Introduction
According to cancer statistics of 2022, ovarian
cancer ranks as the fifth major cause of cancer-associated
mortality for females in the United States, with a 5-year survival
rate <50% for all stages (1).
In addition, ~60% of newly diagnosed patients typically present
with distant metastasis, resulting in a 30% 5-year survival rate
(2). Due to the anatomical
location of the ovaries in the deep pelvic cavity, symptoms of this
cancer include abdominal or pelvic pain or distension, bloating,
urinary frequency and urgency (3,4).
However, they are only perceptible after metastatic malignancy has
been developed or after the volume of the primary tumor has reached
a size that can obstruct the gastrointestinal or pelvic organs
(3,4). Genetic heterogeneity is widespread in
ovarian cancer (5-7).
In ovarian cancer, genomic variation, invasive ability and
sensitivity to platinum profiles are all heterogeneous among
intratumor subclones, subtypes or between primary and recurrent
malignancy (8-10).
This poses a challenge to the development of applicable genetic
biomarkers for early diagnosis, dynamic disease monitoring and
therapeutic target identification in the clinic. Identification of
novel biomarkers and deeper understanding of the genomic profile of
ovarian cancer will likely increase the survival rate from this
disease through metastasis monitoring and finding potential
mutations for guiding individualized treatment.
Compared with traditional tissue biopsies, liquid
biopsy is a relatively non-invasive sample collection method.
Liquid biopsy specimens can be obtained with ease and used to
monitor diseases in real time dynamically at multiple sampling
times (11-13).
The most common type of liquid biopsy is circulating tumor DNA
(ctDNA), which is a fragmented form of single-stranded or
double-stranded DNA that is released into the bloodstream following
the necrosis and apoptosis of primary, metastatic or recurrent
tumor cells (14,15). ctDNA application is promising in
the field of precision oncology due to its potential for use in
reflecting the tumor genetic profile and the wide availability of
techniques, such as PCR and next-generation sequencing technology
(NGS) (16).
However, the accuracy of using ctDNA to reflect all
mutational information in the primary tumor lacks direct
experimental evidence. Previous studies compared the genetic
information between ctDNA and primary or metastatic tumor tissues.
Comparison analysis of genetic profiles in prostate, breast and
lung cancer showed that the concordance between ctDNA and tissues
might depend on alteration types, with considerable concordance in
driver DNA alterations but limited concordance in gene
amplification and hotspot mutations (17-20).
Furthermore, previous analyses suggested that the concordance of
hotspot mutations or specific mutations in ctDNA and tumor tissues
depends on gene types, for example, 90% concordance of the PIK3CA
mutation and 78% concordance of the KRAS mutation were observed in
colorectal cancer, and 50% concordance of EGFR mutation was
observed in lung cancer (21-24).
DNA methylation status and intratumor heterogeneity were also
examined in ctDNA. ctDNA could partially reflect the methylation
status of a specific gene in breast cancer but had limited value of
reflecting intratumor heterogeneity in lung cancer (25,26).
These studies revealed that the integrated analysis with both
liquid and tumor biopsy might help to obtain a more comprehensive
genetic profile of tumors.
ctDNA has exhibited a considerable clinical value as
a tumor biomarker in diagnosis and disease monitoring of ovarian
cancer (27). However, few studies
compared the sequencing results of ctDNA, paired ascites and tumor
tissue samples in ovarian cancer (28,29).
A parallel analysis of these three types of samples mainly
explained the potential of ascites and ctDNA to reflect the
mutational landscape in 10 patients (30), although these results require
further validation.
Therefore, the present study aimed to detect
potential mutant genes in ctDNA samples for clinical application,
the data of which were then used to compare against the mutational
spectrum in plasma, matching ascites and tumor tissue samples using
NGS. The rationale was to further understand the underlying
mechanism of the inter-tumor and intra-tumor heterogeneity in
ovarian cancer, in addition to its genomic landscape. Overall, the
utility of ctDNA to dynamically reflect the overall condition of
ovarian cancer and its prospective value in clinical applications,
such as metastasis and targeted therapy, were examined.
Materials and methods
Patient enrollment and sample
collection
Patients with suspected ovarian cancer were
recruited at the Obstetrics and Gynecology Hospital of Fudan
University (Shanghai, China) between October 2016 and June 2017.
Patients with pathologically confirmed epithelial ovarian cancer
were included. Patients with borderline ovarian tumors,
malignancies in other organs and uncontrollable infection, and
those without consent were excluded. A total of 18 patients were
included. In total, 8-10 ml peripheral blood was collected for
ctDNA from these 18 patients, ~10 ml matched ascites from 6 of
these patients, and 0.5-cm3 tumor tissues from 8 of
these patients. The present study was approved by the Ethical
Committee of Obstetrics and Gynecology Hospital of Fudan University
(approval no. 2016-48; Shanghai, China).
Nucleic acid isolation and quality
assessment
Peripheral blood samples were first centrifuged at
800 x g for 10 min at room temperature within 4 h of blood
collection to obtain the plasma. The plasma samples were further
centrifuged at 16,000 x g for 10 min at room temperature to remove
the remaining cellular debris. ctDNA was extracted from the plasma
using the QIAamp Circulating Nucleic Acid kit (cat. no. 55114;
Qiagen China Co., Ltd.). The quantity and quality of the extracted
DNA were assessed using an UV-vis spectrophotometer (Thermo Fisher
Scientific, Inc.) and the Agilent 2100 Bioanalyzer.
DNA was extracted from tumor tissue or ascite
samples after cell lysis and digestion with the QIAamp DNA Mini Kit
(cat. no. 51304; Qiagen China Co., Ltd.) according to the
manufacturer's protocols. The quantity and quality of the extracted
DNA were assessed as for ctDNA. The extracted DNA from tumor
tissues and ascites was sheared using Covaris sonicator (Covaris
LLC) with the following settings: 4-8˚C, 10% duty factor, 200
cycles per burst and 360 sec, after further quantification using a
Qubit dsDNA HS Assay Kit (cat. no. Q32851; Thermo Fisher
Scientific, Inc.). The extracted DNA was stored at -80˚C.
