Introduction
Preeclampsia (PE) is a pregnancy-specific condition
that affects ~10% pregnancies worldwide and is one of the leading
causes of maternal and neonatal morbidity and mortality (1,2).
Trophoblast cells form the external tissues of embryos and serve an
important role in embryo development (3,4).
Insufficient invasion of cytotrophoblasts (CTBs) into the uterine
artery has been suggested to be an important factor in PE
pathogenesis (3,4). Despite improvements in perinatal
care, PE remains to be a serious clinical problem. Therefore, it is
of clinical and scientific significance to identify novel
therapeutic targets associated with this disease.
Peptidyl arginine deiminase 4 (PAD4) is an enzyme
that mediates the citrullination (post-translational deamination)
of arginine residues to citrullines, which reverses chromatin
condensation (5,6). A previous study has proposed that
PAD4 knockdown can reduce inflammation and susceptibility to
pregnancy loss in mice (7). In
addition, it has been reported that PAD4 could promote the nuclear
translocation of NF-κB and activate NF-κB signaling through NF-κB
essential modulator (NEMO) citrullination, which contributed to
inflammation and transformed the expression of several genes
involved in the pathogenesis of PE (8-10).
The NEMO gene, which induces the activation of NF-κB, is
located on the sex chromosome Xq28 and has been reported to be
notably decreased in pregnancies complicated by PE (11). Upregulated NEMO expression in the
maternal and fetal blood has been reported to be closely associated
with the development of PE (12).
Furthermore, total NEMO (1A, 1B and 1C transcripts) in maternal
blood and placentas of the PE subgroup were previously found to be
significantly increased compared with the controls (13). Therefore, this suggested that PAD4
knockdown can reduce the inflammation by regulating the NEMO/NF-κB
signaling pathway, thereby inhibiting the progression of PE.
In the present study, lipopolysaccharide (LPS) was
used to treat human trophoblast cells (HTR8/SVneo) to simulate the
PE model in vitro to evaluate the role of PAD4 in the
inflammation, invasion and migration of HTR8/SVneo cells and
investigate its potential regulatory effects on the NEMO/NF-κB
signaling pathway (14). These
findings might provide a theoretical and experimental basis for
exploring a novel target for the treatment of PE.
Materials and methods
Cell culture
The human trophoblast cell line HTR8/SVneo was
obtained from American Type Culture Collection. HTR8/SVneo cells
were previously developed by Graham et al (15). It was generated using freshly
isolated extravillous CTB from first trimester placenta and
transfected with a plasmid containing the simian virus 40 large T
antigen (SV40). A previous study demonstrated that this cell line
contains two populations, one of epithelial and the other of
mesenchymal origin (16). In the
present study, HTR-8/SVneo cells were cultured in RPMI-1640 medium
(Gibco; Thermo Fisher Scientific, Inc.) containing 10% fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific, Inc.). The incubator
was set as having 5% CO2 with a humidified atmosphere at
37˚C. To determine the effects of LPS on HTR-8/SVneo cells, LPS
(200 ng/ml; Sigma-Aldrich; Merck KGaA) was added to the culture
medium for 48 h at 37˚C, which was according to the previous study
(17).
Cell transfection
The pLVX lentiviral plasmid containing the short
hairpin RNA (shRNA) against PAD4 (shPAD4#1 or shPAD4#2) or a
scrambled shRNA (shNC) were synthesized by Shanghai GenePharma Co.,
Ltd. The transfection (50 ng plasmid) was done using Lipofectamine
3000 (Invitrogen; Thermo Fisher Scientific, Inc.) at 37˚C for 48 h,
in accordance with the manufacturer's protocols. Transfection
efficiency was evaluated using reverse transcription-quantitative
PCR (RT-qPCR) following 48 h of transfection. After 48 h
transfection, HTR8/SVneo cells were also harvested for further
experiments. The targeting sequences were as follows: shPAD4#1,
5'-GGTGACCCTGACGATGAAAGT-3'; shPAD4#2, 5'-GCAGCTCTTCAAGCTCAAAGA-3';
and shNC, 5'-TTCTCCGAACGTGTCACGT-3'.
Cell viability assay
For assessment of cell viability, cells were seeded
into 96-well plates and analyzed using Cell Counting Kit-8 (CCK-8;
Beijing Solarbio Science & Technology Co., Ltd.). Briefly,
after PAD4 knockdown and treatment with LPS (0, 100, 500, 1,000 and
1,500 ng/ml) for 12 h and TNF-α for 4 h, 10 µl CCK-8 working
solution was added to each well, followed by incubation with normal
cell culture medium for 2 h at 37˚C. Optical density in each well
was then evaluated at the 450 nm using a microplate reader (Bio-Rad
Laboratories, Inc.).
Measurement of inflammatory
factors
The contents of inflammatory cytokines IL-6 (cat.
no. F01440), IL-12 (cat. no. F01380) and monocyte chemoattractant
protein (MCP)-1 (cat. no. F01700) in the culture media were
measured using ELISA according to the manufacturer's protocol
(Shanghai Xitang Biotechnology Co., Ltd.). The optical density
values were then read on a plate reader (BioTek Instruments,
Inc.).
Transwell assay
Transwell assay was performed using 24-well culture
plates with 8-mm pore-size Falcon Transwell inserts (BD
Biosciences). HTR8/SVneo cells (1x105 cell/well) were
suspended in 200 µl serum-free RPMI 1640 and seeded into the upper
chamber of the Matrigel-coated Transwell insert for 8 h at 37˚C.
RPMI 1640 medium containing 10% FBS was placed into the lower
chamber. After 24 h of incubation at 37˚C, cells were fixed with 4%
paraformaldehyde (Sigma-Aldrich; Merck KGaA) for 30 min at room
temperature and then stained with 0.1% crystal violet
(Sigma-Aldrich; Merck KGaA) for 20 min at room temperature. The
stained cells were photographed under an inverted light microscope
(magnification, x100; Olympus Corporation). Image J software v1.8.0
(National Institutes of Health) was used to calculate the numbers
of cells invaded in ≥5 random images.
