Introduction
Diabetic retinopathy (DR) is considered as an
important risk factor for ablepsia in developed countries.
Mitochondrial dysfunction and hyperglycemia-mediated excessive
apoptosis of microvascular cells are the most critical causes of
diabetic microangiopathy. In addition, mitochondria play a key role
in the vascular system via regulation of cellular energy
production, and in the microvascular system via regulation of ATP
formation and superoxide production (1). In particular, it has been reported
that reactive oxygen species (ROS) are involved in cellular
dysfunction such as apoptosis (2).
It has been suggested that the hyperglycemia-induced production of
mitochondrial ROS can be a key factor in initiating pathogenic
signaling in DR (3). The
aforementioned studies indicated that mitochondrial function and
ROS could play a critical role in DR. Therefore, maintaining
mitochondrial function and attenuating ROS production in retinal
endothelial cells could be considered a significant approach for
treating DR.
Prohibitin (PHB) is a negative regulator of
mitochondrial ROS production (4).
In addition, PHB, a conserved mitochondrial chaperone, plays a
significant role in maintaining normal mitochondrial development
and the subunits of mitochondrial proteins (5). A previous study showed that PHB could
not only be translocated into the mitochondria, but also to other
cellular areas, including the plasma and nucleus, and could
therefore be involved in other cellular processes, including
apoptosis and growth (6).
Furthermore, previous studies demonstrated that PHB exerted a key
role in cellular oxidative homeostasis and could protect intestinal
epithelial cells and cardiomyocytes against oxidative stress, thus
acting as potent antioxidant (7,8).
However, the exact mechanism underlying the regulatory effect of
PHB on retinal vascular cells in DR remains elusive.
Therefore, the present study aimed to investigate
the expression levels of PHB in mitochondria of human retinal
capillary endothelial cells (HRCECs) to further evaluate its effect
on regulating mitochondrial function, ROS generation and high
glucose (HG)-induced apoptosis, thus supporting its critical role
in the development of DR.
Materials and methods
Reagents
CM-H2-DCFDA, JC-1, rotenone, antimycin-A
and both reduced and non-reduced Mito Tracker were obtained from
Invitrogen (Thermo Fisher Scientific, Inc.). The antibodies against
PHB1 and cyclooxygenase (COX) IV were purchased from Abcam, while
the Annexin V-FITC apoptosis kit was obtained from Becton,
Dickinson and Company. The antibodies against β-actin, cleaved
poly(ADP-ribose) polymerase (PARP) and cleaved caspase-3 were
obtained from Cell Signaling Technology, Inc. and DAPI from
MilliporeSigma.
HRCEC culture and treatment
HRCECs, obtained from the BeNa Culture Collection
(Shanghai, China), were cultured in Endothelial Cell Medium (ECM;
ScienCell Research Laboratories) supplemented with 5% FBS (Gibco;
Thermo Fisher Scientific, Inc.) in 5% CO2. The cells
were used for subsequent experiments after 4-5 passages. When cells
reached 50-60% confluence, they were treated with 5.5 mmol/l
[normal glucose (NG)] or 30 mmol/l glucose (HG) for 4-6 days.
Subsequently, the cells were divided into the following four
groups: The NG group; the HG group; the HG + negative control (NC)
group; and the HG + PHB-transfected group.
PHB overexpression
HRCECs were first cultured for 24 h and were then
transfected with the pCMV6-XL5 plasmid (Clontech) encompassing the
PHB sequence. When cells reached 50-60% confluency, they were
transfected with the PHB overexpression plasmid (0.2 µg) using a
X-tremeGENE HP DNA transfection reagent (Roche Applied Science) at
37˚C for 24 h according to the manufacturer's instructions.
Following transfection for 24 h the medium was replaced with normal
medium supplemented or not with HG (30 mmol/l). The transfection
efficiency was ~70-80%.
PHB silencing
HRCECs were cultured in ECM supplemented with 5% FBS
at 37˚C and 5% CO2 for 24 h. When cells reached 50-60%
confluency, they were transfected with small interfering RNA
(siRNA, 50 nM) clones against PHB (Qiagen) using HiPerFect
transfection reagent (Qiagen) for 10 h at 37˚C according to the
manufacturer's instructions. Cells were collected at 48 h after
transfection for various assays. The sequence of PHB-siRNA:
5'-AATGTGGATGCTGGGCACAGA-3'; the sequence of negative control:
5'-AAGAGTGTCGGATGCAGGATC-3' (Qiagen).
Preparation of subcellular fractions
of HRCECs
The subcellular fractions of HRCECs were separated
as previously described (9).