Detection of mutations in DNA
sequences using NGS
The SureSelectXT Reagent Kit (cat. no. G9611A;
Agilent Technologies, Inc.) was used to construct the NGS libraries
according to the SureSelect Target Enrichment workflow
instructions. Briefly, after end repair, A-tailing and ligation
with adaptors using a SureSelect Library Prep Kit (cat. no. G9684A;
Agilent Technologies, Inc.), the DNA was amplified by precapture PCR
(1 cycle of 98˚C for 2 min; 10 cycles of 98˚C for 30 sec, 65˚C for
30 sec and 72˚C for 1 min; and then 72˚C for 10 min). The products
were then hybridized and captured using the reagents in the
aforementioned SureSelectXT Reagent Kit. The captured DNA fractions
were amplified by post-capture PCR (1 cycle at 98˚C for 2 min; 16
cycles at 98˚C for 30 sec, 57˚C for 30 sec and 72˚C for 1 min; and
then 72˚C for 10 min) using a Herculase II Fusion DNA Polymerase
kit (cat. no. 600677; Agilent Technologies, Inc.). Agencourt AMPure
XP Beads (cat. no. A63881; Beckman Coulter, Inc.) were used to
purify the samples throughout these processes. The libraries were
assessed with the DNA 1000 Assay (cat. no. 5067-1504; Agilent
Technologies, Inc.) on the Agilent 2100 Bioanalyzer. After quality
assessment, the concentration of PCR products was quantified using
an Agilent qPCR NGS Library Quantification Kit (cat. no. G4880A;
Agilent Technologies, Inc.).
The generated libraries were sequenced with a
concentration of 8 pM for 150-bp paired end reads on an Illumina
MiSeq sequencer using a MiSeq Reagent Kit (cat. no. MS-102-2002;
Illumina, Inc.) according to the manufacturer's recommendations.
All samples were sequenced via the OncoGxOne cancer panel by
Shanghai Liwen Biotechnology Co., Ltd. After Illumina sequencing,
the reads were aligned to the human HG19 reference genome (GRCh37;
https://www.ncbi.nlm.nih.gov/grc) using
Burrows-Wheeler Aligner v0.7.12 (http://bio-bwa.sourceforge.net). Variant calling was
performed using the Genome Analysis Toolkit v3.4 (https://gatk.broadinstitute.org), and was
annotated using ANNOVAR (last update, 06/01/17; https://annovar.openbioinformatics.org).
The variants with a minor allele frequency <5% were filtered
out.
Functional analysis of the mutant
genes
Functional analysis of the potential mutant genes
was performed using public bioinformatics tools and databases.
WebGestalt (2019 version; http://www.webgestalt.org) was used with the following
parameters: The functional databases including Gene Ontology (GO),
Wikipathway and Kyoto Encyclopedia of Genes and Genomes (KEGG) were
selected. The enrichment method was over-representation analysis,
where the false discovery rate (FDR) was determined using the
Benjamini-Hochberg method. The top 10 categories with FDR <0.05
were identified to be among the enriched categories. The cBioPortal
for Cancer Genomics (version 2.0.1; http://www.cbioportal.org/) was used to analyze the
gene and drug-target interaction network. The network, which showed
the query genes, neighbor genes and drug-target information, was
generated by submitting the query genes and adding the drug data in
the Network tab (31).
‘metastasis’ and ‘ovarian cancer’ were used as key words in
Chilibot (last update 06/18/17; http://www.chilibot.net/) to analyze mutant genes, and
the overlap of these two sets was considered as the genes related
to ovarian cancer metastasis. Search Tool for the Retrieval of
Interacting Genes/Proteins (STRING; version 11.0; https://string-db.org) was used to analyze functional
interactions with a medium confidence of 0.4. Comparative analysis
of the peripheral blood, tumor tissue and ascite samples was
performed using Venn diagram (version 2.1; https://bioinfogp.cnb.csic.es/tools/venny/index.html).
Statistical analysis
The data were analyzed using GraphPad Prism 6.0
(GraphPad Software, Inc.) and SPSS 16.0 (SPSS, Inc.). Pearson
correlation was used to identify the correlation coefficient and
significant difference. P<0.05 was considered to indicate a
statistically significant difference.
Results
Patient characteristics
The clinicopathological characteristics of the 18
patients with ovarian cancer are summarized in Table I. The mean age of patients was
51.9±11.1 years. Preoperative serum cancer antigen 125 levels were
>500 IU/l in 72.2% of the patients. In addition, >80% of the
recruited patients were diagnosed with advanced ovarian cancer, at
International Federation of Gynecology and Obstetrics stage III or
IV. Serous ovarian carcinoma (77.8%) was the most common
pathological type, followed by 2 patients with clear cell
carcinoma, 1 with mucinous carcinoma and 1 with endometrioid
carcinoma. In total, 22.2% of the patients were found with lymph
node metastasis.
 | Table ISummary of clinicopathological
characteristics of patients with ovarian cancer. |
Table I
Summary of clinicopathological
characteristics of patients with ovarian cancer.
| Clinicopathological
characteristics | Value |
|---|
| Age range,
years | 24-70 |
| FIGO stage, n
(%) | |
|
II | 1 (5.6) |
|
III | 15 (83.3) |
|
IV | 2 (11.1) |
| Histological type,
n (%) | |
|
Serous
carcinoma | 14 (77.8) |
|
Clear cell
carcinoma | 2 (11.1) |
|
Endometrioid
carcinoma | 1 (5.6) |
|
Mucinous
carcinoma | 1 (5.6) |
| Lymph nodes
metastasis, n (%) | |
|
Positive | 4 (22.2) |
|
Negative | 4 (22.2) |
|
Unknown | 10 (55.6) |
| Preoperation cancer
antigen 125 level, n (%) | |
|
>1000
IU/l | 8 (44.4) |
|
500-1000
IU/l | 5 (27.8) |
|
<500
IU/l | 5 (27.8) |
Mutant genes detected in patients with
ovarian cancer
A panel of 333 potential oncogenes was used for the
targeted NGS of DNA isolated from 18 peripheral blood samples,
eight tumor tissue samples and six ascites samples. This panel
covers a number of reported genes associated with tumor initiation,
those in the clinical investigation phase of cancer drug
application, such as AZD6738 against ATR, and those that have been
clinically applied for targeted therapy, such as larotrectinib
against neurotrophin receptor kinase (Data S1). For example, the small-molecule
or antibody drugs targeting MET, SRC, KRAS and BRCA1/2 were
approved or in clinical trials. In the present study, for the
target regions of the included samples, the coverage reached
>99%, whereas the mean sequencing depth was >200X and the
alignment rate was >95%.