Wound healing assay
For wound healing assay, HTR8/SVneo cells were
seeded into six-well plates at a density of 5x105
cells/well and cultured to 90% confluence. A 200-µl sterile pipette
gun was used to create a wound on the cell surface. The cells were
then cultured in serum-free RPMI 1640 medium and cell migration was
measured at 0 and 48 h and 37˚C by detecting the average distance
of cells migrating into the wound as observed using inverted light
microscopy (magnification, x100; Olympus Corporation). Relative
cell migration rate=(scratch width at 0 h-scratch width after
culture)/scratch width at 0 h.
RT-qPCR analysis
Total RNA was extracted from HTR8/SVneo cells using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) in
accordance with the manufacturer's protocol. Complementary DNA
(cDNA) was synthesized using the PrimeScript™ RT Reagent
Kit (Takara Bio, Inc.). The temperature protocol was as follows:
16˚C for 30 min, 42˚C for 30 min and 85˚C for 5 min, followed by
storage at 4˚C. qPCR was then performed using 2 µg cDNA as the
template and Power SYBR® Green Master Mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.) on the ABI 7500 PCR
system (Applied Biosystems; Thermo Fisher Scientific, Inc.) with
the following thermocycling conditions: 30 sec at 95˚C for 1 cycle;
3 sec at 95˚C, 30 sec at 60˚C and 30 sec at 72˚C for 40 cycles; and
5 min at 72˚C for 1 cycle. The following primer sequences were used
in the present study: PAD4 forward, 5'-CCCAAACAGGGGGTATCAGT-3' and
reverse, 5'-CCACGGACAGCCAGTCAGAA-3' and GAPDH forward,
5'-TGTGGGCATCAATGGATTTGG-3' and reverse,
5'-ACACCATGTATTCCGGGTCAAT-3'. GAPDH was used as the internal
reference gene. Relative gene expression levels were calculated
using the 2-ΔΔCq method (18).
Western blot analysis
Total HTR-8/SVneo cell protein extracts were
obtained using RIPA lysis buffer (Beyotime Institute of
Biotechnology). Nuclear and cytosolic proteins were extracted using
the Nucleoprotein and Cytoplasmic Protein Extraction Kit obtained
from Nanjing KeyGen Biotech Co., Ltd. Concentrations of proteins
were quantified using a BCA kit (Beyotime Institute of
Biotechnology). Equal amounts of each protein sample (40 µg) were
added per lane and separated by electrophoresis in 10% SDS-PAGE and
transferred onto PVDF membranes. Possible non-specific binding was
blocked by 5% skimmed milk for 1 h at room temperature and then
incubated overnight at 4˚C with specific primary antibodies:
Anti-PAD4 (cat. no. ab214810; 1:1,000; Abcam), anti-NEMO (cat. no.
2685S; 1:1,000; Cell Signaling Technology, Inc.), anti-NF-κB p65
(cat. no. 8242T; 1:1,000; Cell Signaling Technology, Inc.),
anti-Histone H3 (cat. no. 4499T; 1:1,000; Cell Signaling
Technology, Inc.) and anti-GAPDH (cat. no. 5174T; 1:1,000; Cell
Signaling Technology, Inc.). The next day, goat anti-rabbit
HRP-conjugated secondary antibodies (cat. no. 31460; 1:10,000;
Thermo Fisher Scientific, Inc.) were added to the membranes and
incubated for 1 h at room temperature. Protein bands were scanned
and visualized using an enhanced chemiluminescence detection system
(Life Technologies; Thermo Fisher Scientific, Inc.). The relative
intensity of each band was quantified using Image J software v1.8.0
(National Institutes of Health). The protein expression was
normalized to either GAPDH or Histone H3 levels.
Immunoprecipitation
HTR-8/SVneo cells were lysed using lysis buffer for
immunoprecipitation (cat. no. 26146; Pierce; Thermo Fisher
Scientific, Inc.). Citrulline was immunoprecipitated from 50 µg
samples using an anti-citrulline antibody (cat. no. ab240908;
Abcam) and incubated for 1 h at 4˚C. This antibody-citrulline
protein immunocomplex was captured from the solution using 50 µg
Protein A/G Plus agarose beads (Santa Cruz Biotechnology, Inc.) and
incubated overnight at 4˚C. Samples were then centrifuged at 1,000
x g for 1 min at 4˚C and the pellet was washed with PBS for three
times followed by boiling in Laemmli buffer (Cell Signaling
Technology, Inc.) for 5 min at 100˚C. Total lysates and
immunoprecipitated samples were then subjected to western blotting
analysis.
Statistical analysis
GraphPad Prism software (version 8.0; GraphPad
Software, Inc.) was applied for data analysis. Data were expressed
as the mean ± standard deviation from three independent
experiments. Comparison between two independent groups was
conducted using unpaired Student's t test. Comparisons among
multiple groups were analyzed using one-way ANOVA followed by
Tukey's post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
PAD4 silencing attenuates the
inflammatory response by LPS-induced HTR8/SVneo cells
The expression of PAD4 was measured after
transfection with shPAD4#1 or shPAD4#2. Significantly decreased
PAD4 expression was observed in HTR8/SVneo cells transfected with
shPAD4#1 or shPAD4#2 compared with that in the shNC group (Fig. 1A). shPAD4#2 was selected for
subsequently experiments due to the lower PAD4 expression levels
induced. To evaluate the role of PAD4 following LPS-induced
inflammation in HTR8/SVneo cells, PAD4 expression was silenced by
transfection with shPAD4#2. As shown in Fig. 1B and C, LPS stimulation led to a significant
increase in PAD4 mRNA and protein expression compared with that in
the control group, which was significantly reversed by PAD4
knockdown. Subsequently, the levels of inflammatory factors were
measured using ELISA. The levels of IL-6, MCP-1 and IL-12 were
significantly elevated after HTR8/SVneo cells exposed to LPS
(Fig. 1D-F). By contrast, PAD4
knockdown partially reversed the promoting effects of LPS on the
secretion of IL-6, MCP-1 and IL-12 (Fig. 1D-F). These findings suggest that
PAD4 knockdown can attenuate the inflammatory response in
LPS-induced HTR8/SVneocells.
