblings who may have inherited the
variant from the affected father. Functional studies of the
identified variants were not performed, but the authors speculated
that the pathogenic mechanism linked to altered KMT2E binding
function was the result of haploinsufficiency.
Case report
The present study reports the case of a 5-year-old
male patient diagnosed with ODLURO syndrome at the age of 4 years,
who was the first reported patient with ODLURO in Romania. The
patient was the only child of non-consanguineous parents and the
family history was negative. The patient was born at term after an
uncomplicated pregnancy and delivery, and displayed normal
parameters at birth.
Gross motor development was delayed, as the patient
began sitting at 7 months and walking at 15 months of age. Verbal
skill communication was never acquired and the mother noticed
behavioral anomalies, including avoidance of gaze, retreating into
repetitive, self-centered games, and no interaction with other
children of his age. After the age of 2 years, the patient was
consulted by several pediatric neurologists and psychiatrists, but
in the absence of specific magnetic resonance imaging anatomical
and electrophysiological functional abnormalities of the CNS, the
patient was diagnosed with an idiopathic autistic spectrum
disorder. At the age of 5 years, the patient was referred for
medical genetic assessment. Overall development showed the-standard
deviation values of height and weight for the age. The patient
recognized only a few basic, short words, but could not reproduce
them or verbally communicate in any way. The patient's father also
faced challenges with social interactions, living isolated from the
family and displaying autistic behaviors, but had not undergone a
psychological evaluation.
Intellectual disability was objectified by the
psychologist using specific evaluation tools, and the patient
presented with an IQ of 52. The patient also displayed the
following characteristics: Self-isolation, repetitive and unusual
body movements, paroxysmal crying, reduced attention span to 6-7
sec and difficulty making eye contact. All of these, in addition
with the presentations of anxiety and a tendency to self-harm
indicated an autism spectrum disorder.
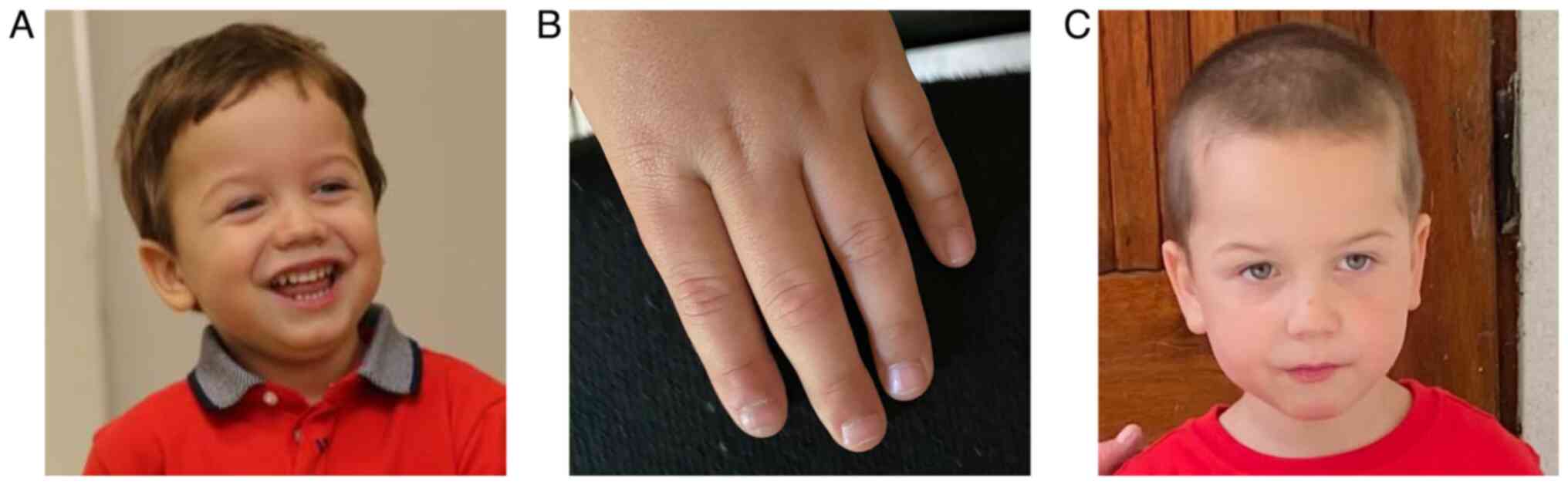
The patient had light, sensitive skin, blonde hair,
blue eyes, and presented macrocephaly and mild facial dysmorphia,
including antimongoloid slanting palpebral fissures, prominent
nasolabial folds and frontal bossing. Another interesting finding
was the narrowing of the distal phalanx of the patient's fingers
(Fig. 1).
