Introduction
Sepsis is a systemic inflammatory syndrome caused by
infection that provides a major cause of mortality in children
(1). A previous study has
indicated that there are ~48.9 million patients with sepsis
worldwide, including ~11 million sepsis deaths, which account for
19.7% of the total global death (2). Patients with severe sepsis can
experience a complicated pathogenesis that also comprises
functional impairment of multiple organs (3). The lung is the main organ of sepsis,
and the most common complication of sepsis is acute lung injury
(ALI) (4). The clinical
manifestations of ALI are progressive hypoxemia and respiratory
distress, which may lead to acute respiratory distress syndrome
(ARDS) if the disease progresses (5). Due to the complex pathogenesis of
pediatric sepsis, which causes great damage to the body tissues and
organs and is difficult to treat, sepsis remains a difficult
problem that needs to be resolved in the medical field in the world
at present (6).
Proviral integrations of Moloney virus 2 (PIM2) is a
serine/threonine protein kinase that exists on the X chromosome in
three isotypes (34 kDa, 37 kDa and 40 kDa) and is highly expressed
in lymphatic and brain tissues (7). PIM2 fulfills important roles in cell
proliferation, apoptosis and other biological processes including
migration, invasion and autophagy (8). At present, the majority of studies on
PIM2 focus on its role in hematological malignancies and solid
tumors, and it has been revealed that PIM2 exerts a promoting
effect on the occurrence and development of multiple myeloma
(9), prostate cancer (10), liver cancer (11) and other tumors, including lung
cancer, ovarian cancer. However, previously published studies have
indicated that PIM2 may exert certain immune regulation effects
(12,13). Yang et al (14) demonstrated that PIM2 can induce
interleukin (IL)-6 production through the IL-1β or TNF-α pathway,
thereby regulating the occurrence of inflammatory diseases. A
previous study revealed that PIM2 is a prognostic marker of
pediatric sepsis (15). Inhibition
of PIM2 has also been indicated to alleviate asthma symptoms and to
improve airway hyper-responsiveness and airway inflammation
(16). Collectively, these data
have indicated that PIM2 may exhibit a role in pediatric
sepsis.
Moreover, low doses of PIM2 protein have been
demonstrated to have a protective effect on oxidative
stress-induced neuronal cell death (17), and PIM2 expression is significantly
upregulated in lipopolysaccharide (LPS)-induced mouse macrophages
(18). Knockdown of PIM2 causes a
marked reduction in the expression of IL-1β and NLR family pyrin
domain-containing 3 (NLRP3) inflammasome in LPS-induced
macrophages, thereby alleviating LPS-induced ARDS (18). In addition, PIM2 expression has
been revealed to be elevated in lung cancer tissue, which may have
a specific regulatory role in the occurrence and development of
lung cancer (19). Therefore, it
is reasonable to hypothesize that PIM2 is involved in LPS-induced
injury of pulmonary epithelial cells.
The present study explored the role of PIM2 in
LPS-induced in vitro ALI cell model in bronchial epithelial
cells and its underlying mechanisms so as to provide a theoretical
basis for the treatment of lung injury caused by pediatric
sepsis.
Materials and methods
Cell culture
Human BEAS-2B pulmonary epithelial cells, obtained
from American Type Culture Collection, were cultured in
Gibco® DMEM with 10% FBS (both Thermo Fisher Scientific,
Inc.) in a humidified incubator at 37˚C and an atmosphere of 5%
CO2. BEAS-2B cells were induced by LPS (at
concentrations of 0, 2.5, 5 and 10 µg/ml) at 37˚C for 12 h to
create an in vitro ALI model.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA from the cells was isolated using
Invitrogen® TRIzol® reagent (Thermo Fisher
Scientific, Inc.). cDNA was synthesized using a
Fermentas® First-Strand cDNA Synthesis kit (Thermo
Fisher Scientific, Inc.). A SYBR Green Master Mix (20 µl;
Invitrogen; Thermo Fisher Scientific, Inc.) was used to perform the
PCR. The reaction thermocycling conditions were as follows: 95˚C
for 5 min, followed by 40 cycles of 94˚C for 15 sec, 60˚C for 20
sec and 72˚C for 40 sec. RT-qPCR using an Applied
Biosciences® ABI StepOnePlus Real-time PCR system
(Thermo Fisher Scientific, Inc.) was used to detect the
transcripts. The expression level was calculated using the
2-ΔΔCq method (20),
and the levels were normalized against GAPDH as the housekeeping
gene. The results are expressed as the fold-changes. The sequences
were designed by Guangzhou RiboBio Co., Ltd. PIM2 forward,
5'-CGTGGAGTTGTCCATCGTG-3', and reverse, 5'-AAGGGAATGTCCCCACACAC-3';
Toll-like receptor 2 (TLR2) forward, 5'-TTGTGACCGCAATGGTATCT-3',
and reverse, 5'-TGTTGGACAGGTCAAGGCT-3'; GAPDH forward,
5'-CATGAGAAGTATGACAACAGCCT-3', and reverse,
5'-AGTCCTTCCACGATACCAAAGT-3'.
Western blotting
Cells were washed with PBS and collected in RIPA
lysis buffer (Cell Signaling Technology, Inc.). Subsequently, the
cells were centrifuged at 16,000 x g for 10 min at 4˚C, and the
supernatant was collected for western blotting. A bicinchoninic
acid protein assay kit (Thermo Fisher Scientific, Inc.) was used to
detect the protein concentrations. Protein (20 µg per lane) samples
were analyzed by (either 10 or 12%) SDS-polyacrylamide gel
electrophoresis and subsequently transferred onto a PVDF membrane.
The membranes were then blocked in 5% skimmed milk powder for 1 h
at room temperature and subsequently incubated overnight at 4˚C
with primary antibodies, including anti-PIM2 (cat. no. ab129057),
anti-Bcl-2 (cat. no. ab32124), anti-Bax (cat. no. ab182733),
anti-cleaved caspase-3 (cat. no. ab32042), anti-caspase-3 (cat. no.
ab32351), anti-cleaved caspase-9 (cat. no. ab2324), anti-caspase-9
(cat. no. ab32539), anti-monocyte chemoattractant protein-1 (MCP-1;
cat. no. ab214819), anti-inducible nitric oxide synthase (iNOS;
cat. no. ab178945), anti-cyclooxygenase (COX)-2 (cat. no.
ab179800), anti-TLR2 (cat. no. ab68159), anti-myeloid
differentiation primary response 88 (MyD88; cat. no. ab133739),
anti-phosphorylated (p)-p65 (cat. no. ab76302), anti-p65 (cat. no.
ab32536) and anti-GAPDH (cat. no. ab9485) all from Abcam (all
dilution, 1:1,000). Membranes were subsequently incubated with
horseradish peroxidase-conjugated secondary antibody (1:5,000
dilution; cat. no. ab150077, Abcam) at room temperature for 1 h.
The protein bands were colored using an enhanced chemiluminescence
kit (Bio-Rad Laboratories, Inc.) and ImageJ 1.50i (National
Institutes of Health) gel analysis software was used to analyze the
bands. GAPDH was used as the loading control.