Briefly, HRCECs were rinsed in PBS and were then resuspended into
Buffer A (20 mmol/l HEPES pH 7.5, 10 mmol/l KCl, 1.5 mmol/l MgCl2,
1 mmol/l EGTA, 1 mmol/l EDTA, 1 mmol/l DTT, 0.1 mmol/l PMSF, 250
mmol/l sucrose) supplemented with protease inhibitors. The cells
were homogenized by 10 strokes in a Dounce homogenizer.
Subsequently, cells were centrifuged at 1,000 x g for 4 min at 4˚C
to isolate cell nuclei and DNA fragments. The supernatant was then
collected and centrifuged at 8,000 x g for 20 min at 4˚C to isolate
heavy cellular fragments rich in mitochondria. The resultant
supernatant was centrifuged at 10,000 x g for 15 min at 4˚C to
obtain the cytosolic fractions.
Western blot analysis
Western blot analysis was performed as previously
described (10). Briefly, equal
amounts of protein (20 µg) were separated by SDS-polyacrylamide gel
electrophoresis and then transferred onto polyvinylidene difluoride
membranes (Millipore, Sigma). The membranes were blocked with 5%
bovine serum albumin (Sigma-Aldrich; Merck KGaA) in Tris-buffered
saline Tween-20 (0.1%) for 2 h at room temperature and then
incubated with primary antibodies at 4˚C overnight. The following
primary antibodies were used: Anti-PHB (dilution, 1:500; product
code ab75766; Abcam), anti-COX IV (dilution, 1:700; product code
ab202554; Abcam), anti-cleaved-caspase 3 (dilution, 1:400; product
no. 9661; Cell Signaling Technology, Inc.), anti-cleaved PARP
(dilution, 1:500; product no. 5625; Cell Signaling Technology,
Inc.) and anti-β-actin (dilution, 1:1,000; cat. no. sc-8432; Santa
Cruz Biotechnology, Inc.). Immunoreactive proteins were detected
with horseradish peroxidase-conjugated secondary antibodies
(dilution, 1:5,000; cat. no. sc-2357; Santa Cruz Biotechnology,
Inc.). Quantification was performed according to the NIH Image
version-1.61 software (National Institutes of Health).
Immunofluorescence staining and live
imaging
HRCECs were cultured on coverslips and were then
fixed with 4% paraformaldehyde for 10 min at room temperature and
0.2% Triton X-100. Subsequently, cells were blocked with PBS for 1
h at room temperature supplemented with 0.2% Tween-20, 10% BSA and
10% serum (Gibco; Thermo Fisher Scientific, Inc.). Following
blocking, the cells were first incubated with the indicated primary
antibody (PHB; dilution, 1:100; product code ab75766; Abcam) and
then with Alexa Fluor 488-conjugated secondary antibody (dilution,
1:1,000; product code ab150077; Abcam). The cell nuclei were
stained for 5 min at room temperature with 2 µg/ml DAPI. For Mito
Tracker staining, HRCECs were stained for 20 min at room
temperature with 0.02 µM Mito Tracker (Invitrogen; Thermo Fisher
Scientific, Inc.). For JC-1 staining, HRCECs were rinsed with PBS
and incubated with JC-1 (Invitrogen) staining solution at 37˚C for
20 min. Pictures were obtained with an FV-1000 confocal laser
scanning microscope (Olympus). For live cell imaging, images were
captured under a phase contrast microscope (Carl Zeiss GmbH).
Analysis of mitochondrial complex
I/III activity
Following HRCEC trypsinization, cells
(1x106/ml) were resuspended and oxygen consumption was
measured using an oxygen probe in the presence or absence of 10 µM
rotenone (an inhibitor of complex I) 10 µM antimycin-A (an
inhibitor of complex III) and 0.05% azide, an inhibitor of
mitochondrial respiration. Finally, the activity of complex I/III
was compared between PHB-knocked down and control cells.
FACS analysis of reactive oxygen
species (ROS)
Firstly, HRCECs (1x106/ml) cultured in
DMEM supplemented with 0.2% Fetal Calf Serum (GIBCO) were incubated
with 5 µM CM-H2-DCDA and 5 µg/ml propidium iodide (PI)
at room temperature for 20 min. HRCECs treated with 100 µM
H2O2 for 20 min served as a positive control
group. H2O2 levels were detected using
CM-H2-DCDA. Subsequently, PHB-depleted cells were
treated with 10 µg/ml PEG-catalase for 2 h at room temperature.
Living cells were analyzed using PI staining for 10 min at room
temperature and the fluorescence intensity in the FL-2 and FL-1
channels was measured using a Becton-Dickinson FACSCalibur system
(BD Biosciences) and FlowJo 7.6 software (Tree Star, Inc.).