Among the most debilitating symptoms, severe
gastroesophageal reflux with recurrent nausea, vomiting and
abdominal pain was reported. Nutrigenomic testing was recommended,
and the results revealed a tendency for compulsive eating
behaviors, especially for calorie-rich foods. Moreover, the
specific HLA DQ 2.5 haplotype defines a predisposition to
developing celiac disease due to gluten intolerance.
Due to the heterogeneous, non-specific clinical
manifestations, whole-exome sequencing (WES) was performed with
parental informed consent. The investigator reported a heterozygous
mutation in the KMT2E gene (KMT2E NM_018682.3: c.498-11T>C),
which was characterized by a deletion that resulted in the
inclusion of a pathogenic exon in the mature mRNA, thus altering
the structure and function of the synthesized polypeptide. Targeted
genetic testing was performed in both parents, and the variant was
identified in the father (Table
I).
 | Table IClinical features of the presented
patient in comparison with other reported patients with ODLURO
syndrome (12-14). |
Table I
Clinical features of the presented
patient in comparison with other reported patients with ODLURO
syndrome (12-14).
| Clinical features in
ODLURO syndrome | Manifestations in
previously reported cases | Manifestations in the
presented case |
|---|
| Facial dysmorphic
features | Dolichocephaly, large
forehead, deep-set eyes, antimongoloid palpebral fissures,
periorbital fullness, prominent cheeks and prominent nasolabial
folds | Macrocephaly, frontal
bossing, antimongoloid palpebral fissures, prominent cheeks and
prominent nasolabial folds |
| Height, weight, and
developmental abnormalities | Short stature and
delayed development | Delayed physical
development |
| Osteoarticular
abnormalities | Tapering fingers | Tapering fingers |
| Neurological and
psychiatric involvement | Hypotonia, seizures,
intellectual disability, delayed speech, anxiety and autism | Autism, intellectual
disability, lack of verbal communication, anxiety and eating
disorders |
| Cardiovascular
involvement | Septal defects | N/A |
| Gastroenterological
involvement | Nausea, vomiting,
motility disorder and gastroesophageal reflux | Nausea, vomiting,
motility disorder and gastroesophageal reflux |
| Immunological
involvement | N/A | Positive for HLA DQ
2.5, which results in a predisposition to developing celiac
disease |
Discussion
ODLURO is a newly defined genetic condition, and as
with most rare genetic diseases, there is little information in the
literature on its etiopathogenesis. The authors who defined this
syndrome revealed an association between KMT2E gene mutations and a
clinical phenotype with several common elements, including
neurodevelopmental delay with autistic behavior and intellectual
disability, non-specific facial dysmorphia and various functional
abnormalities (4).
The patient reported in the present study was a
5-year-old male who was diagnosed with ODLURO based on clinical
manifestations and a heterozygous mutation in the KMT2E gene (KMT2E
NM_018682.3: c.498-11T>C), which is currently classified as a
variant of uncertain significance (VUS) due to the lack of reported
cases with this genetic variant in the medical literature related
to ODLURO. The same variant was identified in the patient's father,
who also presented with psychiatric symptoms, but not to the same
severity as the patient. In this case, VUS management and
evaluating a possible etiopatogenic link with the phenotype was
challenging. The label VUS emerges if no reports connect the
variant to the specific disease. Also, if a variant is extremely
rare, it might take time to conduct comprehensive studies and
catalog enough cases that associate it with a medical condition.