Cell transfection
Cells were transfected either with PIM2-small
interfering (si)RNAs or with scrambled negative control (NC) siRNA
(200 nM; Shanghai GenePharma Co., Ltd.). Transfection of pcDNA3.1
overexpression vector (GenScript) encoding the full-length TLR2 for
overexpression of TLR2 or the empty plasmid negative control (NC)
produced by Shanghai GenePharma Co., Ltd all at the concentration
of 20 nM was performed using the transfection reagent
Lipofectamine™ 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) at
37˚C for 24 h. The sequences of the si-PIM2-1 and si-PIM2-2 primers
were as follows: Si-PIM2-1, 5'-ACCUUCUUCCCGACCCUCAdTdT-3' (sense),
and 5'-UGAGGGUCGGGAAGAAGGUdTdT-3'(antisense); si-PIM2-2,
5'-CUUGGUUUUACAGGUCAUUdTdT-3' (sense), and
5'-AAUGACCUGUAAAACCAAGdCdT-3' (antisense); si-NC,
5'-UUCUCCGAACGUGUCACGUTT-3' (sense), and
5'-ACGUGACACGUUCGGAGAATT-3' (antisense). At 48 h after
transfection, subsequent experiments were conducted.
Cell Counting Kit-8 (CCK-8) assay
Cells were seeded and cultured at a density of
5x103/well in 96-well microplates (Corning, Inc.).
Subsequently, the cells were treated with LPS for 12 h, and
transfected accordingly as described above. CCK-8 reagent (10 µl;
Beyotime Institute of Biotechnology) was added to each well, and
the cells were subsequently cultured for 2 h. A microplate reader
(Bio-Rad Laboratories, Inc.) was used to analyze the absorbance at
450 nm.
TUNEL staining
Apoptosis was assessed using a TUNEL assay kit
(Beyotime Institute of Biotechnology) in accordance with the
manufacturer's protocol. Cells were washed with PBS and then fixed
with 4% paraformaldehyde for 30 min at room temperature.
Subsequently, cells were incubated with permeabilization solution
for 5 min at room temperature followed by TUNEL solution for 1 h at
37˚C. Subsequently, 50 µl DAB substrate was added for 10 min at
15˚C, and hematoxylin was used to re-stain the cells for 10 sec at
room temperature. Slides were mounted using glycerol. The cells
were observed under glass coverslip with PBS under an inverted
microscope (magnification, x200; Olympus Corporation). A total of
three fields were randomly selected for analysis of the images.
Detection of reactive oxygen species
(ROS) production
Intracellular ROS levels were analyzed using the
fluorescence intensity of 2,7-dichlorofluorescein. BEAS-2B cells
cultured on 6-well chamber slides (1x106) were washed
with PBS three times for 5 min/wash, and the slides were incubated
with the fluorescent probe 2,7-dichlorodihydrofluorescindiacetate
(1:1,000 dilution) at 37˚C for 30 min in the dark. The fluorescence
signals were analyzed on a BD FACSCanto™ Clinical Flow Cytometry
System (BD Biosciences), and FlowJo version 7.6 software (FlowJo
LLC) was used to analyze the data.
Detection of superoxide dismutase
(SOD) and plasma glutathione peroxidase (GSH-Px) production
BEAS-2B cells were centrifuged at 1,600 x g for 10
min at 4˚C and the cellular supernatant was collected in order to
detect the levels of SOD (cat. no. HM10163) and GSH-Px (cat. no.
HM10129) using ELISA kits (Bio-Swamp; Wuhan Bienle Biotechnology
Co., Ltd.) according to the manufacturer's instructions. A
microplate reader was subsequently used to measure the absorbance
at 450 nm absorbance wavelength.
ELISA
Briefly, BEAS-2B cells were seeded into 96-well
plates (5x103 cells/well). Following treatment, cell
supernatant was collected after centrifugation at 2,000 x g for 5
min at 4˚C. The intracellular concentrations of IL-1β (cat. no.
557953), IL-6 (cat. no. 555220) and TNF-α (cat. no. 555212) were
assessed using BD OptEIA™ Human IL-1β ELISA Set Ⅱ, BD OptEIA™ Human
IL-6 ELISA Set and BD OptEIA™ Human TNF ELISA Set (BD Biosciences),
and all procedures were performed according to the manufacturer's
instructions.
Statistical analyses
Statistical analyses were performed using SPSS
software (version 18.0; SPSS, Inc.). The data are presented as the
mean ± SD from at least three independent experiments. Comparisons
between two groups were performed using unpaired Student's t-test,
whereas comparisons among multiple groups were performed using
one-way ANOVA followed by a Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Interference with PIM2 increases the
viability of LPS-induced BEAS-2B cells
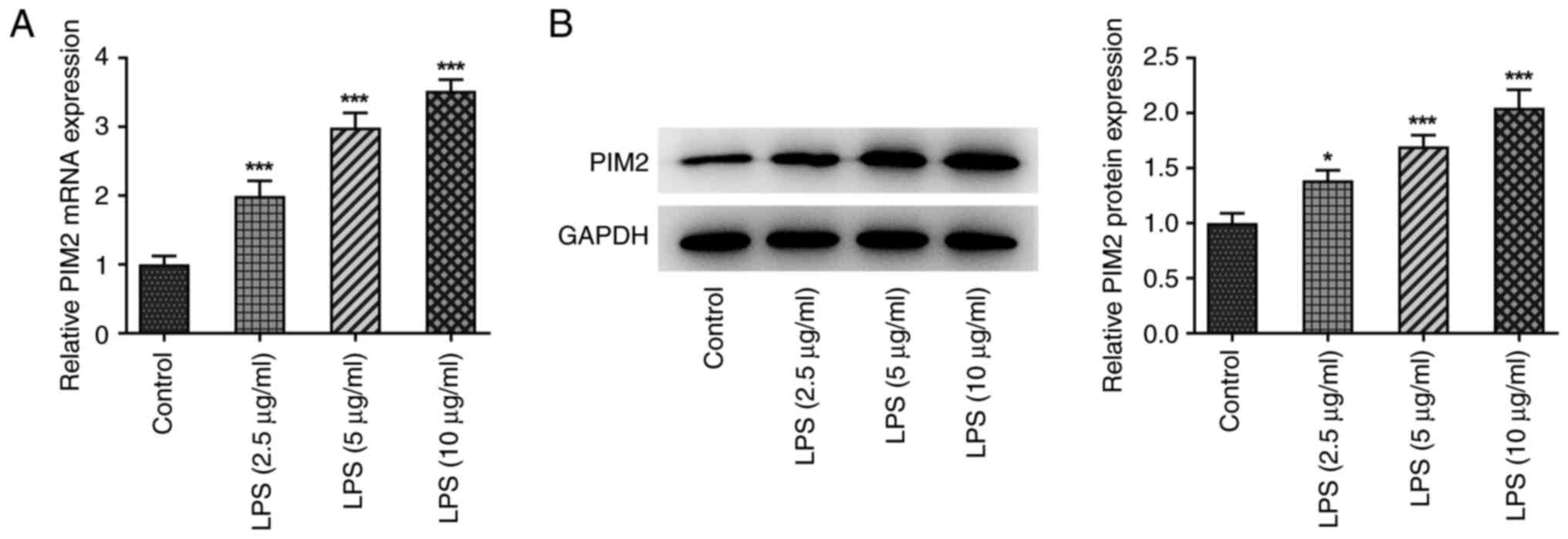
First, expression of PIM2 was detected using both
RT-qPCR and western blotting in BEAS-2B cells induced by different
concentrations of LPS (0, 2.5, 5 and 10 µg/ml). The results
obtained demonstrated that, compared with the control group, the
expression level of PIM2 increased markedly in concert with the
increases in concentration of LPS, which was used to induce the
cells (Fig. 1A and B). Since PIM2 expression was the most
significantly increased when induced using 10 µg/ml LPS, LPS at
this concentration was consequently selected for subsequent
experiments. PIM2 expression was then inhibited using the cell
transfection technique. RT-qPCR and western blotting were used to
determine the efficiency of siRNA interference of the cells. The
results demonstrated that the expression level of PIM2 in the
si-PIM2-1 and si-PIM2-2 groups were significantly decreased
compared with the si-NC group, indicating that the interference was
successful. In addition, as the expression of PIM2 in the si-PIM2-1
group was decreased more obviously compared with the si-PIM2-2
group, si-PIM2-1 was selected to be used in the follow-up
experiments (Fig. 2A and B).