Flow cytometric analysis
Flow cytometric analysis was performed as previously
described (10). In brief, cells
(1x106/ml) were used to assess cell apoptosis with an
Annexin V/propidium iodide (PI) kit (Invitrogen, Thermo Fisher
Scientific, Inc.) according the manufacturer's protocol. Flow
cytometry was used determine the proportion of early apoptotic
cells. Flow cytometry was performed using a FACSCalibur system (BD
Biosciences).
Statistical analysis
Experiments were performed at least three times. All
data are presented as the mean ± SD. Differences between multiple
groups were assessed using a post hoc (Tukey's HSD) test used for
one-way ANOVA in GraphPad Prism 6.0 (GraphPad Software, Inc.) and
SPSS 17.0 (SPSS, Inc.). Comparison between non-parametric variables
was performed using a χ2 test. P<0.05 was considered
to indicate a statistically significant difference.
Results
PHB localizes to the mitochondria and
is downregulated under HG conditions
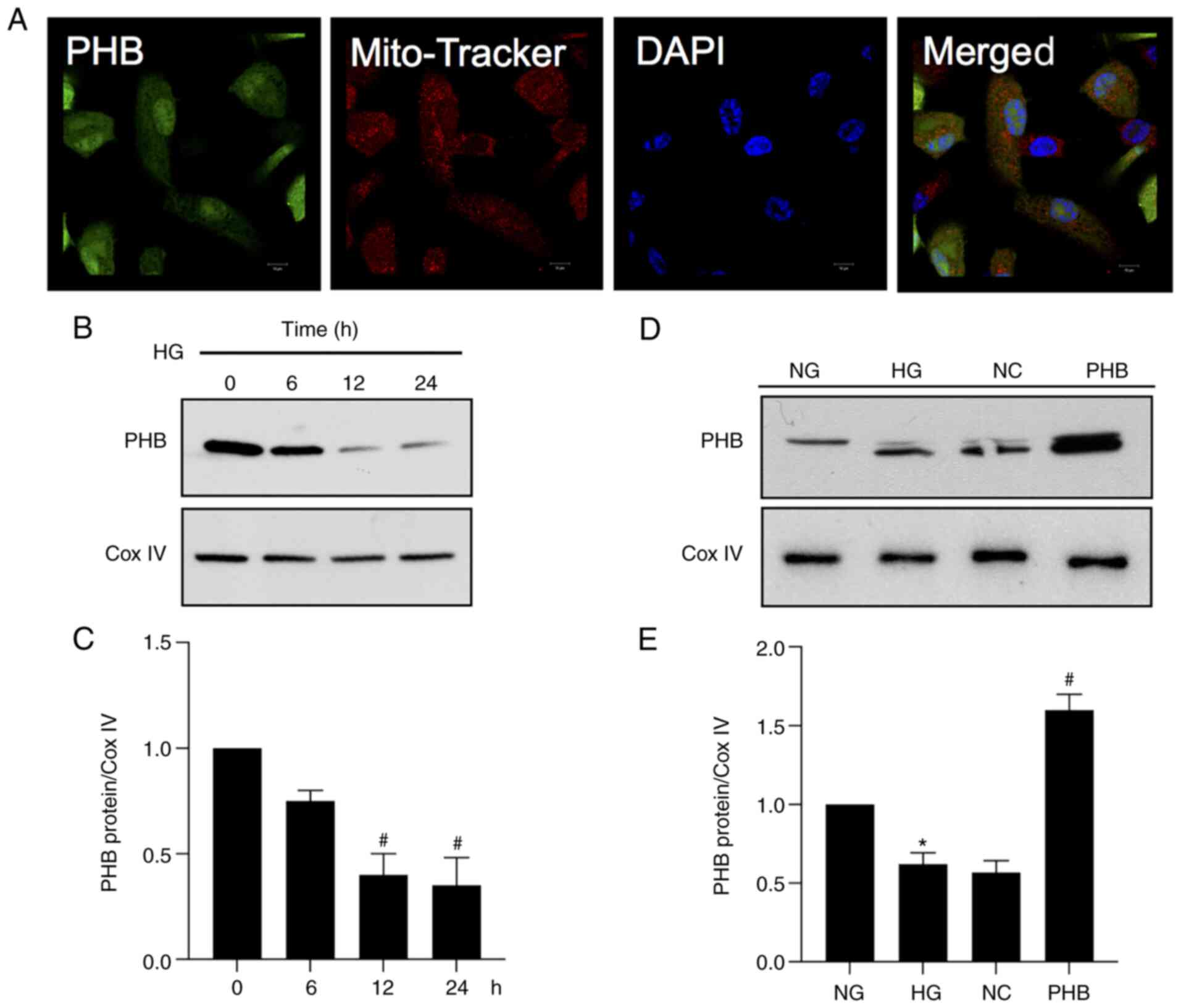
To detect the localization of PHB,
immunofluorescence analysis was carried out in HRCECs. The analysis
revealed that a small proportion of PHB was distributed in the
nuclear compartment, while the majority of PHB was detected in the
mitochondria, as shown using Mito Tracker (Fig. 1A). Furthermore, to determine the
expression levels of PHB in the mitochondria under HG conditions,
HRCECs were exposed to 30 mmol/l glucose (HG). The results revealed
that PHB was downregulated in the mitochondria of HRCECs in the HG
group (Fig. 1B and C). In addition, the expression levels of
PHB were measured in all the different groups and the results
demonstrated that PHB was successfully overexpressed in cells
transfected with PHB overexpression plasmid (Fig. 1D and E). Collectively, these findings indicated
that PHB could be involved in the regulation of mitochondrial
function in HRCECs.
