Introduction
Pseudohypoparathyroidism (PHP) is a rare hereditary
disease and historically the first hormone resistance syndrome
(1). It is estimated that the
prevalence of PHP is approximately 0.34-1.1 per 100,000 (2-4).
In patients with normal renal function, hyperphosphatemia as well
as hypocalcemia are related to a decreased calcemic and
phosphaturic response to injection of bovine parathyroid extract,
in contrast to those with primary hypoparathyroidism, triggering
the hypothesis of resistance to PTH action (2-4).
PHP can be classified into pseudohypoparathyroidism type 1A
(PHP1A), pseudoPHP (PPHP) and PHP type 1B (PHP1B), caused by
maternal and paternal GNAS mutations and abnormal
methylation at maternalguanine nucleotide-binding protein α
stimulating (GNAS) promoter(s), respectively (5). Maternal loss-of-function mutations at
GNAS exons 1-13 cause PHP type 1A (PHP1A). When patients
have a similar phenotype to PHP1A but do not have a mutation in
GNAS, other related disorders need to be considered.
Phenotypic studies in patients with PHP1A revesal the presence of
Albright Hereditary Osteodystrophy (AHO), including brachydactyly,
subcutaneous ossifications, round facies as well as short stature
(2). Due to the variable
manifestations of early clinical of PHP1A, the diagnosis of PHP1A
is often easily overlooked, with frequent misdiagnosis or missed
diagnosis. To this end, this case report described a girl who was
initially diagnosed with hereditary multiple exostoses (HMEs), but
was afterwards confirmed with PHP1A. Moreover, genetic analysis
indicated a new mutation (c2277deIC) of GNAS gene from
maternal GNAS mutations.
Case report
A 12-year-old girl presented to the inpatient
department of Hangzhou Children's Hospital in February 2019,