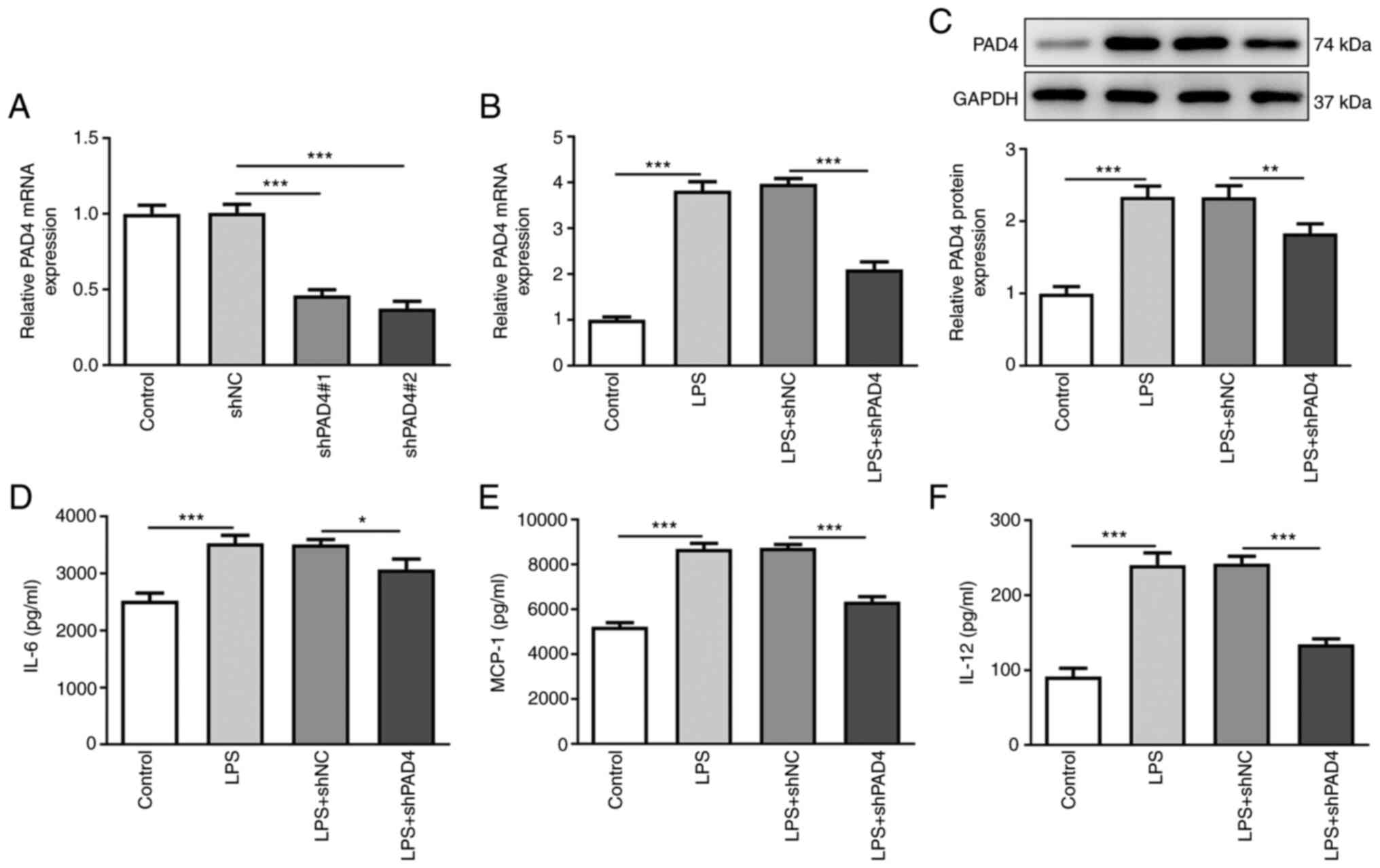 | Figure 1PAD4 knockdown alleviates the
inflammatory response in LPS-treated HTR8/SVneo cells. (A) PAD4
expression was measured by RT-qPCR after transfection with shNC,
shPAD4#1 or shPAD4#2 in HTR8/SVneo cells. The expression of (B)
PAD4 mRNA and (C) protein in LPS-stimulated HTR8/SVneo cells
transfected with PAD4 shPAD4 was measured with RT-qPCR and western
blot analysis, respectively. The concentrations of inflammatory
factors including (D) IL-6, (E) MCP-1 and (F) IL-12 were examined
using ELISA kits. *P<0.05, **P<0.01 and
***P<0.001. PAD4, peptidyl arginine deiminase 4; LPS,
lipopolysaccharide; RT-qPCR, reverse transcription-quantitative
PCR; sh, short hairpin; NC, negative control; MCP-1, monocyte
chemoattractant protein-1. |
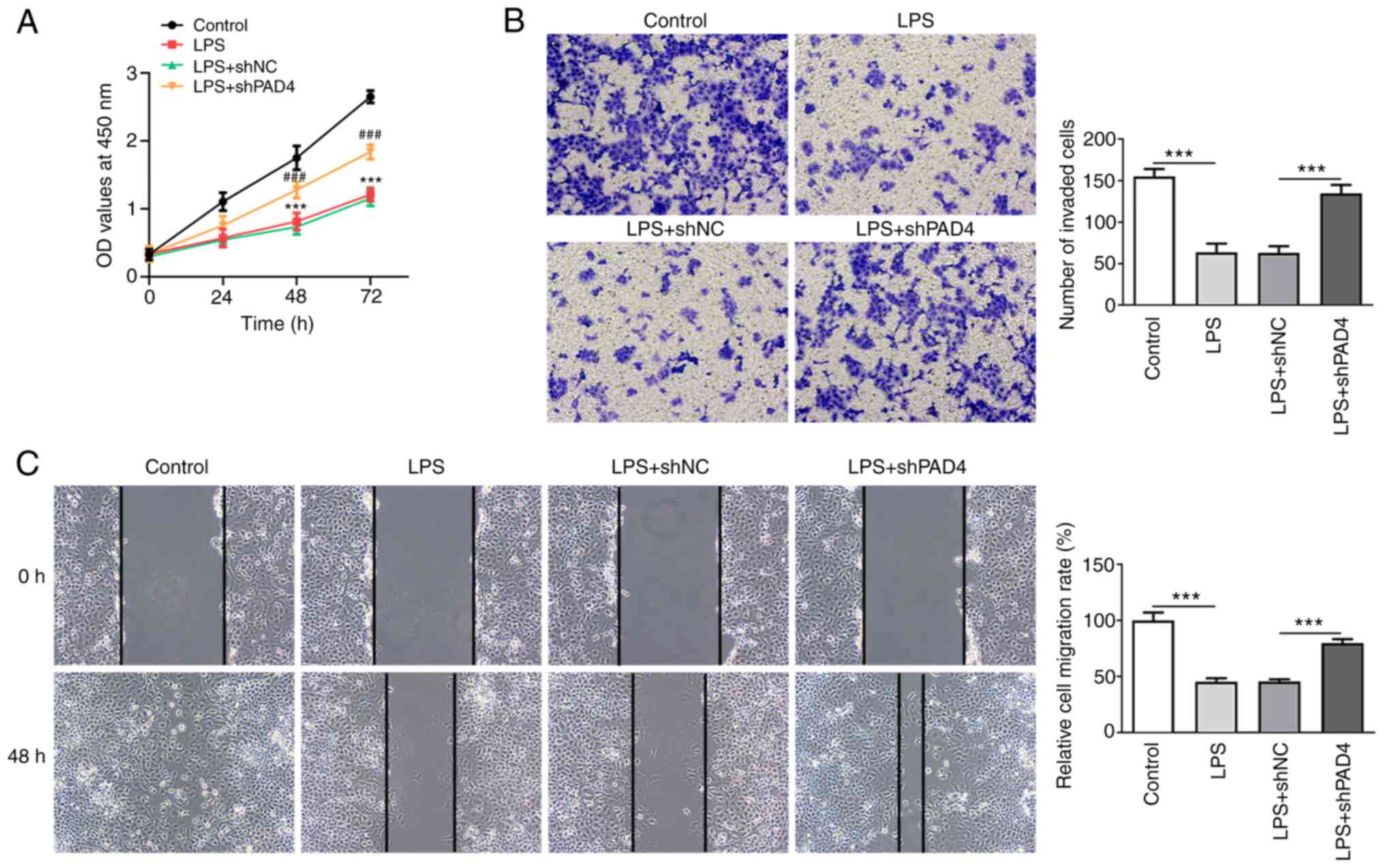
PAD4 silencing promotes the invasion
and migration of HTR8/SVneo cells following exposure to LPS
Cell viability was measured using CCK-8 assay. A
significantly deceased HTR8/SVneo cell viability was observed in
the LPS-induced group compared with that in the control group,