PHB knockdown alters mitochondrial
function in HRCECs
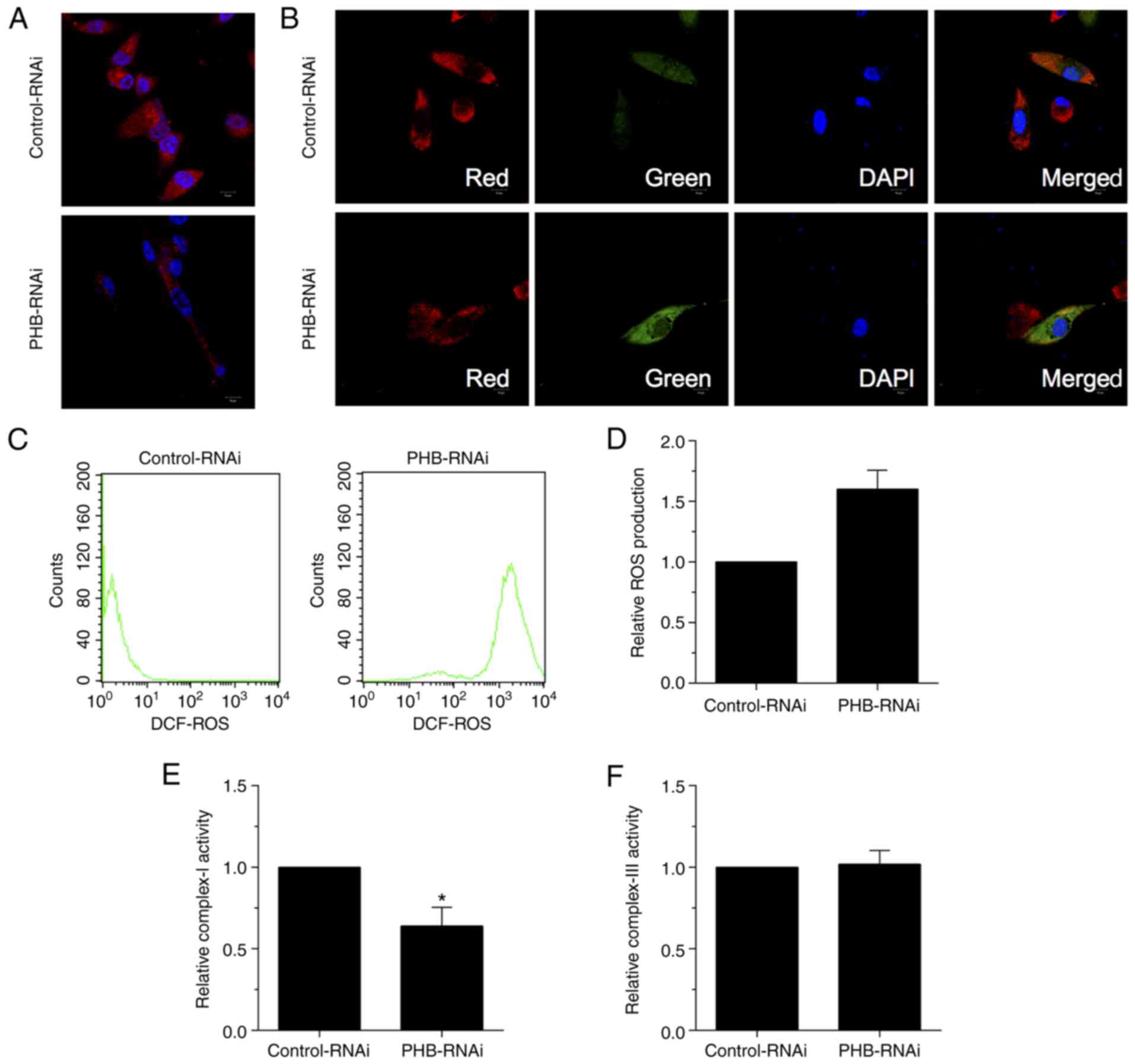
To investigate the role of PHB in mitochondria,
PHB-depleted mitochondria were established in HRCECs and the levels
of ROS were then measured. Immunofluorescence staining in HRCECs
revealed reduced Mito Tracker positive staining, indicating that
mitochondria were inactive with regard to respiration (Fig. 2A). This finding clearly suggested
that the increased ROS production in PHB-depleted HRCECs was due to
mitochondrial respiration. Furthermore, both immunofluorescence
(Fig. 2B) and ROS production
(Fig. 2C and D) analyses revealed reduced red and
enhanced green fluorescence signals in PHB-depleted HRCECs, thus
suggesting that mitochondrial membrane depolarization was promoted
and ROS production was increased, respectively. In addition, the
activity of complex I/III was assessed via measuring oxygen
consumption. The results demonstrated that the activity of complex
I was reduced in PHB-depleted HRCECs compared with the control
group (Fig. 2E). However, the
activity of complex III remained unchanged between both groups
(Fig. 2F). Overall, the
aforementioned results verified that mitochondria mediated ROS
generation in PHB-depleted HRCECs.