complaining of recurrent convulsions for 1.5 months and frequent
episodes of four days. In February 2017, she was admitted at the
Department of orthopedic of Jinhua Central Hospital due to
double-footed mass for more than seven years. At that time, she was
diagnosed with HMEs (double heel) and further received
operation.
The physical examination of the girl showed short
stature (height 135 cm), central obesity (weigh 34 kg), rounded
face and mild mental retardation. The girl denied any family
history of heterotopic ossification or inherited diseases.
Additionally, her parents and two brothers were none of them were
diagnosed with AHO, with normal serum concentration of calcium and
phosphorus.
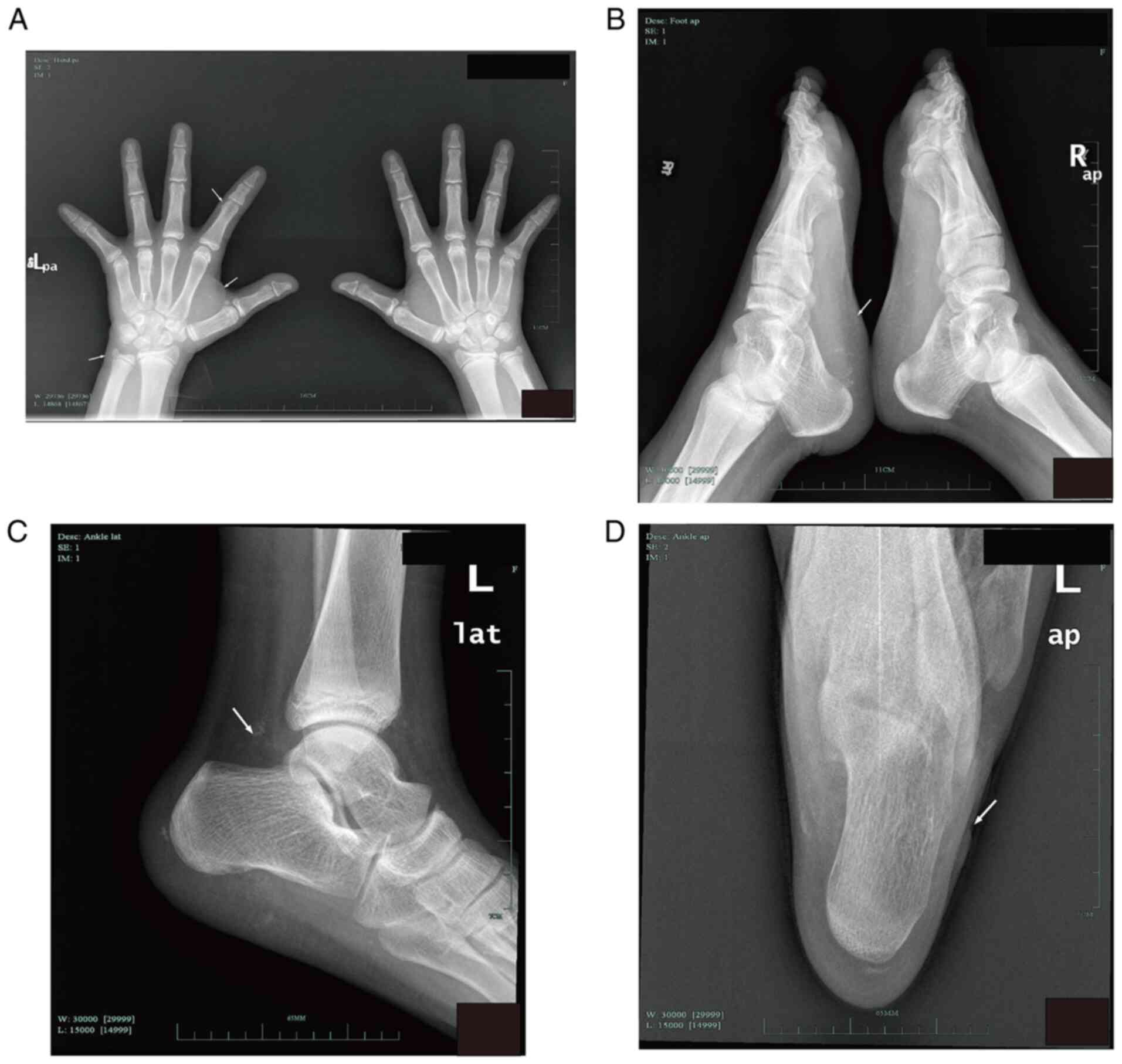
X-rays of hand and foot indicated: i) local cortical
rough defect in left ulna and distal humerus, local mild periosteal
reaction; ii) exogenous small osteophytes on the lateral side of
the left heel and the ulnar side of left hand and iii) multiple
small soft-tissue calcifications of the left palm, wrist and ankle
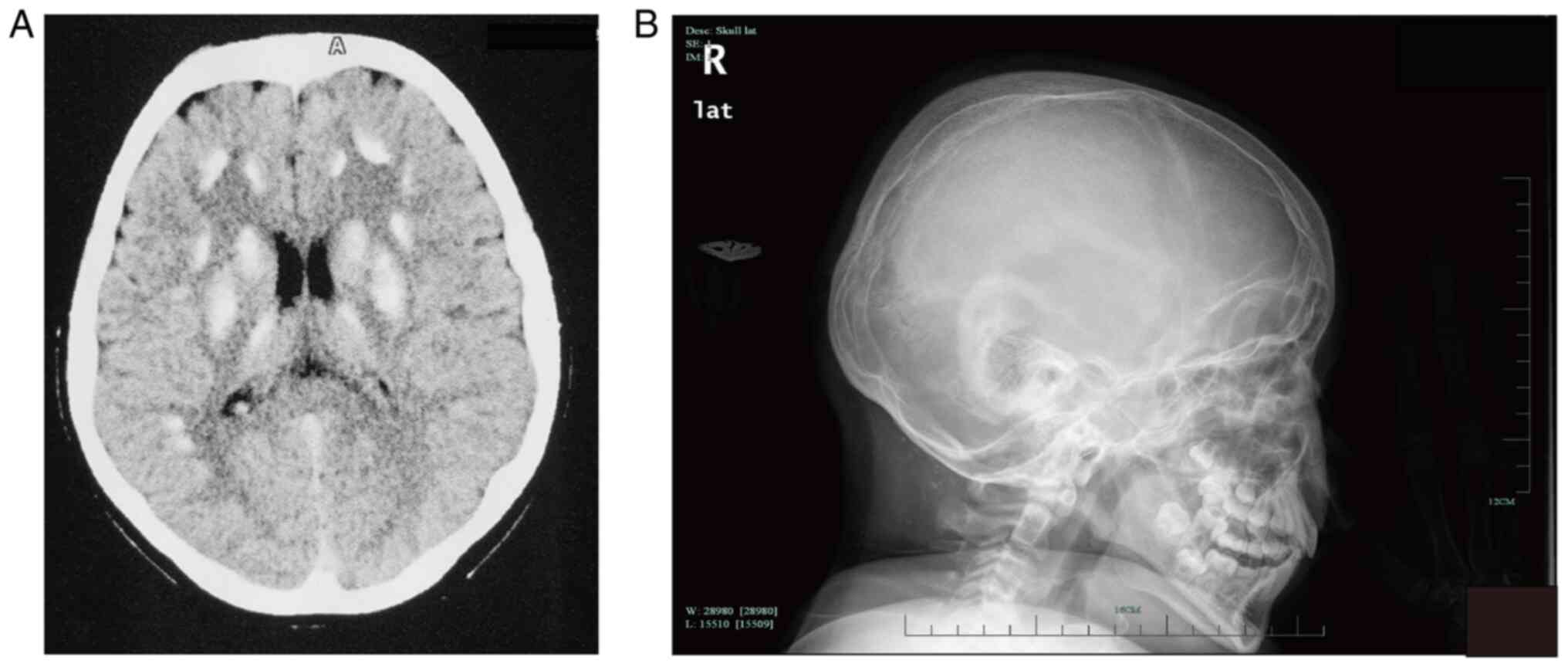
(Fig. 1). Cerebral computed
tomography (CT) revealed extensive symmetric calcifications in the
basal ganglia, thalami and cerebellar hemispheres (Fig. 2). Abdominal ultrasound showed left
kidney crystallization and left liver calcification. Laboratory
examination showed hypocalcemia, blood levels of calcium 1.68
mmol/l (normal range 2.08-2.6 mmol/l), hyperphosphatemia,
phosphorus 2.48 mmol/l (normal range 0.96-1.62 mmol/l), magnesium
0.73 mmol/l (normal range ~0.65-1.25 mmol/l) and parathyroid
hormone 981.00 ng/l (normal range ~0-88.00 ng/l), alkaline
phosphatase (ALP) 318 U/l (normal range ~0-500), 24 h urinary