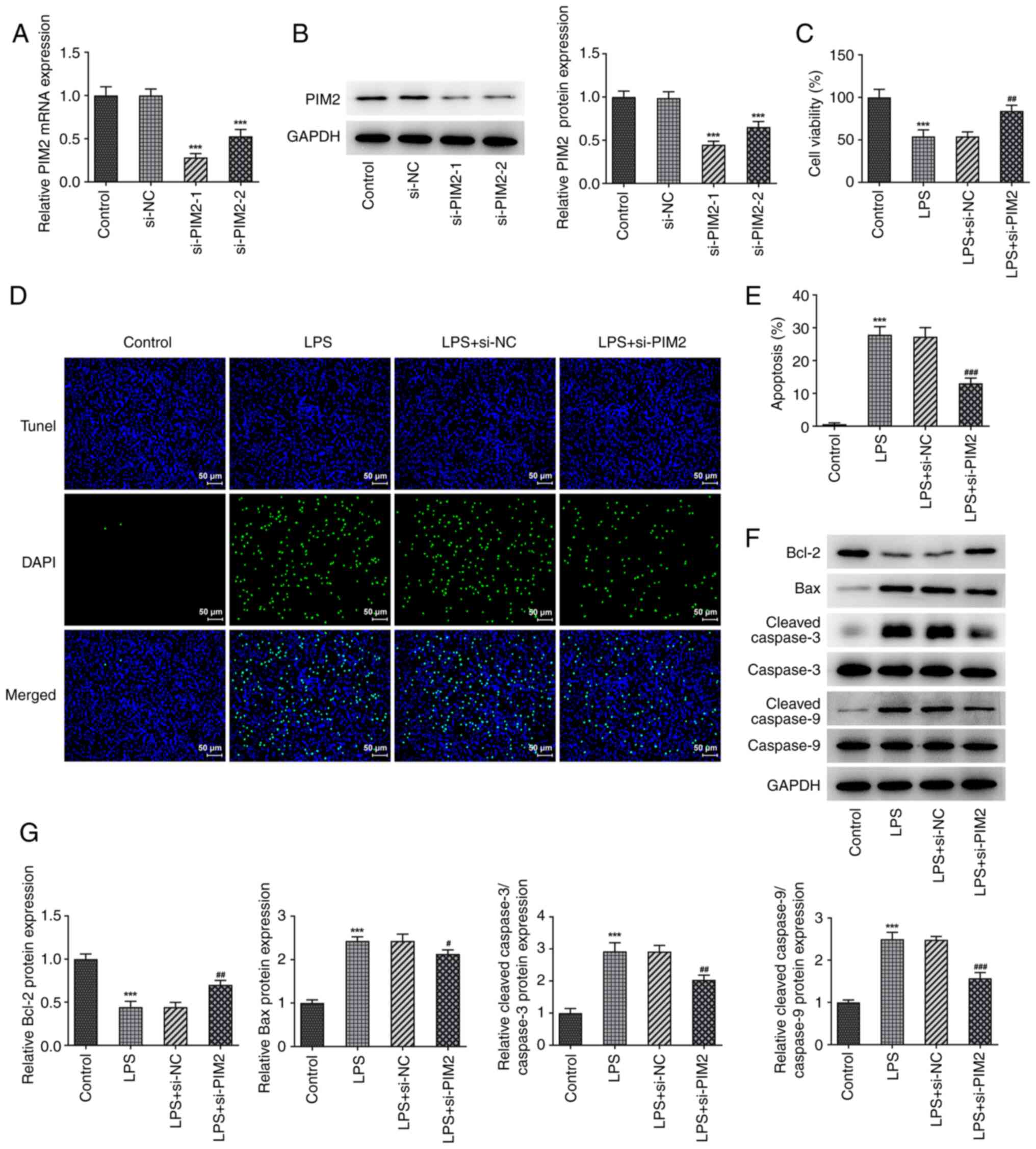 | Figure 2Interference with PIM2 increases
viability of LPS-induced BEAS-2B cells. (A) Reverse
transcription-quantitative PCR and (B) western blotting detected
the expression levels of PIM2 mRNA and protein, respectively, after
transfection of PIM2. ***P<0.001 vs. si-NC. (C) Cell
Counting Kit-8 detected the viability in LPS-induced BEAS-2B cells
after transfection of PIM2. (D and E) TUNEL assay detected the
apoptosis of LPS-induced BEAS-2B cells after transfection of PIM2
(scale bar, 50 µm). (F) Western blotting detected the expression
levels of apoptosis-related proteins (Bcl-2, Bax and cleaved
caspase-3/caspase-3) in LPS-induced BEAS-2B cells after
transfection of PIM2. (G) Statistical analyses of a
apoptosis-related protein bands. ***P<0.001 vs.
control; #P<0.05, ##P<0.01,
###P<0.001 vs. LPS + si-NC. PIM2, Proviral
integrations of Moloney virus 2; LPS, lipopolysaccharide; NC,
negative control; si-, small interfering. |
The CCK-8 assay results demonstrated that cell
viability was significantly decreased after LPS induction compared
with the control group. Compared with the LPS + si-NC treatment
group, the cell viability of the LPS + si-PIM2 group was
significantly increased (Fig. 2C).
Subsequently, the TUNEL assay results indicated that, compared with
the control group, apoptosis was significantly increased in the
BEAS-2B cells induced with LPS only (Fig. 2D and E). In addition, western blotting
demonstrated that LPS-induced cells demonstrated a significant
decrease in Bcl-2 expression and significant increases in Bax and
cleaved caspase-3 and -9 expression levels compared with the
control group (Fig. 2F and
G). Moreover, compared with the
LPS + si-NC group, PIM2 interference significantly reduced the
level of apoptosis LPS + si-PIM2 group, which was accompanied by a
significantly increased level of Bcl-2 expression and decreased
expression levels of Bax and cleaved caspase-3 and -9 (Fig. 2D-G). PIM2 silencing exacerbated the
viability of LPS-treated BEAS-2B cells.