PHB attenuates HG-induced apoptosis in
HRCECs
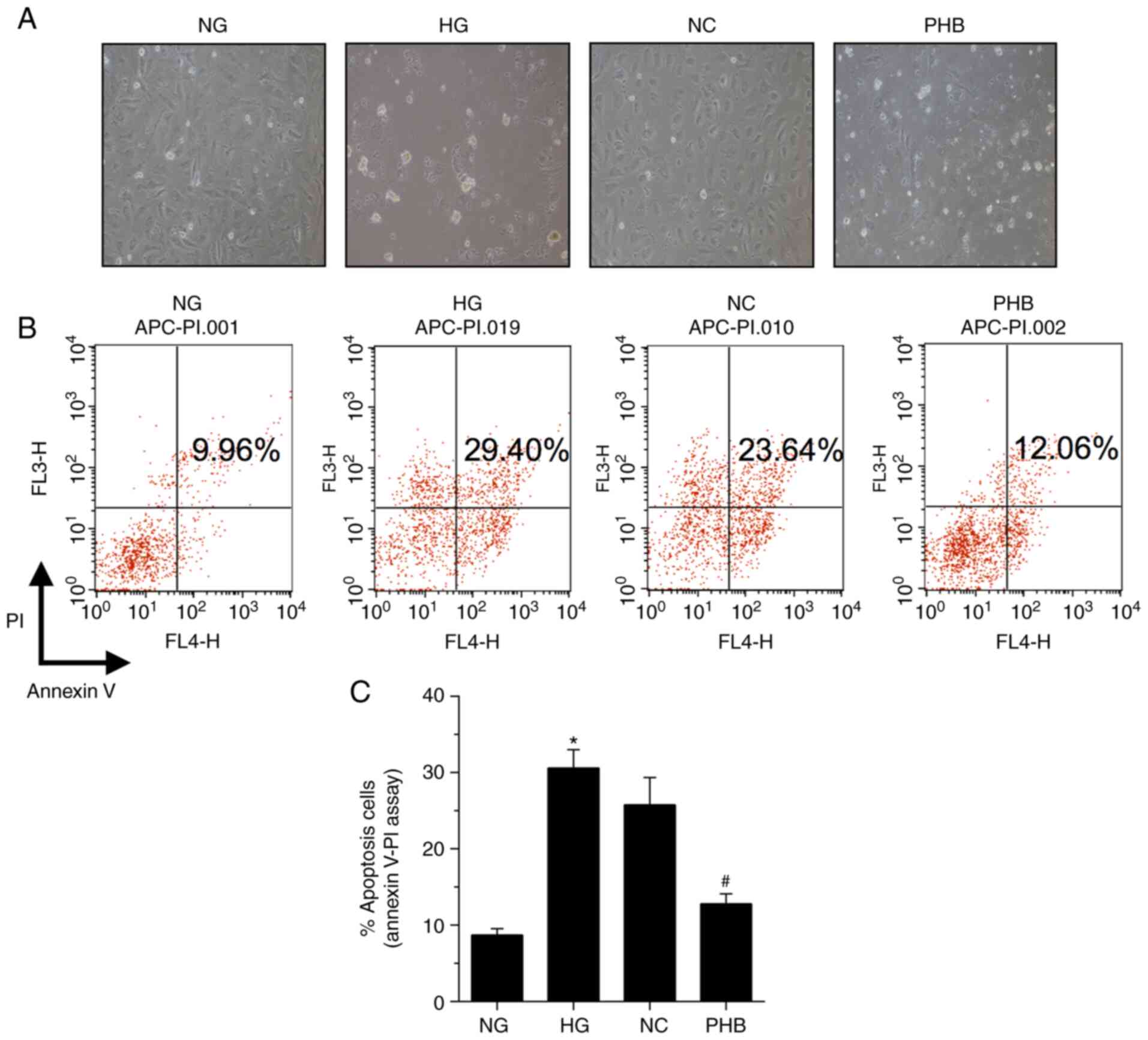
To explore whether PHB could protect HRCECs, the
effect of PHB on HG-induced cell apoptosis was assessed by
evaluating the apoptosis-specific morphological changes in HRCECs.