calcium 0.07 mmol/24 h (normal range ~0-6.25 mmol/24 h), serum
25-hydroxy vitamin D (37.02 nmol/l, normal range: 25-125l nmol/l).
Serum levels of thyroid hormone were normal, the values of TSH
reveal TSH resistance (free T3: 6.01 pmol/l, normal range:
~3.00-7.50 pmol/l; free T4: 11.96 pmol/l, normal range: ~8.37-29.60
pmol/l; TSH:5.69 mU/l, normal range: ~0.4-4.00 mU/l). Serum
insulin-like growth factor-1 (IGF-1) was normal 318.5 ng/ml (normal
range: 126-678 ng/ml). Sex hormones, growth hormone and cortisol
rhythm were also normal.
Genetic counseling and analysis were offered to the
girl and her family members after signing informed consent. DNA
samples isolated from blood specimens were analyzed by Sanger
sequencing as well as multiplex ligation-dependent probe
amplification (MLPA), aiming to determine whether there was a
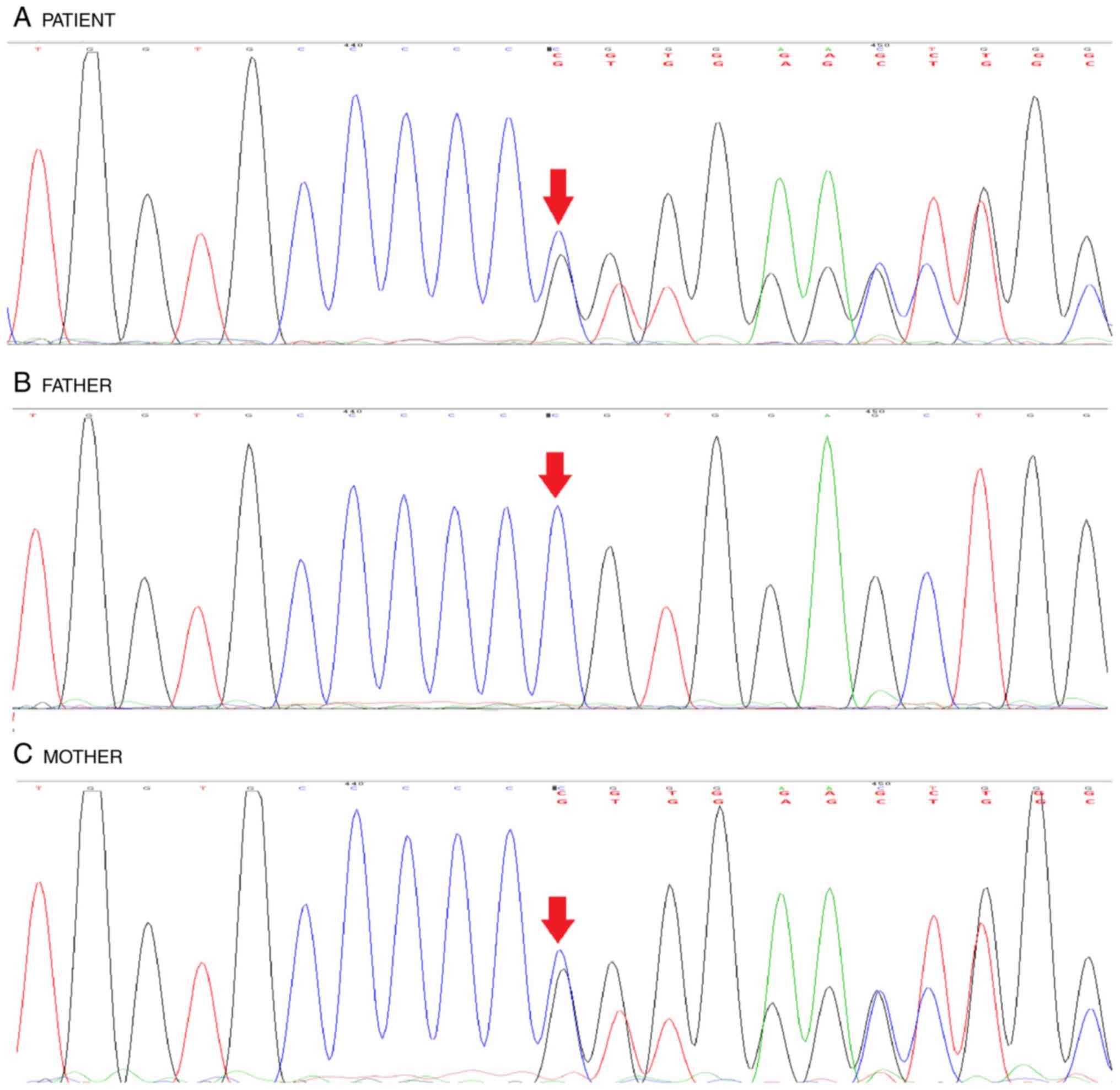
GNAS gene mutation, in line with standard protocols. As a
result, heterozygous c2277delC (p.Val760TrpfsTer16) frameshift
mutation was detected. The targeted mutation analysis of the
GNAS gene revealed a defined mutation with PHP1A, PPHP or
POH, depending of the allele involved. After analysis of clinical
manifestations and gene types, the patient was diagnosed with PHP.
Moreover, her mother was detected to harbor GNAS gene
mutation (Fig. 3).
MLPA analysis further eliminated the additional
pathogenic mechanism with GNAS locus, including deleted or
aberrant methylation profiles. However, the present study was
unable to conduct further research to examine the expression or
activities of Gsα protein.
The patient was treated with intravenous and oral
calcium replacement therapy to target hypocalcemia, with
administration of 1, 25 hydroxyl vitamin D3 (0.04 µg/kg/d). Serum