Interference with PIM2 inhibits
oxidative stress and the inflammatory response of LPS-induced
BEAS-2B cells
ROS expression levels were detected using a
fluorescence kit, and the expression levels of SOD and GSH-Px were
detected using corresponding kits. The results obtained
demonstrated that, ROS expression in the LPS-induced BEAS-2B cells
significantly increased following LPS induction compared with the
control group; whereas the expression levels of SOD and GSH-Px were
significantly decreased. Compared with the LPS + si-NC group, the
ROS expression levels in the LPS + si-PIM2 group were significantly
decreased after interfering with PIM2 expression, whereas those of
SOD and GSH-Px were significantly increased (Fig. 3A-C). In addition, the expression
levels of TNF-α, IL-6 and IL-1β were significantly increased
following LPS induction compared with the control group. However,
following PIM2 interference, the expression levels of these factors
were significantly decreased in the LPS + si-PIM2 group compared
with the LPS + si-NC group (Fig.
3D). Finally, the expression levels of the
inflammation-associated proteins MCP-1, iNOS and COX-2 were
revealed to be consistent with the trends of TNF-α, IL-6 and IL-1β
as determined from western blotting (Fig. 3E). In conclusion, PIM2 knockdown
ameliorated LPS-evoked oxidative stress and inflammatory response
in BEAS-2B cells.
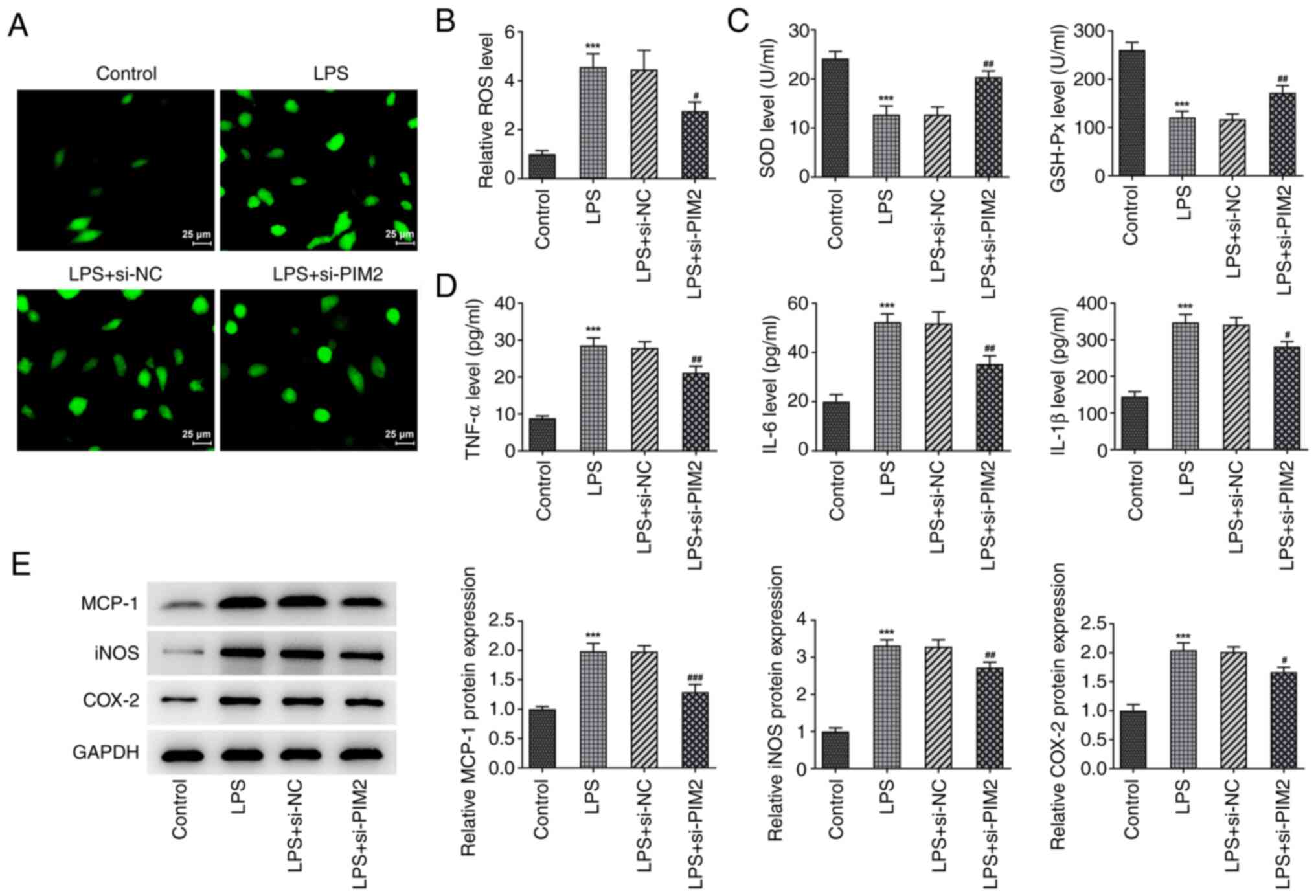 | Figure 3Interference with PIM2 inhibits
oxidative stress and inflammatory response of LPS-induced BEAS-2B
cells. (A) A DCFH-DA kit was used to detect the expression of ROS
and (B) was quantified. (C) SOD and GSH-Px kits were used to detect
the expression of SOD and GSH-Px. (D) ELISA was used to detect the
expression of inflammation factors, TNF-α, IL-6 and IL-1β. (E)
Western blotting detected the expression of inflammation-related
proteins (MCP-1, iNOS and COX-2) in LPS-induced BEAS-2B cells after
transfection. ***P<0.001 vs. control;
#P<0.05, ##P<0.01,
###P<0.001 vs. LPS + si-NC. PIM2, Proviral
integrations of Moloney virus 2; LPS, lipopolysaccharide; NC,
negative control; si-, small interfering; ROS, reactive oxygen
species; SOD, superoxide dismutase; GSH-Px, glutathione peroxidase;
IL, interleukin; MCP-1, monocyte chemoattractant protein-1; iNOS,
inducible nitric oxide synthase; COX-2, cyclooxygenase. |
Interference with PIM2 inhibits
activation of the TLR2/MyD88 signaling pathway in LPS-induced
BEAS-2B cells
The western blotting results demonstrated that the
expression levels of TLR2, MyD88 and p-P65 in BEAS-2B cells were
significantly increased following LPS induction compared with the
control, indicating that LPS could induce activation of the
TLR2/MyD88 pathway. Compared with the LPS + si-NC treatment group,
the expression levels of TLR2, MyD88 and p-P65 were significantly
inhibited following PIM2 expression (Fig. 4). These results preliminarily
suggested that PIM2 interference was able to inhibit the activation
of TLR2/MyD88 pathway. Taken together, PIM2 depletion inactivated
TLR2/MyD88 signaling in LPS-treated BEAS-2B cells.