Therefore, HG markedly enhanced HRCEC apoptosis, which was restored
by PHB (Fig. 3A). Additionally,
HRCEC apoptosis was determined by Annexin V/PI staining. The
apoptosis rate in the NG group was 9.96%, which was significantly
different compared with that in the HG, NC or PHB groups, with
apoptosis rates of 29.40, 23.64 and 12.06%, respectively (Fig. 3B and C). Collectively, the aforementioned
findings suggested that PHB could attenuate HG-induced HRCEC
apoptosis.
Effect of PHB on the expression of
apoptosis-related proteins
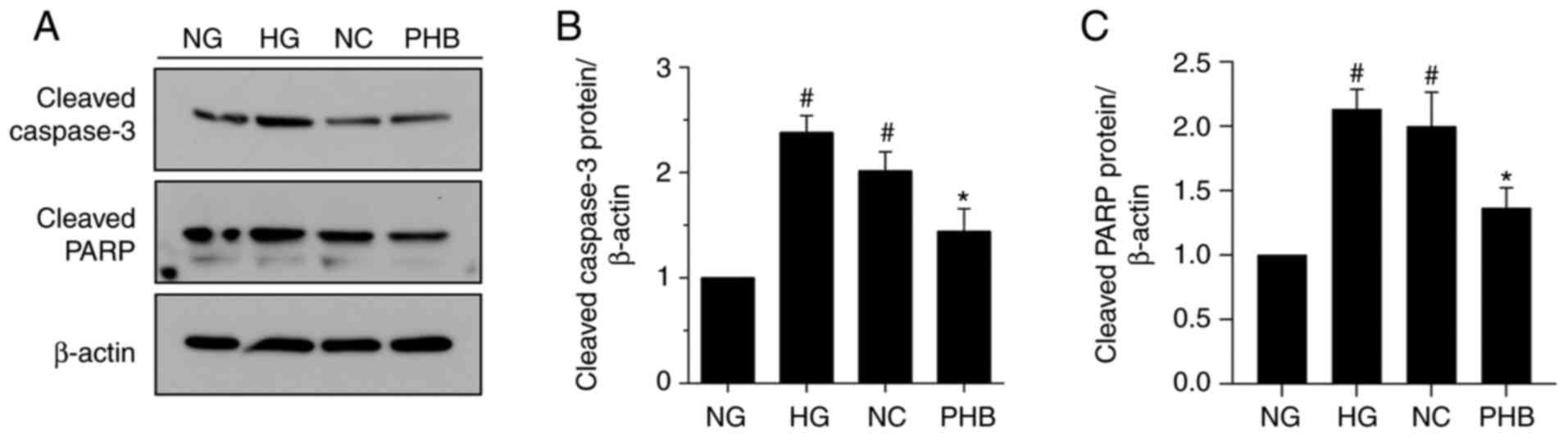
Subsequently, the effect of PHB on the expression
levels of the apoptosis-related proteins, PARP and caspase-3, in
NG-, HG-, NC- and PHB-treated cells was evaluated by western blot
analysis. Consistent with the Annexin V/PI staining results,
western blot analysis revealed that the expression levels of both
caspase-3 and PARP were significantly increased in the HG and NC
groups compared with NG group. However, the expression levels of
both proteins were not notably different between the HG and NC
groups. Notably, caspase-3 and PARP were markedly downregulated in
the PHB group (Fig. 4A-C).
Collectively, the aforementioned findings suggested that PHB could
protect HRCECs against HG-induced apoptosis via downregulating
caspase-3 and PARP.
Discussion
The present study aimed to evaluate the role of PHB
in HRCECs and demonstrated that HRCEC apoptosis was associated with
mitochondrial function. The results showed that PHB was
significantly upregulated in mitochondria, whereas PHB knockdown
inhibited the activity of complex I and enhanced the production of
mitochondrial ROS in HRCECs. In particular, PHB overexpression
could protect HRCECs against HG-induced apoptosis. These results
indicated that PHB could maintain mitochondrial function, inhibit
HRCEC apoptosis and reduce mitochondrial ROS generation in DR.