calcium level, pituitary hormones as well as thyroid function were
regularly monitored until no episodes of hypothyroidism,
hypocalcemia or seizure.
Discussion
Target-organ resistance to other hormones are
commonly observed in patients with PHP1A, acting through
Gsα-coupled receptors, particularly TSH (6). There are also studies concerning
resistance to gonadotrophins as well as GHRH (6,7).
There are diverse presentation and different degrees of severity in
PHP1A as well as its relevance among different individuals, in
which there are considerable overlapping of clinical and molecular
features between the different types (3). Patients with PHP1A are burdened with
half decreased activity of Gsα subunit, due to decreased amounts of
Gsα (2). The human the Gsα gene
(GNAS), contains13 exosmic located at 20q13, with cDNA
length of approximately 1.2 kb (4). Heterozygous mutation of the coding
region of GNAS gene can lead to attenuated bioactivity of
Gsα protein levels (8). In the
present study, a heterozygous c2277delC (p.Val760TrpfsTer16)
frameshift mutation was detected in the patient. However, her
mother harbored a heterogeneous mutation in the GNAS gene
and yet had no signs or symptoms of AHO. These discrepancies in the
phenotype depending on the transmitting parent are explained by a
tissue-specific imprinting of GNAS. When the mutation is
carried by the maternal allele, there is a partial to complete
deficit in Gsα depending on the severity of the mutation (9).
Another feature of PHP1A is the development of
heterotopic subcutaneous ossifications. When in isolation, the
condition is termed osteoma cutis, which is the first sign of a
more severe PHP disorder. There is no definitive treatment and
removal can cause regrowth that is worse (10,11).