which was significantly reversed after transfection with shPAD4#2
(Fig. 2A). LPS stimulation also
significantly inhibited the invasive ability of HTR8/SVneo cells
compared with that in the control group (Fig. 2B). By contrast, PAD4 knockdown
significantly increased cell invasion compared with that in the LPS
+ shNC group (Fig. 2B). In
addition, LPS significantly suppressed the migration of HTR8/SVneo
cells, though PAD4 silencing partially but significantly reversed
this inhibitory effect on the migration of HTR8/SVneo cells after
LPS challenge (Fig. 2C). These
results suggest that silencing of PAD4 expression can promote the
invasion and migration of HTR8/SVneo cells after treatment with
LPS.
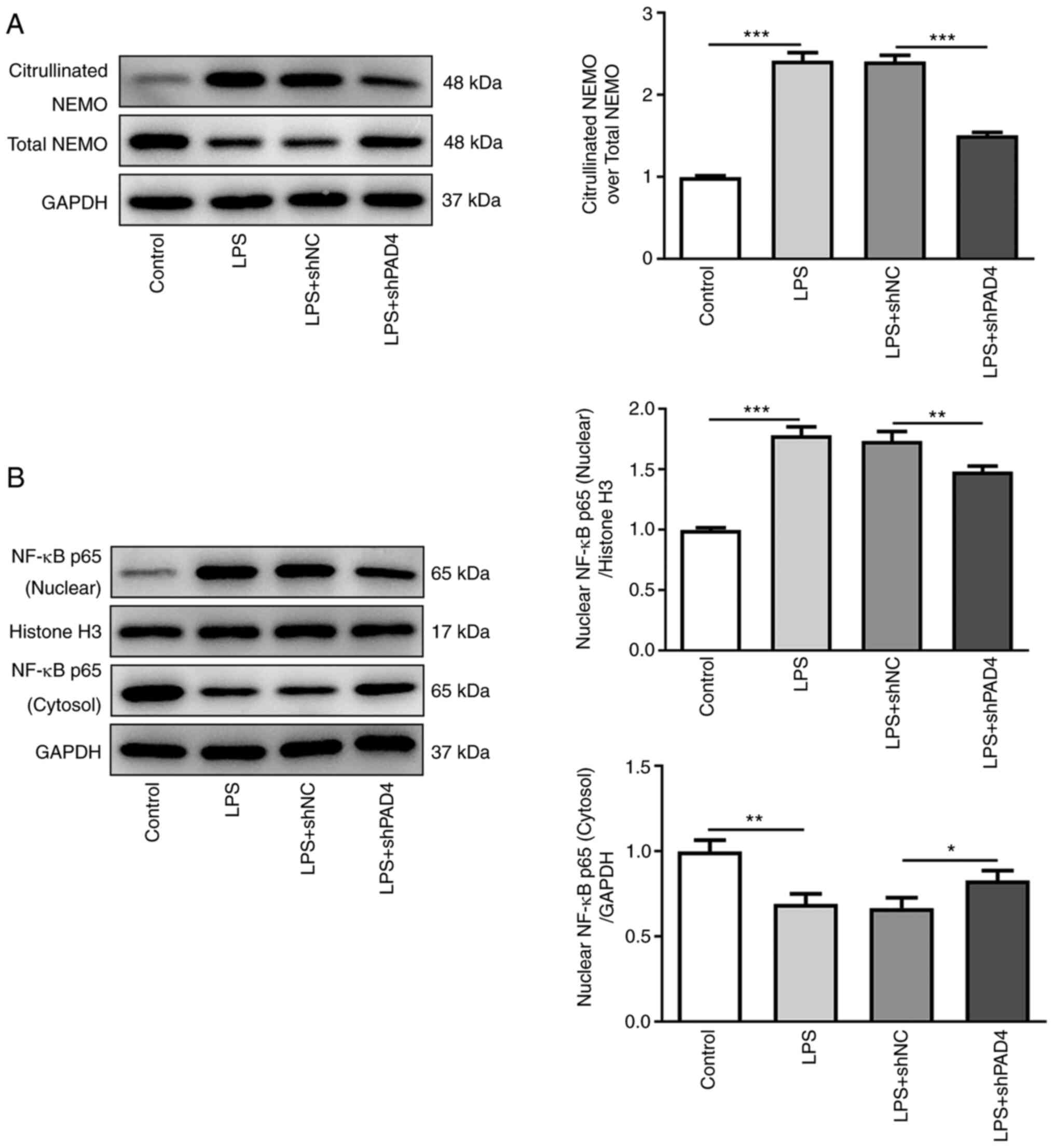
Knocking down PAD4 expression inhibits
NEMO citrullination and reduces NF-κB nuclear translocation in
LPS-treated HTR8/SVneo cells
To explore the potential mechanism of PAD4 in the
progression of PE, the expression of proteins in the NEMO/NF-κB
signaling pathway was measured by western blot analysis. Compared
with that in the control group, LPS treatment significantly
upregulated the expression of citrullinated NEMO whilst