The arguments that the identified variant may be pathogenic are due
to its molecular characteristics; According to the investigator,
in silico analysis shows that the mutation is predicted to
disrupt the highly conserved acceptor splice site and thus may have
a negative effect on the phenotype, as it alters the AG-3' intronic
acceptor site, with the synthesis of an incomplete mRNA and
defective unstable polypeptide synthesis (7).
Although the majority of cases of ODLURO are a
consequence of a de novo mutation, inheritance was identified in a
family with three affected siblings (8) Parental WES analysis identified the
same variant in the father. Interestingly, the father presented
with autistic-like behavior and his wife stated that he has a
childhood history for a psychiatric disorder, but could not provide
details related to the diagnosis. Phenotypic differences may be
explained by incomplete penetrance and variable expressivity, an
already well-known aspect for dominant inheritance (9).
Autism spectrum disorder is common in patients with
ODLURO, especially in males, together with speech delay,
intellectual disability, anxiety and aggressive behavior (8). The KMT2E gene is known to serve an
important role in neurodevelopment, and mutations in this gene have
been linked to several cases of epilepsy, autism spectrum disorder,
intellectual disability and schizophrenia (10). The patient described in the present
study also presented a complete lack of verbal communication, as
well as delayed speech.
Facial dysmorphic features like dolichocephaly,
large forehead, deep-set eyes, antimongoloid palpebral fissures,
periorbital fullness, prominent cheeks and prominent nasolabial
folds have been described in patients with ODLURO (11). The patient described in the present
study presented with macrocephaly, frontal bossing, antimongoloid
palpebral fissures, prominent cheeks and prominent nasolabial
folds.
Gastrointestinal symptoms have also been reported in
numerous patients with ODLURO (4).
However, predisposition to developing coeliac disease and being
positive for HLA DQ 2.5 have not yet been reported in correlation
with ODLURO, and might be independent traits. Although eating
disorders, such as compulsive overeating, have not previously been
reported to be linked to ODLURO, the patient reported in the
present study required nutritional and psychological guidance for
this condition to restore proper nutritional values.
In conclusion, based on the aforementioned genetic
findings and taking into consideration the phenotype of the
patient, the c.498-11T>C, KMT2E VUS was a potential cause for
the patient's symptoms. However, further studies are required to
confirm the pathogenicity of the variant. It appeared that the
mutation was inherited from the father and decreased penetrance may
explain the milder phenotype, restricted to autistic-like behavior,
therefore further functional and clinical studies are necessary and
will allow precise pinpointing of the pathologic significance of
this variant. The case reported in the present study is an example
of rare disease challenges for diagnostic uncertainty when
considering the unknown clinical significance of a specific
variant. Until further evidence for the identified variant has been
identified, the case reported in the present study should remain in
focus as the first case of ODLURO diagnosed in Romania.
ODLURO is a rare neurodevelopmental disorder caused
by a germline mutation in the KMT2E gene that is primarily de
novo, but also inheritable. Common symptoms include delayed
development, low IQ, poor verbal communication, autism spectrum
disorders, anxiety and seizures. Other manifestations include
hypotonia, feeding difficulties and mild physical deformities. The
present study reported a 5-year-old male patient who presented with
macrocephaly and mild facial dysmorphia, including antimongoloid
slanting palpebral fissures, prominent nasolabial folds and frontal
bossing, tapering fingers, and severe gastroesophageal reflux with
recurrent nausea, vomiting and abdominal pain in association with
KMT2E NM_018682.3: c.498-11T>C, heterozygous variant, with
gluten intolerance as an atypical finding.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
AC and MSM conducted the genetic consult and
counselling. AC, ZCB, EK, DM and II interpreted the genetic test
results and confirmed the authenticity of all the raw data. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
The patient's family provided consent for
publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Omim.org: OMIM Entry-# 618512-
O'Donell-Luria-Rodan Syndrome; ODLURO. https://www.omim.org/entry/618512> Accessed August
3, 2021.