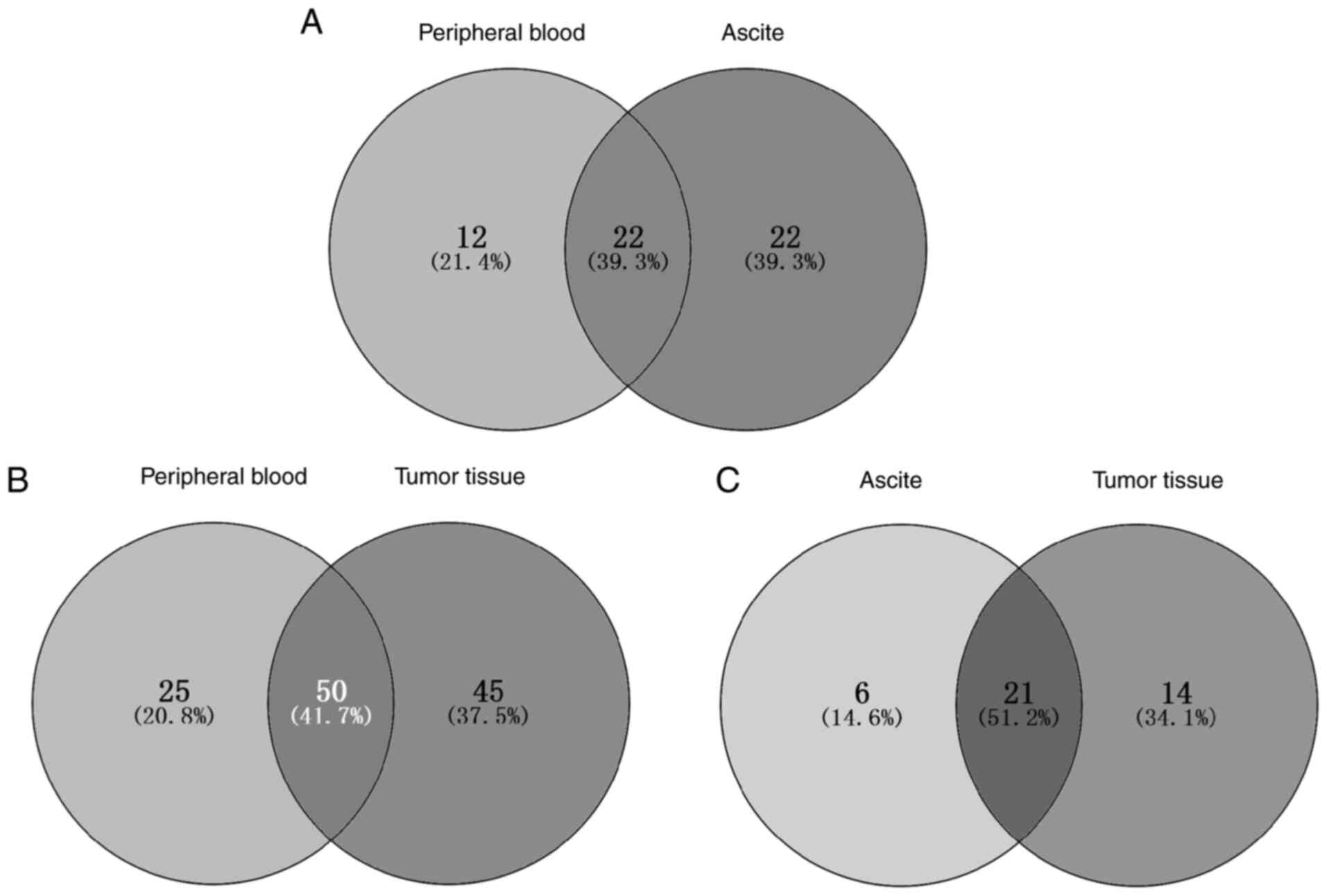
A total of 104 genes were found to be mutated in the
ctDNA samples from ≥1 patient (Fig.
1A). Of these genes, 29 were detected in ≥3 patients, 40 were
detected in ≥2 patients, whereas the remaining 64 genes were
mutated in only 1 patient. In the ovarian cancer tissue samples, 95
mutant genes were found, among which mutations in 34 genes were
found in ≥2 patients (Fig. 1B). A
total of 44 mutant genes were found in ascites samples, 23 of which
were shared by ≥2 patients (Fig.
1C).
Mutational heterogeneity in patients
with ovarian cancer
The mutant genes found in the peripheral blood,
tumor tissue and ascites samples were compared to understand the
concordance among the mutation spectra and genomic landscapes of
the different samples from patients with ovarian cancer. Mutational
heterogeneity was widely observed among the specimens and patients.
The mutant genes shared by different patients and different sample
types within the same individuals are shown in Figs. 1 and 2. The highest similarity was found
between the ascites and tumor tissues (51.2%), followed by that
between peripheral blood and tumor tissue samples (41.7%). The
similarity between peripheral blood and ascite samples was the
lowest (39.3%).
In addition, the mutation sites and types in the
same gene were different among patients. For example, a missense
mutation (D268E) was detected in the PTEN gene in the ctDNA
from patients H3 and H14, whilst a deletion mutation (T321fs) in
the same gene was detected in the ctDNA from patient H13.