significantly downregulating total NEMO expression, which was
significantly reversed by PAD4 silencing (Fig. 3A). In addition, significantly
increased nuclear NF-κB p65 protein levels and significantly
decreased cytoplasmic NF-κB p65 protein levels were noted in the
LPS group compared with those in the control group, which was also
significantly reversed by PAD4 knockdown (Fig. 3B). These observations suggest that
knocking down PAD4 expression can suppress NEMO citrullination and
decrease nuclear NF-κB translocation in LPS-induced HTR8/SVneo
cells.
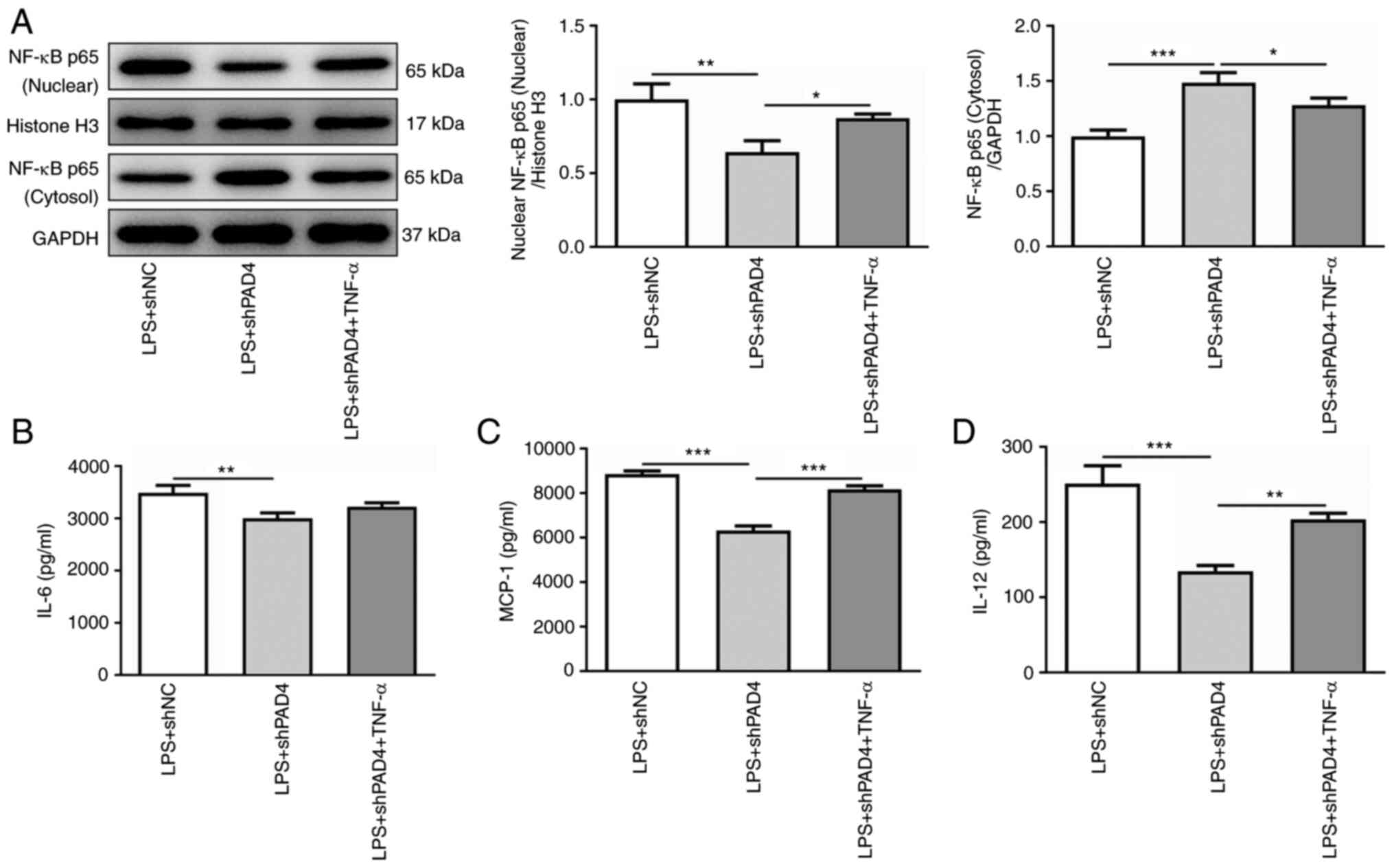
TNF-α reverses the effects of PAD4
silencing on inflammation, invasion and migration in LPS-induced
HTR8/SVneo cells
TNF-α was subsequently used to treat HTR8/SVneo
cells to activate NF-κB signaling to evaluate the effects of PAD4
silencing on inflammation, invasion and migration was mediated by
NEMO/NF-κB signaling. TNF-α intervention significantly elevated the
level of nuclear NF-κB p65 expression and reduced that of
cytoplasmic NF-κB p65 expression compared with that in the
untreated LPS + shPAD4 group (Fig.