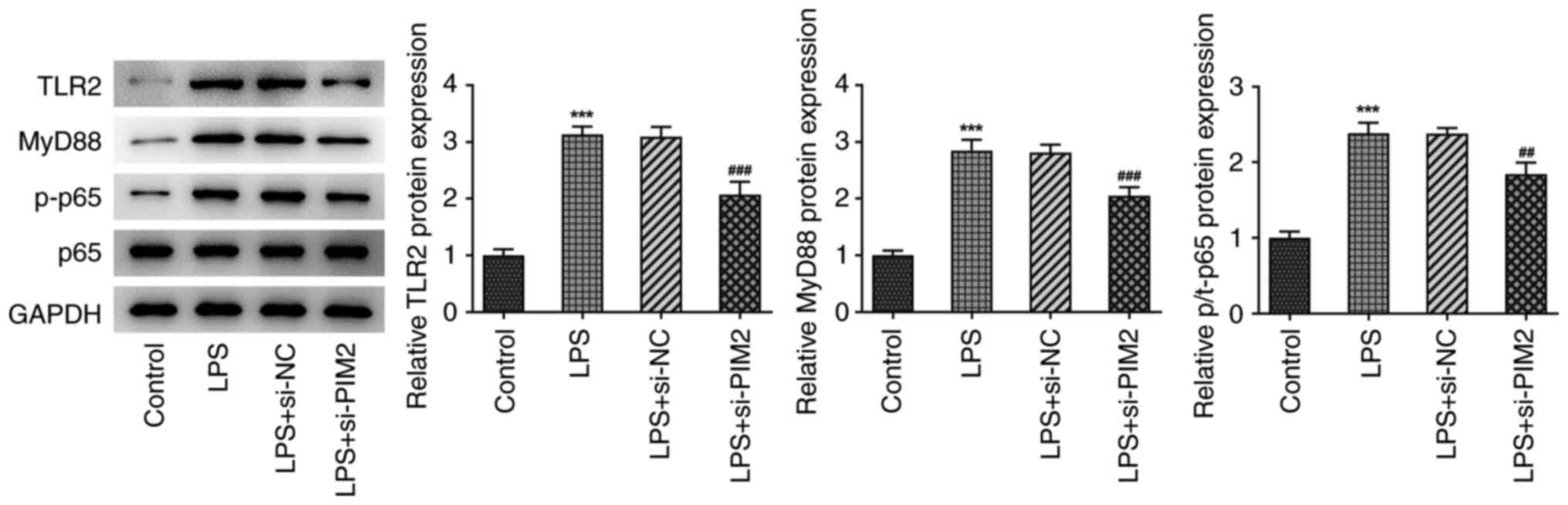 | Figure 4Interference with PIM2 inhibited the
activation of the TLR2/MyD88 signaling pathway in LPS-induced
BEAS-2B cells. Western blotting detected the expression of
TLR2/MyD88 signaling pathway associated proteins, TLR2, MyD88 and
p/t-P65. ***P<0.001 vs. control;
##P<0.01, ###P<0.001 vs. LPS + si-NC.
p-, phosphorylated; t-, total; PIM2, Proviral integrations of
Moloney virus 2; LPS, lipopolysaccharide; NC, negative control;
si-, small interfering; TLR2, Toll-like receptor 2; MyD88, myeloid
differentiation primary response 88. |
Upregulation of TLR2 reverses the
inhibitory effect of PIM2 interference on BEAS-2B cell damage
Subsequently, the cells were grouped into control,
LPS, LPS + si-PIM2, LPS + si-PIM2 + overexpression (Oe)-NC and LPS
+ si-PIM2 + Oe-TLR2 treatment groups to further explore the
mechanism of PIM2 in LPS-induced BEAS-2B cell injury. First, TLR2
was overexpressed using the cell transfection technique, and the
transfection efficiency was confirmed using RT-qPCR and western
blotting assays (Fig. 5A and
B). It was revealed that, compared
with the LPS + si-PIM2 + Oe-NC treatment group, the LPS + si-PIM2 +
Oe-TLR2 group had a significantly decreased cell viability
(Fig. 5C) and significantly
increased levels of apoptosis (Fig.
5D and E); this was
accompanied by a significant decrease of Bcl-2 expression, and
significantly increased levels of Bax and cleaved/total caspase-3
and -9 expression (Fig. 5F).
Subsequently, the levels of the oxidative stress-associated
indicators ROS, SOD and GSH-Px (Fig.
6A-C), as well as the inflammatory indicators TNF-α, IL-6 and
IL-1β, were examined (Fig. 6D).
The results demonstrated that the expression levels of ROS were
significantly increased in the LPS + si-PIM2 + Oe-TLR2 group
compared with the LPS + si-PIM2 + Oe-NC group (Fig. 6A and B), while the expression levels of SOD and
GSH-Px were significantly decreased (Fig. 6C). In addition, the expression
levels of TNF-α, IL-6 and IL-1β were significantly increased in the
LPS + si-PIM2 + Oe-TLR2 group compared with the LPS + si-PIM2 +
Oe-NC group (Fig. 6D). The
expression trends of MCP-1, iNOS and COX-2 were revealed to be
consistent with those of the inflammatory factors TNF-α, IL-6 and
IL-1 (Fig. 6E). Collectively,
these results suggested that upregulation of TLR2 could reverse the
inhibitory effect of PIM2 interference on cell damage. Overall,
TLR2 elevation offset the mitigated BEAS-2B cell injury due to PIM2
deficiency.
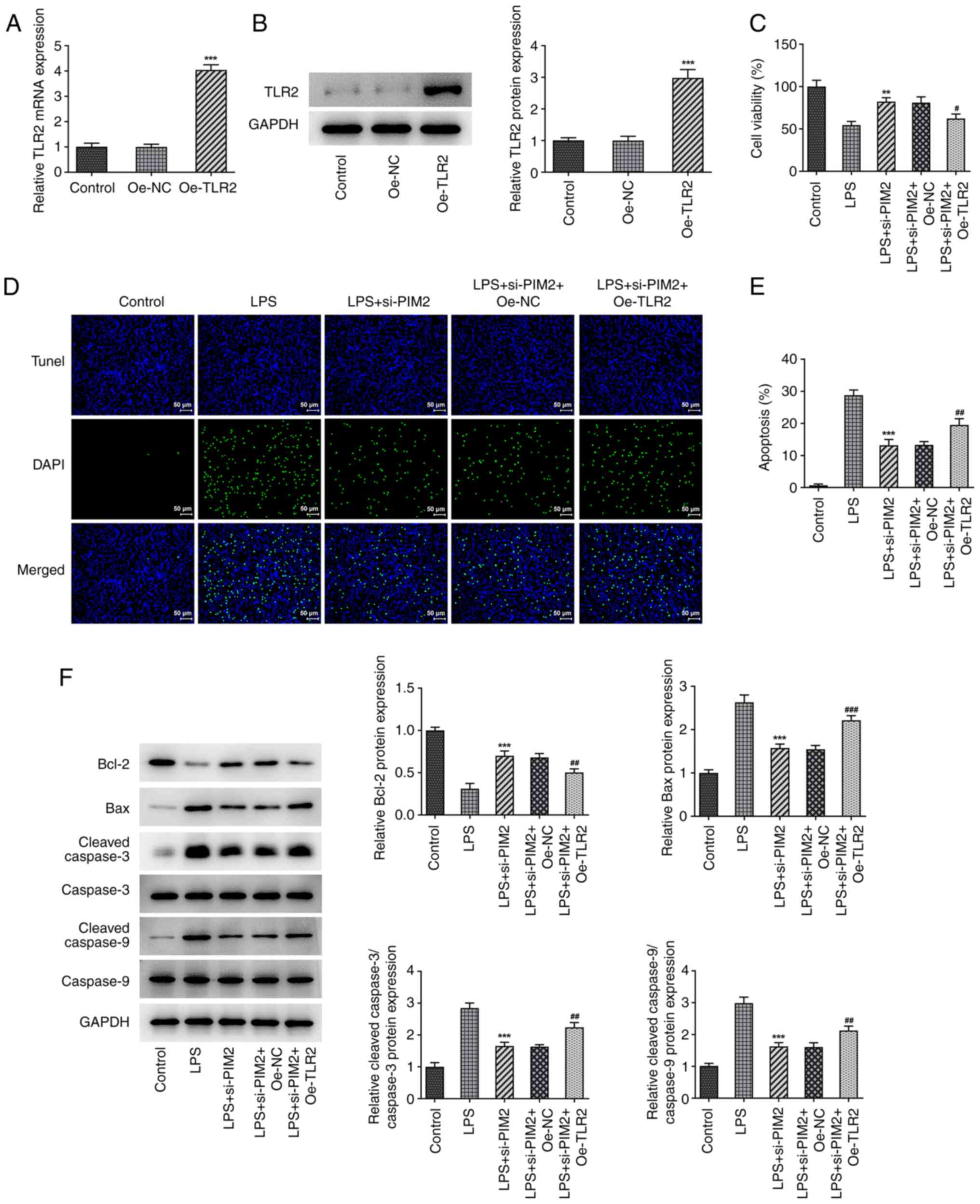 | Figure 5Upregulation of TLR2 reverses the
inhibitory effect of PIM2 interference on BEAS-2B cell viability.