PHB is a conserved and widely expressed protein
that, is distributed in several cellular compartments, such as in
the mitochondria, and lipid rafts in the plasma membrane and
nucleus (11). The present study
revealed that PHB was expressed in HRCECs. Furthermore,
co-localization assays in HRCECs showed that PHB was mainly
expressed in mitochondria. However, the mechanism underlying the
effects of PHB under HG conditions remains elusive. To evaluate the
function of PHB in HRCECs, cells were transfected with siRNA clones
targeting PHB to silence its expression. Therefore, PHB knockdown
affected mitochondrial function and promoted ROS production.
It has been reported that the production of ROS from
mitochondria can be promoted by the depolarization of the
mitochondrial membrane and blockade of electrons at complexes I and
III, which are widely accepted as the source of ROS in mitochondria
(12). The activity of complex III
and the contribution of the mitochondrial connection observed in
PHB-depleted cells suggested that a large number of electrons in
complex I could be used for the production of ROS when the
expression of PHB was knocked down. Furthermore, it was
hypothesized that the function of cytochrome oxidase and ATP
generation could be maintained through compensatory signaling
pathways. These effects could be associated with the increase of
electrons in the II/III complex. Additionally, ROS generation could
be promoted in PHB-depleted HRCECs due to the lack of mitochondrial
respiration in these cells.
Furthermore, PHB attenuated HG-induced apoptosis in
HRCECs. A previous study revealed that treatment of endothelial
cells with HG for three days enhanced cell apoptosis (13). Another study showed that the
apoptosis rate of retinal pericytes, cultured under HG conditions
for seven days, was higher compared with those cultured under NG
conditions (14,15). Additionally, compared with
endothelial cells cultured under NG conditions, cells exposed to 23
and 30 nM glucose for 25 and 45 days, respectively, exhibited
enhanced apoptotic rates (16).
Interestingly, the apoptosis rate in cells treated with 23 or 30 nM
HG was not significantly different. The results of the present
study were consistent with the aforementioned studies, as the
apoptosis rate in the HG and NC groups was significantly increased
compared with the NG group. However, the apoptosis rate was notably
reduced in the PHB group. No statistically significant difference
was observed in apoptosis between the HG and NC groups. Therefore,
the present study indicated that PHB could attenuate HG-induced
HRCEC apoptosis.
It has been reported that PHB exerts its
anti-apoptotic effects through the caspase signaling pathway
(17,18). Herein, to evaluate the
anti-apoptotic effect of PHB on HRCECs, the protein expression
levels of caspase-3 and PARP were determined by western blot
analysis. The aforementioned proteins play a crucial role in
regulating mitochondrial cell death (19). The results showed that caspase-3
and PARP were notably upregulated in the HG and NC groups compared
with the NG group. However, the levels of both proteins were not
statistically different between the HG and NC groups.
Interestingly, the protein levels of PARP and caspase-3 were
notably decreased in the PHB group compared with the NC group.
Overall, PHB attenuated HG-induced apoptosis in HRCECs via
downregulating caspase-3 and PARP.
PHB may be a promising approach for DR. However, the
more detailed mechanism of PHB in DR need to be done in
vivo. In summary, the results of the present study suggested
that PHB could be necessary for maintaining mitochondrial function
via regulating ROS generation and apoptosis in HRCECs. These
findings highlighted the widely accepted notion that mitochondria
are a significant cellular compartment and that ROS serves a
crucial role in DR (20). Both
apoptosis and mitochondrial function are associated with several
pathological processes during microvascular damage, including DR
(21). Overall, the results of the
present study could provide an experimental basis for the
protective effect of PHB in the management of DR. PHB could become
an interesting candidate for some possible clinical implications in
DR.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by Wuhan Municipal
Health Commission Medical Research (grant no. WX19Q29).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Author's contributions
LZ wrote the original draft, performed the
experiments and collected the data. YH contributed to the analysis
of data and the design of this study. All authors read and approved
the final manuscript. LZ and YH confirm the authenticity of all the
raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Regina C, Panatta E, Candi E, Melino G,
Amelio I, Balistreri CR, Annicchiarico-Petruzzeli M, Di Danilele N
and Ruvolo G: Vascular ageing and endothelial cell senescence:
Molecular mechanisms of physiology and diseases. Mech Ageing Dev.
159:14–21. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Volpe CMO, Villar-Delfino PH, Dos Anjos
PMF and Nogueria-Machado JA: Cellular death, reactive oxygen
species (ROS) and diabetic complications. Cell Death Dis.