4A). In addition, it was found that the secretion levels of
IL-6, MCP-1 and IL-12 were markedly enhanced after the HTR8/SVneo
cells were treated with TNF-α compared with those in the untreated
LPS + shPAD4 group (Fig.
4B-D).
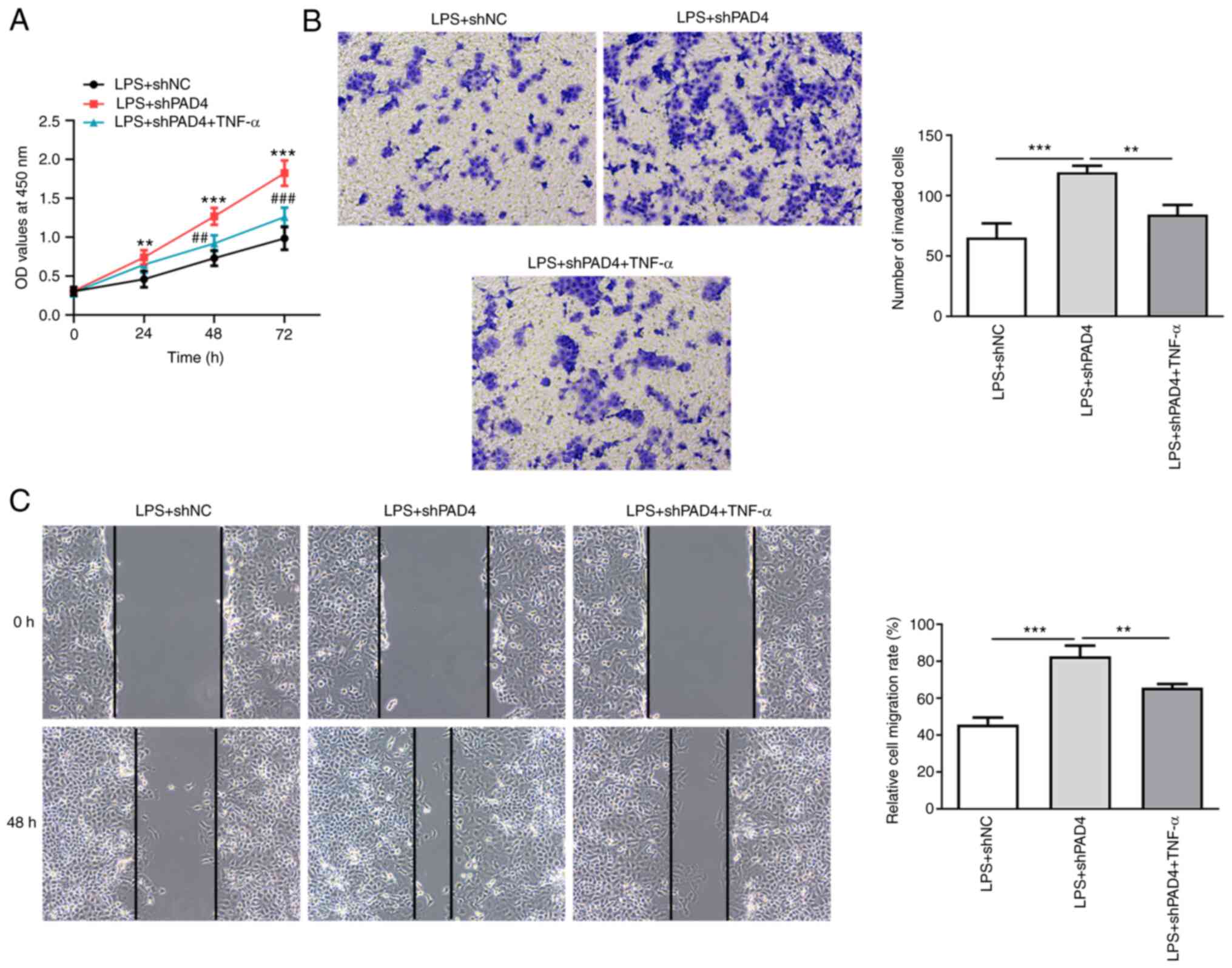
Downstream, TNF-α treatment led to significantly
increased cell viability compared with that in the untreated LPS +
shPAD4 group (Fig. 5A). In
addition, the abilities of HTR8/SVneo cell invasion and migration
were significantly reduced after treatment with TNF-α, LPS exposure
and PAD4 silencing compared with that in the untreated LPS + shPAD4
group (Fig. 5B and C). Collectively, these data suggest that
PAD4 silencing inhibits inflammation and promotes the invasion and
migration of trophoblast cells by inactivation of the NEMO/NF-κB
signaling pathway.
Discussion
PE is a pregnancy-specific syndrome that originates
from the placenta with a number of systems involved, including the
liver, kidney and cardiovascular systems (19,20).
It is potentially life-threatening for both mother and baby
(21). It is widely accepted that
PE is typically caused by insufficient trophoblastic infiltration
into the endometrium during embryonic development, leading to
severe endothelial malfunction in the uterine placenta (22). The present study demonstrated that
PAD4 expression is increased following LPS treatment in HTR8/SVneo
cells in vitro. Mechanistically, PAD4 silencing could
inhibit inflammation whilst promoting invasion and migration of
trophoblast cells by inactivating the NEMO/NF-κB signaling
pathway.
Previous studies have reported that an altered
immune system response and excessive inflammation can contribute to
the development of PE (23,24).
Inflammatory responses at the utero-placental interface were
associated with deficient extravillous trophoblast invasion during
placentation in transgenic preeclamptic rat models and patients
with PE (18,25). Several in vitro studies
previously suggested that exposure to inflammatory stimuli can
trigger the secretion of proinflammatory cytokines from trophoblast
cells, such as IL-6, IL-12 and MCP-1, to mediate PE (26,27).