(A) Reverse transcription-quantitative PCR and (B) western blotting
detected the expression of TLR2 mRNA and protein, respectively,
after transfection of TLR2. ***P<0.001 vs. si-NC. (C)
Cell Counting Kit-8 detected the viability in LPS-induced BEAS-2B
cells after transfection. (D and E) TUNEL assay detected the
apoptosis of LPS-induced BEAS-2B cells after transfection (scale
bar, 50 µm). (F) Western blotting detected the expression levels of
apoptosis-related proteins in LPS-induced BEAS-2B cells after
transfection. **P<0.01, ***P<0.001 vs.
LPS; #P<0.05, ##P<0.01,
###P<0.001 vs. LPS + si-PIM2 + Oe-TLR2. PIM2,
Proviral integrations of Moloney virus 2; LPS, lipopolysaccharide;
NC, negative control; si-, small interfering; TLR2, Toll-like
receptor 2; oe, overexpression. |
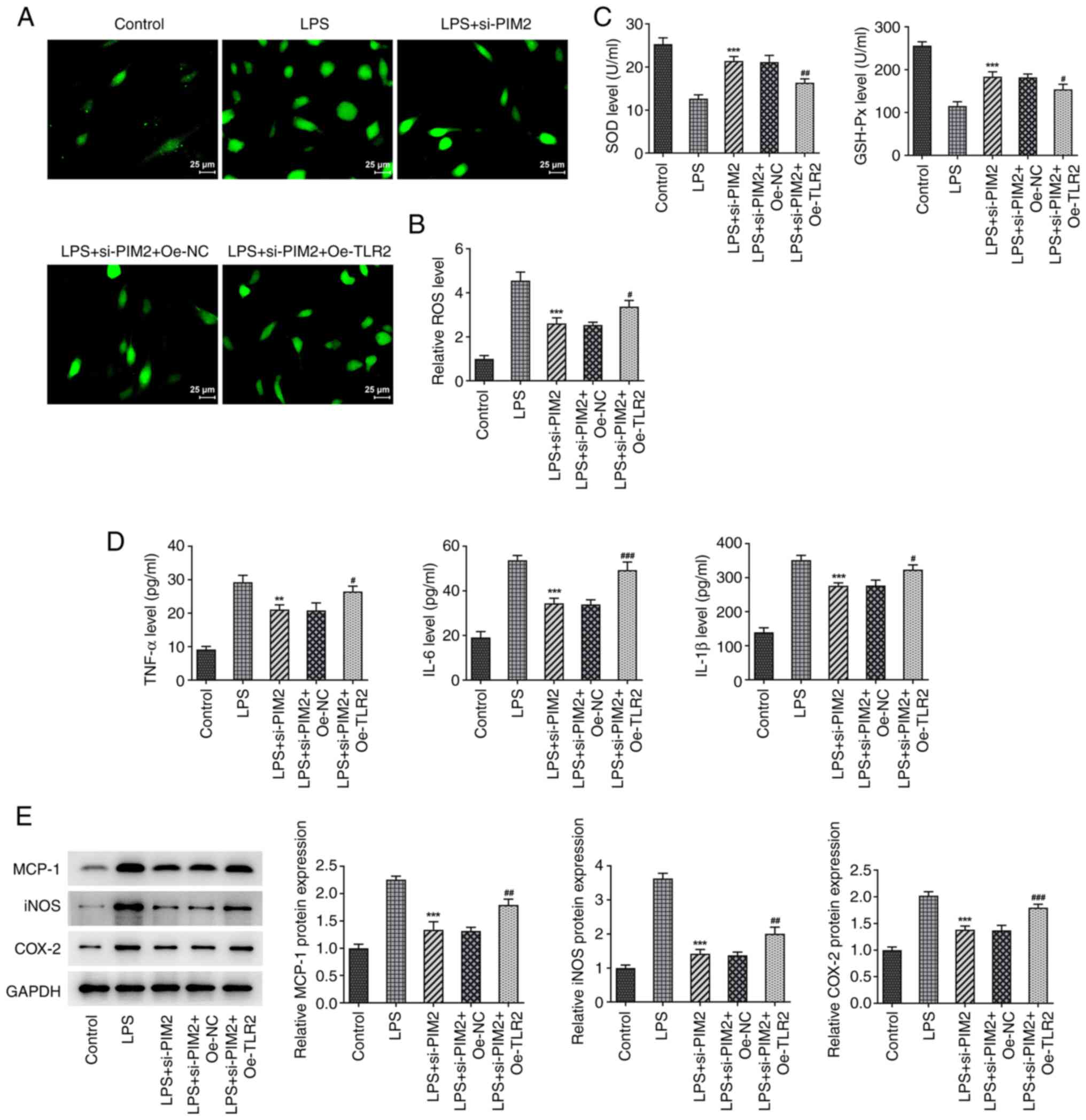 | Figure 6Upregulation of TLR2 reverses the
inhibitory effect of PIM2 interference on BEAS-2B cell oxidative
stress and inflammation. (A) DCFH-DA kit was used to detect and (B)
quantify the expression levels of ROS (scale bar, 25 µm). (C) SOD
and GSH-Px kits were used to detect the expression levels of SOD
and GSH-Px. (D) ELISA was used to detect the expression of
inflammation factors TNF-α, IL-6 and IL-1β. (E) Western blotting
detected the expression of inflammation-related proteins (MCP-1,
iNOS and COX-2) in LPS-induced BEAS-2B cells after transfection.
**P<0.01, ***P<0.001 vs. LPS;
#P<0.05, ##P<0.01,
###P<0.001 vs. LPS + si-PIM2 + Oe-TLR2. PIM2,
Proviral integrations of Moloney virus 2; LPS, lipopolysaccharide;
NC, negative control; si-, small interfering; ROS, reactive oxygen
species; SOD, superoxide dismutase; GSH-Px, glutathione peroxidase;
IL, interleukin; MCP-1, monocyte chemoattractant protein-1; iNOS,
inducible nitric oxide synthase; COX-2, cyclooxygenase; Oe,
overexpression. |
Discussion
A large number of inflammatory factors produced by
sepsis are able to enter the lung tissue via the blood circulation,
and accumulate in lung tissue to produce pulmonary inflammatory
reactions, thereby damaging the pulmonary capillary endothelial
cells and pulmonary epithelial cells, ultimately causing sepsis
combined with ALI (21,22). Children's bodily functions are not
fully developed, and for them, sepsis is more dangerous; therefore,
pediatric sepsis may lead to more serious organ damage, such as
ALI, which is more difficult to treat (23). Consequently, it is important to
explore the underlying mechanism through which sepsis causes lung
injury in children.