9(119)2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wu MY, Yiang GT, Lai TT and Li CJ: The
oxidative stress and mitochondrial dysfunction during the
pathogenesis of diabetic retinopathy. Oxid Med Cell Longev.
2018(3420187)2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Zheng H and Lu GM: Reduction of prohibitin
expression contributes to left ventricular hypertrophy via
enhancement of mitochondrial reactive oxygen species formation in
spontaneous hypertensive rats. Free Radic Res. 49:164–174.
2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Artal-Sanz M and Tavernarakis N:
Prohibitin and mitochondrial biology. Trends Endocrinol Metab.
20:394–401. 2009.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Mishra S, Ande SR and Nyomba BL: The role
of prohibitin in cell signaling. FEBS J. 277:3937–3946.
2010.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Theiss AL, Idell RD, Srinivasan S,
Klapproth JM, Jones DP, Merlin D and Sitaraman SV: Prohibitin
protects against oxidative stress in intestinal epithelial cells.
FASEB J. 21:197–206. 2007.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Muraguchi T, Kawawa A and Kubota S:
Prohibitin protects against hypoxia-induced H9c2 cardiomyocyte cell
death. Biomed Res. 31:113–122. 2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Chowdhury I, Branch A, Olatinwo M, Thomas
K, Matthew R and Thompson WE: Prohibtin (PHB) acts as a potent
survival factor against ceramide induced apoptosis in rat granulosa
cells. Life Sci. 89:295–303. 2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
He Y, Wang N, Shen Y, Zheng Z and Xu X:
Inhibition of high glucose-induced apoptosis by uncoupling protein
2 in human umbilical vein endothelial cells. Int J Mol Med.
33:1275–1281. 2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Peng YT, Chen P, Ouyang RY and Song L:
Multifaceted role of prohibitin in cell survival and apoptosis.
Apoptosis. 20:1135–1149. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Vinogradov AD and Grivennikov VG:
Oxidation of NADH and ROS production by respiratory complex I.
Biochim Biophys Acta. 1857:863–871. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Beltramo E, Nizheradze K, Berrone E,
Tarallo S and Porta M: Thiamine and benfotiamine prevent apoptosis
induced by high glucose-conditioned extracellular matrix in human
retinal pericytes. Diabetes Metab Res Rev. 25:647–656.
2009.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Naruse K, Nakamura J, Hamada Y, Nakayama
M, Chaya S, Komori T, Kato K, Kasuya Y, Miwa K and Hotta N: Aldose
reductase inhibition prevents glucose induced apoptosis in cultured
bovine retinal microvascular pericytes. Exp Eye Res. 71:309–315.
2000.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Romeo G, Liu WH, Asnaghi V, Kern TS and
Lorenzi M: Activation of nuclear factor-kappaB induced by diabetes
and high glucose regulated a proapoptotic program in retinal
pericytes. Diabetes. 51:2241–2248. 2002.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Cui Y, Xu X, Bi H, Zhu Q, Wu J, Xia X,
Qiushi Ren and Ho PC: Expression modification of uncoupling
proteins and MnSOD in retinal endothelial cells and pericytes
induced by high glucose: The role of reactive oxygen species in
diabetic retinopathy. Exp Eye Res. 83:807–816. 2006.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhou TB, Qin YH, Zhou C, Lei FY, Zhao YJ,
Chen J, Su LN and Huang WF: Less expression of prohibitin is
associated with increased caspase-3 expression and cell apoptosis
in renal interstitial fibrosis rats. Nephrology (Carlton).
17:189–196. 2012.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chowdhury I, Thompson WE, Welch C, Thomas
K and Matthews R: Prohibitin (PHB) inhibits apoptosis in rat
granulosa cells (GCs) through the extracellular signal-regulated
kinase1/2 (ERK1/2) and the Bcl family of proteins. Apoptosis.
18:1513–1525. 2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Mantena SK, Sharma SD and Katiyar SK:
Berberine inhibits growth, induces G1 arrest and apoptosis in human
epidermoid carcinoma A431 cells by regulating Cdki-Cdk-cyclin
cascade, disruption of mitochondrial membrane potential and
cleavage of caspase 3 and PARP. Carcinogenesis. 27:2018–2027.
2006.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Schrier SA and Falk MJ: Mitochondrial
disorders and the eye. Curr Opon Ophthalmol. 22:325–331.
2011.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhang W, Liu H, AI-Shabrawey M, Caldwell
RW and Caldwell RB: Inflammation and diabetic retinal microvascular
complications. J Cardiovasc Dis Res. 2:96–103. 2011.PubMed/NCBI View Article : Google Scholar
|