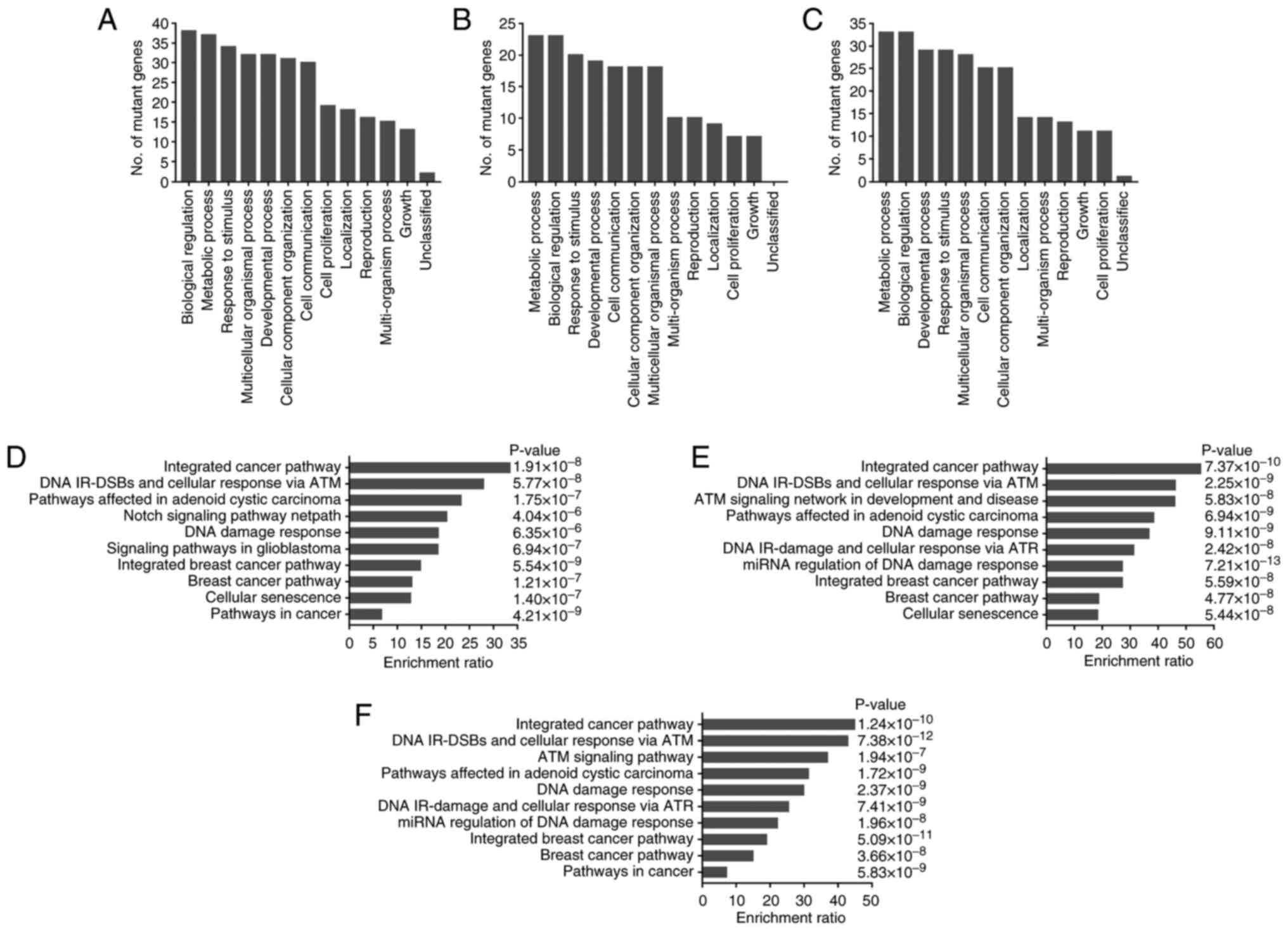
Functional analysis of the mutant
genes
To further explore the potential functions of these
mutated genes in ovarian cancer samples, the mutant genes found in
≥2 patients were analyzed using the WebGestalt tool. GO analysis
revealed that the mutant genes in peripheral blood, tumor tissue
and ascites samples mainly participated in the biological processes
of ‘Response to stimulus’, ‘biological regulation’, ‘metabolic
process’, ‘cell communication’ and ‘cell proliferation’ (Fig. 3A-C). These mutant genes were also
analyzed for KEGG and Wikipathway enrichment. The enriched pathways
in peripheral blood, tumor tissue and ascites samples were similar
and mainly included ‘integrated cancer pathway’, ‘DNA IR-DSBs and
cellular response via ATM’, ‘DNA damage response’, ‘Cellular
senescence’ and ‘Notch signaling pathway netpath’ (Fig. 3D-F).
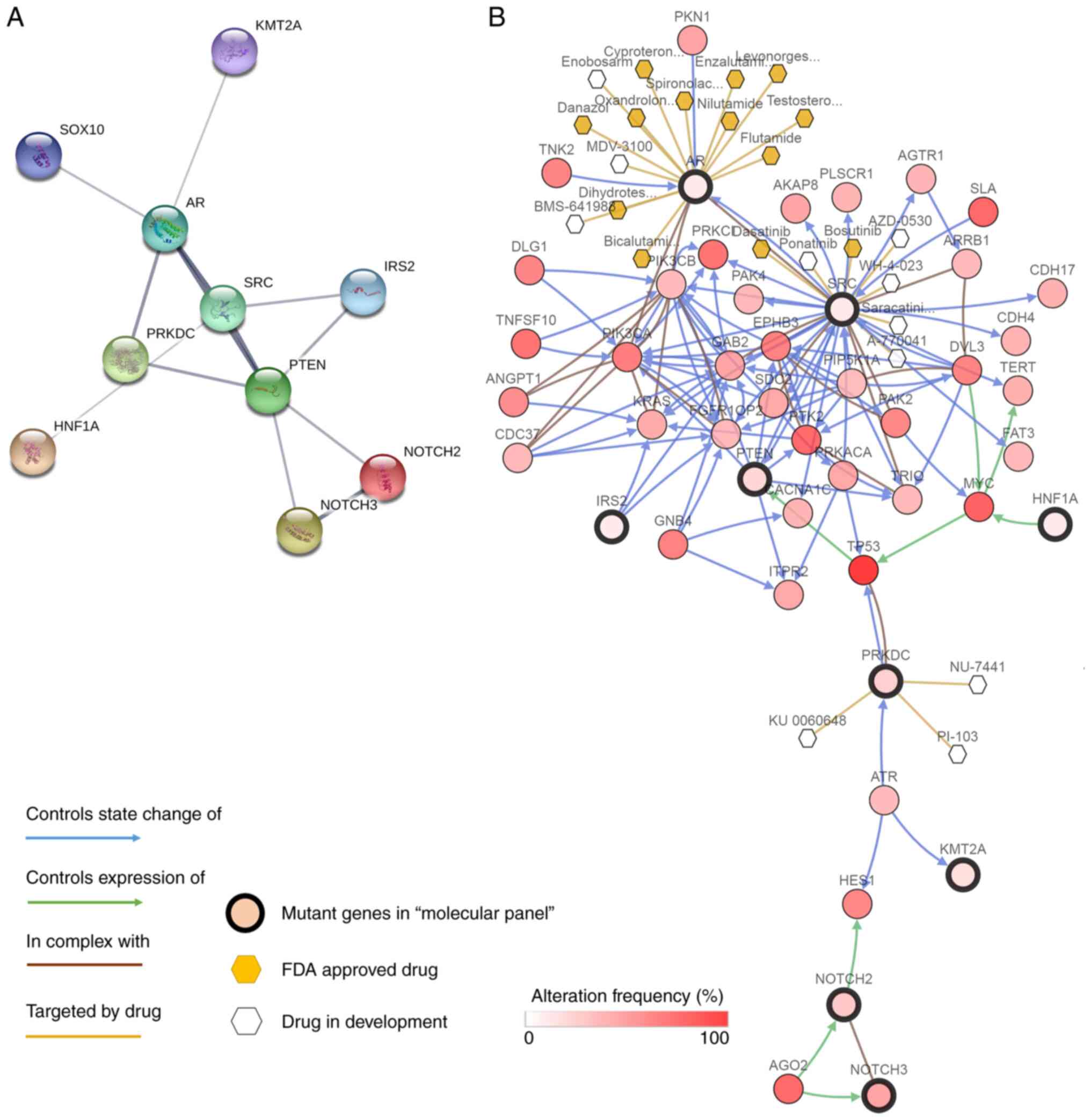
Potential of the mutant genes for
metastasis evaluation
To find the potential of using mutant genes in the
ctDNA samples to monitor metastasis, ‘metastasis’ and ‘ovarian
cancer’ were used as key words in Chilibot to analyze the 40 mutant
genes presented in ≥2 patients. A cluster covering 10 genes, namely
NOTCH2, NOTCH3, lysine methyltransferase 2A (KMT2A), PTEN, androgen
receptor (AR), DNA-activated protein kinase catalytic subunit
(PRKDC), hepatocyte nuclear factor 1 homeobox A (HNF1A), SRC,
insulin receptor substrate 2 (IRS2) and SRY-box transcription
factor 10 (SOX10), was obtained. This cluster was then designated
as the mutant gene panel for performing correlation analysis
between tumor dissemination and metastasis in patients with ovarian
cancer. The coverage of this mutant gene panel in the 18 patients
who underwent ctDNA sequencing in the present study was 94.4%
(17/18). This count was based on tumor lesions >1 cm in the
abdominal and pelvic cavities during intraoperative exploration,
although miliary lesions were not counted. If the number of tumor
lesions was >10 in a patient, then the number of lesions was
counted as 10. Pearson correlation analysis found that the number
of mutant genes in this mutant gene panel was positively correlated
with the number of tumor lesions with a correlation coefficient of
0.851 (P<0.001). This suggests that this panel derived from
ctDNA samples may have the potential to be used in reflecting tumor
dissemination and metastasis. Furthermore, an interaction network
of the mutant genes within this panel was obtained using STRING
(Fig. 4A), which indicated the
direct or indirect interactions among the mutant genes.