In the set of experiments performed in the present
study, LPS-induced BEAS-2B cells were used to simulate a model of
ALI induced by sepsis (24). In a
study by Wang et al (24),
LPS at 10 µg/ml was selected for induction, and LPS at this
concentration could induce the model of extracorporeal pneumonia.
In addition, the present study selected 2.5, 5 and 10 µg/ml LPS for
induction, and demonstrated that PIM2 expression was most
significantly increased after 10 µg/ml LPS induction. Therefore, 10
µg/ml LPS was selected in the present study. The results obtained
revealed that after LPS induction, cell viability decreased
significantly, the levels of apoptosis increased and oxidative
stress and inflammation occurred in the cells. Taken together,
these results demonstrated that the present model had been
successfully induced.
PIM2 is a prognostic marker of pediatric sepsis
(15); although, to the best of
our knowledge, the role of PIM2 both in pediatric sepsis and in
lung injury caused by sepsis has yet to be reported. PIM2 has been
reported to be highly expressed in lung cancer, and microarray
analysis has indicated that it is associated with cell
proliferation (25). In addition,
PIM1 and PIM2 share a high degree of amino acid sequence homology,
and their functions are fundamentally the same (26). PIM1 has been demonstrated to
inhibit the inflammatory response and cell viability of BEAS-2B
cells in obstructive pulmonary disease, thereby inducing cell death
(27). Therefore, it was possible
to hypothesize that PIM2 also fulfills a role in LPS-induced
BEAS-2B cells. In the present study, PIM2 expression was
significantly increased after the BEAS-2B cells had been induced by
LPS. Following the inhibition of PIM2 expression, cell viability
was increased, apoptosis was decreased and the cellular
inflammatory response was also decreased. Moreover, a previously
published study has indicated that PIM2 interference can inhibit
the expression of IL-1β and NLRP3 in LPS-induced macrophages,
thereby inhibiting inflammatory responses (18). Similarly, the present set of
experiments revealed that the expression levels of TNF-α, IL-6 and
IL-1β in LPS-induced BEAS-2B cells were decreased after interfering
with PIM2 expression, and the inflammatory response and oxidative
stress response were both inhibited. Collectively, these results
demonstrated that PIM2 interference could inhibit LPS-induced cell
damage.
Subsequently, the present study sought to unravel
the underlying regulatory mechanism of PIM2. It was revealed that
the TLR2/MyD88 signaling pathway was abnormally expressed during
the entire process. The TLR2/MyD88 inflammatory pathway forms an
important part of the body's inflammatory response (28). TLR2 interacts with MyD88, forming a
complex that activates downstream signal transduction through a
series of phosphorylation processes, which ultimately activates the
expression of various genes, including those of TNF-α, IL-1, IL-6
and adhesion molecules, thereby inducing corresponding inflammatory
responses (29). It has also been
demonstrated that inhibition of the TLR2/MyD88 pathway alleviates
LPS-induced ALI in a rat model (30). It is also hypothesized that TLR2
may bind LPS through the LPS-binding protein (31). In the present set of experiments,
the TLR2/MyD88 signaling pathway was revealed to be activated
following LPS-induced injury of the BEAS-2B cells.
PIM2 has been indicated to regulate the expression
of downstream signaling pathways in a TLR2-dependent manner
(32). In addition, a previously
published study demonstrated that PIM2 is able to activate the
NF-κB signaling pathway to promote the occurrence of liver cancer
(33), and that this activation is
caused by the activation of the TLR2/MyD88 signaling cascade
(34). Growth differentiation
factor 11, a regulator of skin biology, exerts a protective role in
LPS-induced lung injury through inhibiting the TLR2/high mobility
group box 1/NF-κB signaling axis (35). Therefore, it was possible to
hypothesize that PIM2 may exert a role in ALI caused by pediatric
sepsis by effecting downstream regulation of the TLR2/MyD8
signaling pathway. To further confirm this hypothesis, TLR2 was
overexpressed, which resulted in a reversal of the inhibition
mediated by PIM2 on the LPS-induced inflammatory response,
oxidative stress and apoptosis in BEAS-2B cells. These results
revealed that knockdown of PIM2 was able to alleviate LPS-induced
pulmonary epithelial cell injury via inhibiting the TLR2/MyD88
pathway.
The present study has certain limitations. The
conclusions of the current study were not verified in animal
experiments, so these will be verified in the following
experiments. In addition, the BEAS-2B cell line was used in our
experiment, which should be verified in more cell lines. Our
research group will also discuss other cell lines in the
future.
In conclusion, the present study confirmed that
knockdown of PIM2 was able to alleviate LPS-induced pulmonary
epithelial cell injury via inhibiting the TLR2/MyD88 pathway. The
present study has therefore provided a potential novel basis for
the treatment of ALI caused by pediatric sepsis.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JD and XY conceived and designed the study. HH and
BW performed the experiments. XY and HH analyzed the experimental
data. JD and BW wrote and revised the manuscript. XY and HH confirm
the authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cecconi M, Evans L, Levy M and Rhodes A:
Sepsis and septic shock. Lancet. 392:75–87. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Rudd KE, Johnson SC, Agesa KM, Shackelford
KA, Tsoi D, Kievlan DR, Colombara DV, Ikuta KS, Kissoon N, Finfer
S, et al: Global, regional, and national sepsis incidence and
mortality, 1990-2017: Analysis for the Global Burden of Disease
Study. Lancet. 395:200–211. 2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lelubre C and Vincent JL: Mechanisms and
treatment of organ failure in sepsis. Nat Rev Nephrol. 14:417–427.
2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Yan XX, Zheng AD, Zhang ZE, Pan GC and
Zhou W: Protective effect of pantoprazole against sepsis-induced
acute lung and kidney injury in rats. Am J Transl Res.
11:5197–5211. 2019.PubMed/NCBI
|
|
5
|
Li X, Jamal M, Guo P, Jin Z, Zheng F, Song
X, Zhan J and Wu H: Irisin alleviates pulmonary epithelial barrier
dysfunction in sepsis-induced acute lung injury via activation of
AMPK/SIRT1 pathways. Biomed Pharmacother.
118(109363)2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Uhle F, Lichtenstern C, Brenner T and
Weigand MA: Pathophysiology of sepsis. Anasthesiol Intensivmed
Notfallmed Schmerzther. 50:114–122. 2015.PubMed/NCBI View Article : Google Scholar : (In German).
|
|
7
|
Brault L, Gasser C, Bracher F, Huber K,
Knapp S and Schwaller J: PIM serine/threonine kinases in the
pathogenesis and therapy of hematologic malignancies and solid
cancers. Haematologica. 95:1004–1015. 2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Yang T, Ren C, Lu C, Qiao P, Han X, Wang
L, Wang D, Lv S, Sun Y and Yu Z: Phosphorylation of HSF1 by PIM2
Induces PD-L1 expression and promotes tumor growth in breast
cancer. Cancer Res. 79:5233–5244. 2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Fujii S, Nakamura S, Oda A, Miki H,
Tenshin H, Teramachi J, Hiasa M, Bat-Erdene A, Maeda Y, Oura M, et
al: Unique anti-myeloma activity by thiazolidine-2,4-dione
compounds with Pim inhibiting activity. Br J Haematol. 180:246–258.