|
|
2
|
Velmans C, O'Donnell-Luria AH, Argilli E,
Tran Mau-Them F, Vitobello A, Chan MC, Fung JL, Rech M, Abicht A,
Aubert Mucca M, et al: O'Donnell-Luria-Rodan syndrome: Description
of a second multinational cohort and refinement of the phenotypic
spectrum. J Med Genet, 2021 (Epub ahead of print). doi:
10.1136/jmedgenet-2020-107470.
|
|
3
|
Zhang X, Novera W, Zhang Y and Deng LW:
MLL5 (KMT2E): Structure, function, and clinical relevance. Cell Mol
Life Sci. 74:2333–2344. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
O'Donnell-Luria AH, Pais LS, Faundes V,
Wood JC, Sveden A, Luria V, Abou Jamra R, Accogli A, Amburgey K,
Anderlid BM, et al: Heterozygous variants in KMT2E cause a spectrum
of neurodevelopmental disorders and epilepsy. Am J Hum Genet.
104:1210–1222. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Shen E, Shulha H, Weng Z and Akbarian S:
Regulation of histone H3K4 methylation in brain development and
disease. Philos Trans R Soc Lond B Biol Sci.
369(20130514)2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Li Y, Fan L, Luo R, Yang Z, Yuan M, Zhang
J and Gan J: Case Report: De novo variants of KMT2E cause
O'Donnell-Luria-Rodan syndrome: Additional cases and literature
review. Front Pediatr. 9(641841)2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Dong S, Walker MF, Carriero NJ, DiCola M,
Willsey AJ, Ye AY, Waqar Z, Gonzalez LE, Overton JD, Frahm S, et
al: De novo insertions and deletions of predominantly paternal
origin are associated with autism spectrum disorder. Cell Rep.
9:16–23. 2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Schell R, Martin N Mullis, Takeshi Matsui,
Ryan Foree and IM Ehrenreich: Genetic basis of a spontaneous
mutation’s expressivity. BioRxiv, 2020.
doi:10.1101/2020.04.03.024547.
|
|
9
|
Cooper DN, Krawczak M, Polychronakos C,
Tyler-Smith C and Kehrer-Sawatzki H: Where genotype is not
predictive of phenotype: Towards an understanding of the molecular
basis of reduced penetrance in human inherited disease. Hum Genet.
132:1077–1130. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Shulha HP, Cheung I, Whittle C, Wang J,
Virgil D, Lin CL, Guo Y, Lessard A, Akbarian S and Weng Z:
Epigenetic signatures of autism: TrimethylatedH3K4landscapes in
prefrontal neurons. Arch Gen Psychiatry. 69:314–324.
2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zech M, Boesch S, Maier EM, Borggraefe I,
Vill K, Laccone F, Pilshofer V, Ceballos-Baumann A, Alhaddad B,
Berutti R, et al: Haploinsufficiency ofKMT2B, encoding the
lysine-specific histone methyltransferase 2B, results in
early-onset generalized dystonia. Am J Hum Genet. 99:1377–1387.
2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Sharawat IK, Panda PK and Dawman L:
Clinical characteristics and genotype-phenotype correlation in
children with KMT2E gene-related neurodevelopmental disorders:
Report of two new cases and review of published literature.
Neuropediatrics. 52:98–104. 2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Conforti R, Iovine S, Santangelo G,
Capasso R, Cirillo M, Fratta M and Caranci F: ODLURO syndrome:
Personal experience and review of the literature. Radiol Med.
126:316–322. 2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Bosemani T, Orman G, Boltshauser E, Tekes
A, Huisman TA and Poretti A: Congenital abnormalities of the
posterior fossa. Radiographics. 35:200–220. 2015.PubMed/NCBI View Article : Google Scholar
|