In addition, a validation analysis of this mutant
gene panel was performed in matching tumor tissue and ascites
samples isolated from the same patient. The coverage of this panel
in the paired tumor tissues and ascites was 100%. In total,
mutations in six of the genes in the panel, namely NOTCH2, KMT2A,
PTEN, AR, PRKDC and SOX10, were found in ascites. By contrast,
mutations in eight genes in the panel, specifically NOTCH2, KMT2A,
PTEN, AR, PRKDC, HNF1A, IRS2 and SOX10, were found in the ovarian
cancer tissues.
Further exploration of mutations
associated with targeted therapy
To assess whether the mutant genes had the potential
to become targets for ovarian cancer therapy, the cBioPortal online
tool was used for exploration. This integrates gene-centric
drug-target information. It was found that the genes, except SOX10,
in the present panel are either currently under investigation as a
target or can already be targeted therapeutically (Fig. 4B). The hormone therapy class of
therapeutic agents, such as cyproterone and enzalutamide, can be
used for antitumor therapy by targeting AR, whereas there are
numerous small-molecule drugs that can target SRC (32,33).
NOTCH2 and NOTCH3 can also be targeted for ovarian cancer, since
NOTCH3 knockdown has been found to increase the chemosensitivity of
paclitaxel-resistant ovarian cancer cells (34,35).
Discussion
Previous studies on the utility of ctDNA of ovarian
cancer have mainly focused on early detection and screening
(36,37), treatment response evaluation
(38-40)
and prognosis prediction (41).
Higher levels of somatic mutations in ctDNA have been associated
with decreased clinical benefit from treatment and shorter
progression-free survival or overall survival (40,42-44).
By applying NGS or PCR, various types of genetic alterations in
ctDNA and/or ovarian cancer tumor tissues have been found,
including chromosomal rearrangements (45), chromosomal instability (46), DNA methylation (47,48),
and gene mutation and amplification (43,49,50).
In the present study, NGS and public database resources based on
patient characteristics were used to analyze the profile and
function of the mutant genes and mutation patterns in ctDNA, paired
ascites and tumor tissue samples from patients with ovarian cancer.
By investigating the possible concordance and differences among
patients and types of specimens, the mechanism underlying the
heterogeneity of ovarian cancer was studied from multiple
angles.
ctDNA may have the ability to reveal information
regarding the mutation status of all genes involved in the tumor
features (51,52). In the present study, ctDNA
sequencing was performed in patients with ovarian cancer, where 104
mutant genes were found in all plasma samples. Subsequently, a
mutant gene panel that associated the most with tumor metastasis
and dissemination was established after analyzing the 40 genes
present in ≥2 patient among the ctDNA samples. These results
suggest that ctDNA, as a conventional type of non-invasive liquid
biopsy, may have the potential for application in monitoring the
tumor metastasis process dynamically. However, the dynamic changes
in ctDNA expression and mutation profile before and after tumor
relapse were not explored in the present study. In addition, the
dynamic changes in the ctDNA profile and their association with
imaging data and CA125 levels were not analyzed. Therefore, this
mutant gene panel requires further validation in future
studies.
Through the mutual verification and comparative
analysis of sequencing data from ctDNA, ascites and tumor tissues,
an understanding of the overall mutational landscape and the
heterogeneity of ovarian cancer was at least partially revealed.
The heterogeneity of gene mutations in ovarian cancer is commonly
observed, where they are manifested as differences in the mutation
locations and mutation types (53). In the present study, this
heterogeneity was observed from three angles; the inter-tumor
heterogeneity among different patients, the intra-tumor
heterogeneity among different lesions within the same patient and
the clonal heterogeneity in different subclones within the same
lesion were all considered. It was then predicted that gene
mutations in different types of specimens from the same patient
were also heterogeneous in the present study. In particular, some
of the gene mutations that could not be detected in tumor tissues
could be detected in ctDNA samples, which can be explained by the
existence of tumor subclonal heterogeneity and due to the space
restriction of tumor sampling. However, it should be noted that the
source of ctDNA is not only tumor cells. The cells of the tumor
microenvironment, including stromal cells, endothelial cells and
immune cells, may release DNA into the circulatory system in
patients with cancer (54,55).
ctDNA confers advantages in being able to reflect
the overall tumor information compared with the space restrictions
of tissue biopsy, particularly in disease monitoring and management
(52,56). However, this does not suggest that
ctDNA can be used to fully represent all tumor information.
Comparative analysis results showed that mutation detection data
from ctDNA cannot completely cover the mutant genes found in
ascites and tumor tissues. Plasma mutation detection typically has
low positive predictive value, meaning that more sensitive ctDNA
assays are needed due to the low quantity of ctDNA in a number of
patients (57). As a result,
parallel sequencing analysis of liquid biopsies, tumor tissues and
ascites can be used to unravel the overall tumor information more
comprehensively. Combination of clinical, surgical, pathological
and molecular features all facilitates the transformation of linear
treatments into multidisciplinary and integrated approaches, in
turn promoting the advent of individualized therapy to improve the
survival outcome (58-60).