2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ren K, Gou X, Xiao M, He W and Kang J:
Pim-2 cooperates with downstream factor XIAP to inhibit apoptosis
and intensify malignant grade in prostate cancer. Pathol Oncol Res.
25:341–348. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kronschnabl P, Grunweller A, Hartmann RK,
Aigner A and Weirauch U: Inhibition of PIM2 in liver cancer
decreases tumor cell proliferation in vitro and in vivo primarily
through the modulation of cell cycle progression. Int J Oncol.
56:448–459. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Yin G, Li Y, Yang M, Cen XM and Xie QB:
Pim-2/mTORC1 pathway shapes inflammatory capacity in rheumatoid
arthritis synovial cells exposed to lipid peroxidations. Biomed Res
Int. 2015(240210)2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Deng G, Nagai Y, Xiao Y, Li Z, Dai S,
Ohtani T, Banham A, Li B, Wu SL, Hancock W, et al: Pim-2 Kinase
influences regulatory T cell function and stability by mediating
Foxp3 protein N-terminal phosphorylation. J Biol Chem.
290:20211–20220. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Yang J, Li X, Hanidu A, Htut TM, Sellati
R, Wang L, Jiang H and Li J: Proviral integration site 2 is
required for interleukin-6 expression induced by interleukin-1,
tumour necrosis factor-α and lipopolysaccharide. Immunology.
131:174–182. 2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Tonial CT, Costa CAD, Andrades GRH,
Crestani F, Bruno F, Piva JP and Garcia PCR: Performance of
prognostic markers in pediatric sepsis. J Pediatr (Rio J).
97:287–294. 2021.
|
|
16
|
Du W, Chen T, Ni Y, Hou X, Yu Y, Zhou Q,
Wu F, Tang W and Shi G: Role of PIM2 in allergic asthma. Mol Med
Rep. 16:7504–7512. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Woo SJ, Shin MJ, Kim DW, Jo HS, In Yong J,
Ryu EJ, Cha HJ, Kim SJ, Yeo HJ, Cho SB, et al: Effects of low doses
of Tat-PIM2 protein against hippocampal neuronal cell survival. J
Neurol Sci. 358:226–235. 2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Wang F, Xu L, Dong G, Zhu M, Liu L and
Wang B: PIM2 deletion alleviates lipopolysaccharide (LPS)-induced
respiratory distress syndrome (ARDS) by suppressing NLRP3
inflammasome. Biochem Biophys Res Commun. 533:1419–1426.
2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Wang F, Gu T, Chen Y, Chen Y, Xiong D and
Zhu Y: Long non-coding RNA SOX21-AS1 modulates lung cancer progress
upon microRNA miR-24-3p/PIM2 axis. Bioengineered. 12:6724–6737.
2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Malard B, Lambert C and Kellum JA: In
vitro comparison of the adsorption of inflammatory mediators by
blood purification devices. Intensive Care Med Exp.
6(12)2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Wu C, Li H, Zhang P, Tian C, Luo J, Zhang
W, Bhandari S, Jin S and Hao Y: Lymphatic flow: A potential target
in sepsis-associated acute lung injury. J Inflamm Res. 13:961–968.
2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Emr BM, Alcamo AM, Carcillo JA, Aneja RK
and Mollen KP: Pediatric sepsis update: How are children different?
Surg Infect (Larchmt). 19:176–183. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wang J, Yuan X and Ding N: IGF2BP2
knockdown inhibits LPS-induced pyroptosis in BEAS-2B cells by
targeting caspase 4, a crucial molecule of the non-canonical
pyroptosis pathway. Exp Ther Med. 21(593)2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Guo WF, Lin RX, Huang J, Zhou Z, Yang J,
Guo GZ and Wang SQ: Identification of differentially expressed
genes contributing to radioresistance in lung cancer cells using
microarray analysis. Radiat Res. 164:27–35. 2005.PubMed/NCBI View
Article : Google Scholar
|
|
26
|
Liu Z, Han M, Ding K and Fu R: The role of
Pim kinase in immunomodulation. Am J Cancer Res. 10:4085–4097.
2020.PubMed/NCBI
|
|
27
|
de Vries M, Heijink IH, Gras R, den Boef
LE, Reinders-Luinge M, Pouwels SD, Hylkema MN, van der Toorn M,
Brouwer U, van Oosterhout AJ and Nawijn MC: Pim1 kinase protects
airway epithelial cells from cigarette smoke-induced damage and
airway inflammation. Am J Physiol Lung Cell Mol Physiol.
307:L240–L251. 2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wu J, Gan Y, Li M, Chen L, Liang J, Zhuo
J, Luo H, Xu N, Wu X, Wu Q, et al: Patchouli alcohol attenuates
5-fluorouracil-induced intestinal mucositis via TLR2/MyD88/NF-kB
pathway and regulation of microbiota. Biomed Pharmacother.
124(109883)2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Lin HY, Tang CH, Chen YH, Wei IH, Chen JH,
Lai CH and Lu DY: Peptidoglycan enhances proinflammatory cytokine
expression through the TLR2 receptor, MyD88, phosphatidylinositol
3-kinase/AKT and NF-kappaB pathways in BV-2 microglia. Int
Immunopharmacol. 10:883–891. 2010.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Hu JT, Lai J, Zhou W, Chen XF, Zhang C,
Pan YP, Jiang LY, Zhou YX, Zhou B and Tang ZH: Hypothermia
alleviated LPS-induced acute lung injury in Rat models through
TLR2/MyD88 pathway. Exp Lung Res. 44:397–404. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ding PH, Wang CY, Darveau RP and Jin L:
Porphyromonas gingivalis LPS stimulates the expression of
LPS-binding protein in human oral keratinocytes in vitro. Innate
Immun. 19:66–75. 2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Narayana Y, Bansal K, Sinha AY, Kapoor N,
Puzo G, Gilleron M and Balaji KN: SOCS3 expression induced by PIM2
requires PKC and PI3K signaling. Mol Immunol. 46:2947–2954.
2009.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Tang X, Cao T, Zhu Y, Zhang L, Chen J, Liu
T, Ming X, Fang S, Yuan YF, Jiang L, et al: PIM2 promotes
hepatocellular carcinoma tumorigenesis and progression through
activating NF-ĸB signaling pathway. Cell Death Dis.
11(510)2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Bansal K, Kapoor N, Narayana Y, Puzo G,
Gilleron M and Balaji KN: PIM2 Induced COX-2 and MMP-9 expression
in macrophages requires PI3K and Notch1 signaling. PLoS One.
4(e4911)2009.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Xu HB, Qin B, Zhang J, Chen YJ, Shen WW
and Cao LN: Growth differentiation factor 11 relieves acute lung
injury in mice by inhibiting inflammation and apoptosis. Eur Rev
Med Pharmacol Sci. 24:6908–6918. 2020.PubMed/NCBI View Article : Google Scholar
|