PAD4 is an enzyme that catalyzes the citrullination process and
serves a significant role in innate immunity, infection control and
autoimmune diseases (5,28). Shelef et al (29) suggested that PAD4 contributes to
TNF-α-induced inflammatory arthritis. In PAD4-null mice, the renal
neutrophil infiltration and inflammation after ischemia-reperfusion
injury were markedly alleviated compared with those in wild-type
mice (30). In addition, loss of
PAD4 function has been reported to reduce inflammation and
susceptibility to pregnancy loss in a PAD4 knockout mouse model
(7). In the present study, reduced
levels of inflammatory factors IL-6, MCP-1 and IL-12 as a result of
PAD4 silencing in LPS-induced HTR8/SVneo cells provided further
support for the potential role of PAD4 in PE during pregnancy.
During the development of placenta, cytotrophoblast
proliferation, invasion and migration are orchestrated steps
critical for normal pregnancy (31,32).
Efficient trophoblastic infiltration forms the basis for the
effective establishment of uteroplacental circulation and is key to
successful pregnancy (33).
Insufficient invasion and migration of trophoblast cells results in
shallow embryo implantation, inadequate placental invasion and
abnormal spiral artery remodeling, which are generally regarded to
be major mechanisms underlying PE (34). To the best of our knowledge, the
present study is the first to explore the effects of PAD4 on the
invasion and migration of HTR8/SVneo cells following exposure to
LPS, where PAD4 silencing promoted the invasion and migration of
HTR8/SVneo cells.
Subsequently, the potential mechanism of PAD4 in the
development of PE was investigated in the present study. Consistent
with the previous findings (8,9), the
results indicate that PAD4 can promote the NF-κB nuclear
translocation and activate NF-κB activity through NEMO
citrullination. The expression of NEMO was found to be
conspicuously higher in the maternal and fetal blood of PE cases
compared with that in healthy controls (11). Importantly, the NEMO transcription
level in maternal blood of the PE subgroup was significantly
increased (12). NEMO is one of
the regulatory subunits of the IκB kinase (IκK) complex, which
controls activation of NF-κB signaling pathway (35). It has been reported that the NF-κB
and TNF-α in the NF-κB pathway are highly expressed in the
peripheral blood mononuclear cells of patients with PE, compared
with the control group (36). In
the present study, PAD4 silencing downregulated the expression of
citrullinated NEMO and nuclear NF-κB p65 expression, suggesting
that PAD4 may regulate the NEMO/NF-κB pathway in LPS-induced
HTR8/SVneo cells. Furthermore, activation of NF-κB signaling by the
addition of TNF-α reversed the impact of PAD4 silencing on the
inflammation, invasion and migration of HTR8/SVneo cells. The
present study is a preliminary study of the role of PAD4 in PE by
an LPS-induced HTR8/SVneo cell model. The lack of in vivo
experiments to investigate the effects of PAD4 on an animal PE
model and the experiments for the examination of related signaling
pathways that can be regulated by PAD4 are limitations of the
present study. Therefore, further comprehensive and in-depth
analyses are required to validate the findings of the present
study.
Taken together, the present study suggests that PAD4
is highly expressed in LPS-induced HTR8/SVneo cells in
vitro. Mechanistically, PAD4 silencing inhibited inflammation
but promoted the invasion and migration of trophoblasts by
inactivating the NEMO/NF-κB signaling pathway. These findings lay a
foundation for furthering the understanding in the complex
molecular mechanisms underlying PE and provide a promising target
for the treatment of this disease.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MZ, MX and XL searched the literature, designed the
experiments and conducted the experiments. JL and YL analyzed and
interpreted the data. MZ and MX wrote the manuscript. XL and YL
revised the manuscript. MZ and MX confirmed the authenticity of all
the raw data. All authors have read and approved the final version
of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Capriglione S, Plotti F, Terranova C,
Gulino FA, Di Guardo F, Lopez S, Scaletta G and Angioli R:
Preeclampsia and the challenge of early prediction: Reality or
utopia? State of art and critical review of literature. J Matern
Fetal Neonatal Med. 33:677–686. 2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Mazzuca MQ, Li W, Reslan OM, Yu P, Mata KM
and Khalil RA: Downregulation of microvascular endothelial type B
endothelin receptor is a central vascular mechanism in hypertensive
pregnancy. Hypertension. 64:632–643. 2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Li X, Wu C, Shen Y, Wang K, Tang L, Zhou
M, Yang M, Pan T, Liu X and Xu W: Ten-eleven translocation 2
demethylates the MMP9 promoter, and its down-regulation in
preeclampsia impairs trophoblast migration and invasion. J Biol
Chem. 293:10059–10070. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Xie N, Jia Z and Li L: miR-320a
upregulation contributes to the development of preeclampsia by
inhibiting the growth and invasion of trophoblast cells by
targeting interleukin 4. Mol Med Rep. 20:3256–3264. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Rohrbach AS, Slade DJ, Thompson PR and
Mowen KA: Activation of PAD4 in NET formation. Front Immunol.
3(360)2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Wang S and Wang Y: Peptidylarginine
deiminases in citrullination, gene regulation, health and
pathogenesis. Biochim Biophys Acta. 1829:1126–1135. 2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Erpenbeck L, Chowdhury CS, Zsengellér ZK,
Gallant M, Burke SD, Cifuni S, Hahn S, Wagner DD and Karumanchi SA:
PAD4 deficiency decreases inflammation and susceptibility to
pregnancy loss in a mouse model. Biol Reprod.
95(132)2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Rabadi M, Kim M, D'Agati V and Lee HT:
Peptidyl arginine deiminase-4-deficient mice are protected against
kidney and liver injury after renal ischemia and reperfusion. Am J
Physiol Renal Physiol. 311:F437–F449. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Rabadi MM, Han SJ, Kim M, D'Agati V and
Lee HT: Peptidyl arginine deiminase-4 exacerbates ischemic AKI by
finding NEMO. Am J Physiol Renal Physiol. 316:F1180–F1190.
2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Vaughan JE and Walsh SW: Activation of
NF-κB in placentas of women with preeclampsia. Hypertens Pregnancy.
31:243–251. 2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Sakowicz A, Lisowska M, Biesiada L,
Płuciennik E, Gach A, Rybak-Krzyszkowska M, Huras H, Sakowicz B,
Romanowicz H, Piastowska-Ciesielska AW, et al: Placental expression
of NEMO protein in normal pregnancy and preeclampsia. Dis Markers.