Therefore, comprehensive analysis into the genetic landscape will
likely contribute to the realization of multidisciplinary
approaches and personalized medicine in the field of ovarian cancer
treatment.
The present study has a number of limitations. A
small sample size of matched ctDNA, ascite and tumor tissue samples
was evaluated. Low quantities of ascites, insufficient remaining
tumor tissues after pathological sampling and quality problems of
the tissue samples all contributed to the exclusion of some samples
in the present study. As a form of non-invasive examination,
peripheral blood samples for ctDNA detection can be easily
collected. This maybe one of the reasons why the number of cases
with available ctDNA was larger compared with those with paired
tumor tissues or ascites. The correlation analysis between
mutations in the gene panel in the ctDNA samples and patient
survival was lacking in the present study. The cancer panel used in
this study included 333 tumor-related genes covering the exons and
some introns of coding genes, meaning that mutations in non-coding
regions were not explored. This also meant that the ctDNA
sequencing data in the present study were not comparable to the
sequencing data in TCGA. The sequencing data in TCGA were based on
tumor tissues. Therefore, only TCGA was used to assist in screening
for mutations due to the difference in subjects between the present
study and the external dataset. In addition, since all data in
public databases were based on existing reported studies, relying
on public databases to assist in screening for genes would run the
risk of overlooking significant mutations that were not previously
reported.
In conclusion, the present study analyzed the mutant
genes of ctDNA and found a molecular panel that can be used to
reflect the dissemination and metastasis process of ovarian cancer.
In addition, this panel of mutant genes was validated against
external data sets, whereas the sequencing data of this panel of
mutant genes were compared among ctDNA, matching ascites and tumor
tissue samples. A deeper understanding of the overall mutational
landscape and heterogeneity was obtained in ovarian cancer. Results
from the present study suggest that it is not sufficient to rely
solely on the information provided by a specific type of specimens
for investigating the genomic landscape and screening for mutations
in cancers. Combining the information obtained from multiple types
of samples with other diagnostic information facilitates more
comprehensive understanding of the pathophysiology of cancer. For
future clinical applications of liquid biopsy, a standardized and
optimized method for interpreting and utilizing NGS and PCR data is
required.
Supplementary Material
Supplementary Data
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by grants from the
National Natural Sciences Foundation of China (grant no. 82172747)
and the Shanghai Medical Center of Key Programs for Female
Reproductive Diseases (grant no. 2017ZZ01016).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are not publicly available due to patient privacy
protection and the Regulation of the People's Republic of China
Administration of Human Genetic Resources (Document Number: Order
No. 717 of the State Council of the People's Republic of China) but
are available from the corresponding author on reasonable
request.
Authors' contributions
CX and XZ designed the study. XJie performed the
experiments and analyzed the data. MD and MZ performed data
collection and analyzed the data. XJin performed data collection
and part of the data analysis. QC performed part of the data
analysis. XJie, CX and XZ confirmed the authenticity of the raw
data. All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethical
Committee of Obstetrics and Gynecology Hospital of Fudan University
(approval no. 2016-48; Shanghai, China). Written informed consent
was obtained prior to initiation of the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2022. CA Cancer J Clin. 72:7–33.
2022.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2020. CA Cancer J Clin. 70:7–30. 2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Jelovac D and Armstrong DK: Recent
progress in the diagnosis and treatment of ovarian cancer. CA
Cancer J Clin. 61:183–203. 2011.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Sundar S, Neal RD and Kehoe S: Diagnosis
of ovarian cancer. BMJ. 351(h4443)2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sabini C, Sorbi F, Cunnea P and Fotopoulou
C: Ovarian cancer stem cells: Ready for prime time? Arch Gynecol
Obstet. 301:895–899. 2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lee JY, Yoon JK, Kim B, Kim S, Kim MA, Lim
H, Bang D and Song YS: Tumor evolution and intratumor heterogeneity
of an epithelial ovarian cancer investigated using next-generation
sequencing. BMC Cancer. 15(85)2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Schwarz RF, Ng CK, Cooke SL, Newman S,
Temple J, Piskorz AM, Gale D, Sayal K, Murtaza M, Baldwin PJ, et
al: Spatial and temporal heterogeneity in high-grade serous ovarian
cancer: A phylogenetic analysis. PLoS Med.
12(e1001789)2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bai H, Li H, Li W, Gui T, Yang J, Cao D
and Shen K: The PI3K/AKT/mTOR pathway is a potential predictor of
distinct invasive and migratory capacities in human ovarian cancer
cell lines. Oncotarget. 6:25520–25532. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Luo S, Zhang Y, Yang Y, Zhu S, Liu W, Zhu
J, Liang X, Jiang Z, Sun S, Hou X, et al: Clonal tumor mutations in
homologous recombination genes predict favorable clinical outcome
in ovarian cancer treated with platinum-based chemotherapy. Gynecol
Oncol. 158:66–76. 2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Castellarin M, Milne K, Zeng T, Tse K,
Mayo M, Zhao Y, Webb JR, Watson PH, Nelson BH and Holt RA: Clonal
evolution of high-grade serous ovarian carcinoma from primary to
recurrent disease. J Pathol. 229:515–524. 2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Bhardwaj BK, Thankachan S, Venkatesh T and
Suresh PS: Liquid biopsy in ovarian cancer. Clin Chim Acta.
510:28–34. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Asante DB, Calapre L, Ziman M, Meniawy TM
and Gray ES: Liquid biopsy in ovarian cancer using circulating
tumor DNA and cells: Ready for prime time? Cancer Lett. 468:59–71.
2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Tuaeva NO, Falzone L, Porozov YB, Nosyrev
AE, Trukhan VM, Kovatsi L, Spandidos DA, Drakoulis N, Kalogeraki A,
Mamoulakis C, et al: Translational application of circulating DNA
in oncology: Review of the last decades achievements. Cells.
8(1251)2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Sorenson GD, Pribish DM, Valone FH, Memoli
VA, Bzik DJ and Yao SL: Soluble normal and mutated DNA sequences
from single-copy genes in human blood. Cancer Epidemiol Biomarkers
Prev. 3:67–71. 1994.PubMed/NCBI
|
|
15
|
Zhou S, Xu B, Qi L, Zhu D, Liu B and Wei
J: Next-generation sequencing reveals mutational accordance between
cell-free DNA from plasma, malignant pleural effusion and ascites
and directs targeted therapy in a gastric cancer patient. Cancer
Biol Ther. 20:15–20. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Barbosa A, Peixoto A, Pinto P, Pinheiro M
and Teixeira MR: Potential clinical applications of circulating
cell-free DNA in ovarian cancer patients. Expert Rev Mol Med.
20(e6)2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Wyatt AW, Annala M, Aggarwal R, Beja K,
Feng F, Youngren J, Foye A, Lloyd P, Nykter M, Beer TM, et al:
Concordance of circulating tumor DNA and matched metastatic tissue
biopsy in prostate cancer. J Natl Cancer Inst.
109(djx118)2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Xie F, Zhang Y, Mao X, Zheng X, Han-Zhang
H, Ye J, Zhao R, Zhang X and Sun J: Comparison of genetic profiles
among primary lung tumor, metastatic lymph nodes and circulating
tumor DNA in treatment-naïve advanced non-squamous non-small cell
lung cancer patients. Lung Cancer. 121:54–60. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Xu B, Shan G, Wu Q, Li W, Wang H, Li H,
Yang Y, Long Q and Zhao P: Concordance of genomic alterations
between circulating tumor DNA and matched tumor tissue in Chinese
patients with breast cancer. J Oncol. 2020(4259293)2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Imperial R, Nazer M, Ahmed Z, Kam AE,
Pluard TJ, Bahaj W, Levy M, Kuzel TM, Hayden DM, Pappas SG, et al:
Matched whole-genome sequencing (WGS) and whole-exome sequencing
(WES) of tumor tissue with circulating tumor DNA (ctDNA) analysis:
Complementary modalities in clinical practice. Cancers (Basel).
11(1399)2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wan R, Wang Z, Lee JJ, Wang S, Li Q, Tang
F, Wang J, Sun Y, Bai H, Wang D, et al: Comprehensive analysis of
the discordance of EGFR mutation status between tumor tissues and
matched circulating tumor DNA in advanced non-small cell lung
cancer. J Thorac Oncol. 12:1376–1387. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Liu HE, Vuppalapaty M, Wilkerson C, Renier
C, Chiu M, Lemaire C, Che J, Matsumoto M, Carroll J, Crouse S, et
al: Detection of EGFR Mutations in cfDNA and CTCs, and comparison
to tumor tissue in non-small-cell-lung-cancer (NSCLC) patients.
Front Oncol. 10(572895)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Kidess-Sigal E, Liu HE, Triboulet MM, Che
J, Ramani VC, Visser BC, Poultsides GA, Longacre TA, Marziali A,
Vysotskaia V, et al: Enumeration and targeted analysis of KRAS,
BRAF and PIK3CA mutations in CTCs captured by a label-free
platform: Comparison to ctDNA and tissue in metastatic colorectal
cancer. Oncotarget. 7:85349–85364. 2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Beije N, Helmijr JC, Weerts MJA, Beaufort
CM, Wiggin M, Marziali A, Verhoef C, Sleijfer S, Jansen MPHM and
Martens JWM: Somatic mutation detection using various targeted
detection assays in paired samples of circulating tumor DNA,
primary tumor and metastases from patients undergoing resection of
colorectal liver metastases. Mol Oncol. 10:1575–1584.
2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Chimonidou M, Strati A, Malamos N, Kouneli
S, Georgoulias V and Lianidou E: Direct comparison study of DNA
methylation markers in EpCAM-positive circulating tumour cells,
corresponding circulating tumour DNA, and paired primary tumours in
breast cancer. Oncotarget. 8:72054–72068. 2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhang Y, Chang L, Yang Y, Fang W, Guan Y,
Wu A, Hong S, Zhou H, Chen G, Chen X, et al: Intratumor
heterogeneity comparison among different subtypes of non-small-cell
lung cancer through multi-region tissue and matched ctDNA
sequencing. Mol Cancer. 18(7)2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Giannopoulou L and Lianidou ES: Liquid
biopsy in ovarian cancer. Adv Clin Chem. 97:13–71. 2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Jagelkova M, Zelinova K, Laucekova Z,
Bobrovska M, Dankova Z, Grendar M and Dokus K: Comparison of
somatic mutation profiles between formalin-fixed paraffin embedded
tissues and plasma cell-free DNA from ovarian cancer patients
before and after surgery. Biores Open Access. 9:73–79.
2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Du ZH, Bi FF, Wang L and Yang Q:
Next-generation sequencing unravels extensive genetic alteration in
recurrent ovarian cancer and unique genetic changes in
drug-resistant recurrent ovarian cancer. Mol Genet Genomic Med.
6:638–647. 2018.PubMed/NCBI View
Article : Google Scholar
|
|
30
|
Han MR, Lee SH, Park JY, Hong H, Ho JY,
Hur SY and Choi YJ: Clinical implications of circulating tumor DNA
from ascites and serial plasma in ovarian cancer. Cancer Res Treat.
52:779–788. 2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBioPortal. Sci Signal. 6(pl1)2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Mayer EL and Krop IE: Advances in
targeting Src in the treatment of breast cancer and other solid
malignancies. Clin Cancer Res. 16:3526–3532. 2010.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Kim LC, Song L and Haura EB: Src kinases
as therapeutic targets for cancer. Nat Rev Clin Oncol. 6:587–595.
2009.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Kang H, Jeong JY, Song JY, Kim TH, Kim G,
Huh JH, Kwon AY, Jung SG and An HJ: Notch3-specific inhibition
using siRNA knockdown or GSI sensitizes paclitaxel-resistant
ovarian cancer cells. Mol Carcinog. 55:1196–1209. 2016.PubMed/NCBI View
Article : Google Scholar
|
|
35
|
Zhang J, Yin XJ, Xu CJ, Ning YX, Chen M,
Zhang H, Chen SF and Yao LQ: The histone deacetylase SIRT6 inhibits
ovarian cancer cell proliferation via down-regulation of Notch 3
expression. Eur Rev Med Pharmaco. 19:818–824. 2015.PubMed/NCBI
|
|
36
|
Cohen PA, Flowers N, Tong S, Hannan N,
Pertile MD and Hui L: Abnormal plasma DNA profiles in early ovarian
cancer using a non-invasive prenatal testing platform: Implications
for cancer screening. BMC Med. 14(126)2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Phallen J, Sausen M, Adleff V, Leal A,
Hruban C, White J, Anagnostou V, Fiksel J, Cristiano S, Papp E, et
al: Direct detection of early-stage cancers using circulating tumor
DNA. Sci Transl Med. 9(eaan2415)2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Lin KK, Harrell MI, Oza AM, Oaknin A,
Ray-Coquard I, Tinker AV, Helman E, Radke MR, Say C, Vo LT, et al:
BRCA reversion mutations in circulating tumor DNA predict primary
and acquired resistance to the PARP inhibitor rucaparib in
high-grade ovarian carcinoma. Cancer Discov. 9:210–219.