2019(8418379)2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Sakowicz A, Hejduk P, Pietrucha T,
Nowakowska M, Płuciennik E, Pospiech K, Gach A, Rybak-Krzyszkowska
M, Sakowicz B, Kaminski M, et al: Finding NEMO in preeclampsia. Am
J Obstet Gynecol. 214:538.e1–538.e7. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sakowicz A, Pietrucha T,
Rybak-Krzyszkowska M, Huras H, Gach A, Sakowicz B, Banaszczyk M,
Grzesiak M and Biesiada L: Double hit of NEMO gene in preeclampsia.
PLoS One. 12(e0180065)2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Hu J, Zhang J and Zhu B: Protective effect
of metformin on a rat model of lipopolysaccharide-induced
preeclampsia. Fundam Clin Pharmacol. 33:649–658. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Graham CH, Hawley TS, Hawley RG,
MacDougall JR, Kerbel RS, Khoo N and Lala PK: Establishment and
characterization of first trimester human trophoblast cells with
extended lifespan. Exp Cell Res. 206:204–211. 1993.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Abou-Kheir W, Barrak J, Hadadeh O and
Daoud G: HTR-8/SVneo cell line contains a mixed population of
cells. Placenta. 50:1–7. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Fan M, Li X, Gao X, Dong L, Xin G, Chen L,
Qiu J and Xu Y: LPS induces preeclampsia-like phenotype in rats and
HTR8/SVneo cells dysfunction through TLR4/p38 MAPK pathway. Front
Physiol. 10(1030)2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Jena MK, Sharma NR, Petitt M, Maulik D and
Nayak NR: Pathogenesis of preeclampsia and therapeutic approaches
targeting the placenta. Biomolecules. 10(953)2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Calicchio R, Buffat C, Vaiman D and
Miralles F: Endothelial dysfunction: Role in the maternal syndrome
of preeclampsia and long-term consequences for the cardiovascular
system. Ann Cardiol Angeiol (Paris). 62:215–220. 2013.PubMed/NCBI View Article : Google Scholar : (In French).
|
|
21
|
Pauli JM and Repke JT: Preeclampsia:
Short-term and long-term implications. Obstet Gynecol Clin North
Am. 42:299–313. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Dai X and Cai Y: Down-regulation of
microRNA let-7d inhibits the proliferation and invasion of
trophoblast cells in preeclampsia. J Cell Biochem. 119:1141–1151.
2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Shaw J, Tang Z, Schneider H, Saljé K,
Hansson SR and Guller S: Inflammatory processes are specifically
enhanced in endothelial cells by placental-derived TNF-α:
Implications in preeclampsia (PE). Placenta. 43:1–8.
2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Harmon AC, Cornelius DC, Amaral LM,
Faulkner JL, Cunningham MW Jr, Wallace K and LaMarca B: The role of
inflammation in the pathology of preeclampsia. Clin Sci (Lond).
130:409–419. 2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Cotechini T, Komisarenko M, Sperou A,
Macdonald-Goodfellow S, Adams MA and Graham CH: Inflammation in rat
pregnancy inhibits spiral artery remodeling leading to fetal growth
restriction and features of preeclampsia. J Exp Med. 211:165–179.
2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kadam L, Kilburn B, Baczyk D, Kohan-Ghadr
HR, Kingdom J and Drewlo S: Rosiglitazone blocks first trimester
in-vitro placental injury caused by NF-κB-mediated inflammation.
Sci Rep. 9(2018)2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Zaga-Clavellina V, Garcia-Lopez G,
Flores-Herrera H, Espejel-Nuñez A, Flores-Pliego A,
Soriano-Becerril D, Maida-Claros R, Merchant-Larios H and
Vadillo-Ortega F: In vitro secretion profiles of interleukin
(IL)-1beta, IL-6, IL-8, IL-10, and TNF alpha after selective
infection with Escherichia coli in human fetal membranes. Reprod
Biol Endocrinol. 5(46)2007.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Vossenaar ER, Nijenhuis S, Helsen MM, van
der Heijden A, Senshu T, van den Berg WB, van Venrooij WJ and
Joosten LA: Citrullination of synovial proteins in murine models of
rheumatoid arthritis. Arthritis Rheum. 48:2489–2500.
2003.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Shelef MA, Sokolove J, Lahey LJ, Wagner
CA, Sackmann EK, Warner TF, Wang Y, Beebe DJ, Robinson WH and
Huttenlocher A: Peptidylarginine deiminase 4 contributes to tumor
necrosis factor α-induced inflammatory arthritis. Arthritis
Rheumatol. 66:1482–1491. 2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Li H, Han SJ, Kim M, Cho A, Choi Y,
D'Agati V and Lee HT: Divergent roles for kidney proximal tubule
and granulocyte PAD4 in ischemic AKI. Am J Physiol Renal Physiol.
314:F809–F819. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Lorquet S, Pequeux C, Munaut C and Foidart
JM: Aetiology and physiopathology of preeclampsia and related
forms. Acta Clin Belg. 65:237–241. 2010.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Martin E, Ray PD, Smeester L, Grace MR,
Boggess K and Fry RC: Epigenetics and preeclampsia: Defining
functional epimutations in the preeclamptic placenta related to the
TGF-β pathway. PLoS One. 10(e0141294)2015.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Walker JJ: Pre-eclampsia. Lancet.
356:1260–1265. 2000.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Jiang Y and Chen Y and Chen Y: Knockdown
of JARID2 inhibits the viability and migration of placenta
trophoblast cells in preeclampsia. Mol Med Rep. 16:3594–3599.
2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Chen D, Yu M, Chen H, Zeng M, Sun Y and
Huang Q: Identification and functional characterization of NEMO in
Crassostrea gigas reveals its crucial role in the NF-κB activation.
Fish Shellfish Immunol. 80:46–55. 2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Ali Z, Zafar U, Khaliq S and Lone KP:
Elevated tumor necrosis factor (TNF)-α mRNA expression correlates
with nuclear factor kappa B expression in peripheral blood
mononuclear cells in preeclampsia. J Coll Physicians Surg Pak.
30:158–162. 2020.PubMed/NCBI View Article : Google Scholar
|