2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Alves MC, Fonseca FLA, Yamada AMTD, Barros
LADR, Lopes A, Silva LCFF, Luz AS, Melo Cruz FJS and Del Giglio A:
Increased circulating tumor DNA as a noninvasive biomarker of early
treatment response in patients with metastatic ovarian carcinoma: A
pilot study. Tumour Biol. 42(1010428320919198)2020.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Kim YM, Lee SW, Lee YJ, Lee HY, Lee JE and
Choi EK: Prospective study of the efficacy and utility of TP53
mutations in circulating tumor DNA as a non-invasive biomarker of
treatment response monitoring in patients with high-grade serous
ovarian carcinoma. J Gynecol Oncol. 30(e32)2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Charo LM, Eskander RN, Okamura R, Patel
SP, Nikanjam M, Lanman RB, Piccioni DE, Kato S, McHale MT and
Kurzrock R: Clinical implications of plasma circulating tumor DNA
in gynecologic cancer patients. Mol Oncol. 15:67–79.
2021.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Yang F, Tang J, Zhao Z, Zhao C and Xiang
Y: Circulating tumor DNA: A noninvasive biomarker for tracking
ovarian cancer. Reprod Biol Endocrinol. 19(178)2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Noguchi T, Iwahashi N, Sakai K, Matsuda K,
Matsukawa H, Toujima S, Nishio K and Ino K: Comprehensive gene
mutation profiling of circulating tumor DNA in ovarian cancer: Its
pathological and prognostic impact. Cancers (Basel).
12(3382)2020.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Salemi R, Falzone L, Madonna G, Polesel J,
Cinà D, Mallardo D, Ascierto PA, Libra M and Candido S: MMP-9 as a
candidate marker of response to BRAF inhibitors in melanoma
patients with BRAFV600E mutation detected in
circulating-free DNA. Front Pharmacol. 9(856)2018.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Harris FR, Kovtun IV, Smadbeck J, Multinu
F, Jatoi A, Kosari F, Kalli KR, Murphy SJ, Halling GC, Johnson SH,
et al: Quantification of somatic chromosomal rearrangements in
circulating cell-free DNA from ovarian cancers. Sci Rep.
6(29831)2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Vanderstichele A, Busschaert P, Smeets D,
Landolfo C, Van Nieuwenhuysen E, Leunen K, Neven P, Amant F, Mahner
S, Braicu EI, et al: Chromosomal instability in cell-free DNA as a
highly specific biomarker for detection of ovarian cancer in women
with adnexal masses. Clin Cancer Res. 23:2223–2231. 2017.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Giannopoulou L, Mastoraki S, Buderath P,
Strati A, Pavlakis K, Kasimir-Bauer S and Lianidou ES: ESR1
methylation in primary tumors and paired circulating tumor DNA of
patients with high-grade serous ovarian cancer. Gynecol Oncol.
150:355–360. 2018.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Giannopoulou L, Chebouti I, Pavlakis K,
Kasimir-Bauer S and Lianidou ES: RASSF1A promoter methylation in
high-grade serous ovarian cancer: A direct comparison study in
primary tumors, adjacent morphologically tumor cell-free tissues
and paired circulating tumor DNA. Oncotarget. 8:21429–21443.
2017.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Noguchi T, Sakai K, Iwahashi N, Matsuda K,
Matsukawa H, Yahata T, Toujima S, Nishio K and Ino K: Changes in
the gene mutation profiles of circulating tumor DNA detected using
CAPP-Seq in neoadjuvant chemotherapy-treated advanced ovarian
cancer. Oncol Lett. 19:2713–2720. 2020.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Paracchini L, Beltrame L, Grassi T,
Inglesi A, Fruscio R, Landoni F, Ippolito D, Delle Marchette M,
Paderno M, Adorni M, et al: Genome-wide copy-number alterations in
circulating tumor DNA as a novel biomarker for patients with
high-grade serous ovarian cancer. Clin Cancer Res. 27:2549–2559.
2021.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Sidaway P: Prostate cancer: Mutations in
ctDNA reflect features of metastatic disease. Nat Rev Clin Oncol.
14(526)2017.PubMed/NCBI View Article : Google Scholar
|
|
52
|
ctDNA is a specific and sensitive
biomarker in multiple human cancers. Cancer Discov.
4(OF8)2014.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Lawrence MS, Stojanov P, Polak P, Kryukov
GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH,
Roberts SA, et al: Mutational heterogeneity in cancer and the
search for new cancer-associated genes. Nature. 499:214–218.
2013.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Thierry AR, El Messaoudi S, Gahan PB,
Anker P and Stroun M: Origins, structures, and functions of
circulating DNA in oncology. Cancer Metast Rev. 35:347–376.
2016.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Mouliere F and Thierry AR: The importance
of examining the proportion of circulating DNA originating from
tumor, microenvironment and normal cells in colorectal cancer
patients. Expert Opin Biol Ther. 12 (Suppl 1):S209–S215.
2012.PubMed/NCBI View Article : Google Scholar
|
|
56
|
ctDNA reveals targetable alterations.
Cancer Discov. 10(OF4)2020.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Franczak C, Filhine-Tressarieu P, Broséus
J, Gilson P, Merlin JL and Harlé A: Clinical interest of
circulating tumor DNA in oncology. Arch Med Res. 49:297–305.
2018.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Falzone L, Scandurra G, Lombardo V,
Gattuso G, Lavoro A, Distefano AB, Scibilia G and Scollo P: A
multidisciplinary approach remains the best strategy to improve and
strengthen the management of ovarian cancer (Review). Int J Oncol.
59(53)2021.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Dondi G, Coluccelli S, De Leo A, Ferrari
S, Gruppioni E, Bovicelli A, Godino L, Coadă CA, Morganti AG,
Giordano A, et al: An analysis of clinical, surgical, pathological
and molecular characteristics of endometrial cancer according to
mismatch repair status. A multidisciplinary approach. Int J Mol
Sci. 21(7188)2020.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Heudel PE, Devouassoux-Shisheboran M,
Taieb S, Genestie C, Selle F, Morice P, Rouzier R and Ray-Coquard
I: Multidisciplinary management of advanced ovarian cancer for an
optimal therapeutic strategy. Eur J Gynaecol Oncol. 38:175–180.
2017.PubMed/NCBI
|