Nevertheless, the diagnosis might remain uncertain under the
majority of clinical situations, due to the misdiagnosis of these
clinical symptoms as nonspecific in the normal population. By
retrospectively analyzing the medical history concerning the
evolution of disease progression throughout the patient's lifetime:
TSH resistance caused PTH resistance-related hypocalcemia as well
as hypothyroidism in early stage, followed by bilateral heels
disease and worsening hypocalcemia and recently recurrent
seizures.
The clinical manifestations of PHP-1A are variable
and the rate of misdiagnosis is high. Due to different complaints,
the patients were admitted in different departments. The child had
been treated with a double-pedal mass for 7 years. Considering
congenital multiple osteochondroma (double heel), attention should
be paid to blood calcium, blood phosphorus and PTH levels during
the operation and physical examination should be carried out to
determine whether there is physical development. In delayed and AHO
performance; the disease is mostly the first consultation in
neurology and emergency departments, often with convulsions as the
main complaint, so when patients with suspected seizures are
examined, blood calcium, blood phosphorus and brain CT should be
detected. Careful examination of the presence or absence of AHO
performance, for patients with normal renal function but with low
calcium, hyperphosphatemia, should involve checking the blood
calcium, blood phosphorus and PTH level to assist in the diagnosis,
as well as early calcium and active vitamin D3, to correct
hypocalcemia and to ensure the diagnosis as soon as possible, to
prevent death caused by severe low calcium convulsions. The present
study indicated the clinical significance of early diagnosis of
PHP1A, which serves a critical role in proper therapeutic
approaches and long-term management strategies, essential for both
the patient and the family. Notably, proper genetic counseling also
serves a significant role in establishment of effective
communication with the family, rendering efficient information
exchange and the successful performance of genetic analysis.
Therefore, multidisciplinary screenings, along with individualized
therapeutic strategies are strongly recommended to enhance the
clinical outcomes in real-world practice (3,12).
In practice, clinicians should use a combination of phenotypic and
genotypic information to suggest and confirm the diagnosis of
PHP1A.
Acknowledgements
Not applicable.
Funding
Funding: The present study was funded by Grant from Natural
Science Foundation of Zhejiang Province (grant nos. LQ20H040001 and
LY20H130003) and Hangzhou Medical Health Science and Technology
(grant no. 0020190868).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JZ and MG obtained and analyzed the patient's
information and wrote the manuscript. SZ, SW, LW and WS obtained
and analyzed the patient's information and reviewed the discussion
part of the clinical manifestations and imaging features. JZ, MG,
LW and WS confirm the authenticity of all the raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Medical Ethics
Committee of Hangzhou Children's Hospital (approval number 201901,
Hangzhou, China).
Patient consent for publication
Written informed consent to publish this case report
was obtained from the patient and her family.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Germain-Lee EL: Management of
pseudohypoparathyroidism. Curr Opin Pediatr. 31:537–549.
2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Linglart A, Levine MA and Jüppner H:
Pseudohypoparathyroidism. Endocrinol Metab Clin North Am.
47:865–888. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Mantovani G: Clinical review:
Pseudohypoparathyroidism: Diagnosis and treatment. J Clin
Endocrinol Metab. 96:3020–3030. 2011.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Mantovani G, Bastepe M, Monk D, de Sanctis
L, Thiele S, Usardi A, Ahmed SF, Bufo R, Choplin T, De Filippo G,
et al: Diagnosis and management of pseudohypoparathyroidism and
related disorders: First international consensus statement. Nat Rev
Endocrinol. 14:476–500. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Hanna P, Grybek V, Perez de Nanclares G,
Tran LC, de Sanctis L, Elli F, Errea J, Francou B, Kamenicky P,
Linglart L, et al: Genetic and epigenetic defects at the GNAS locus
lead to distinct patterns of skeletal growth but similar
early-onset obesity. J Bone Miner Res. 33:1480–1488.
2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Savaş Erdeve Ş, Berberoğlu M, Şıklar Z,
Evliyaoğlu O, Hiort O and Öcal G: Long-term follow-up of a
pseudohypoparathyroidism type 1A patient with missense mutation
(Pro115Ser) in exon 5. J Clin Res Pediatr Endocrinol. 2:85–88.
2010.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Mantovani G and Spada A: Resistance to
growth hormone releasing hormone and gonadotropins in Albright's
hereditary osteodystrophy. J Pediatr Endocrinol Metab. 19 (Suppl
2):S663–S670. 2006.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Riepe FG, Ahrens W, Krone N, Fölster-Holst
R, Brasch J, Sippell WG, Hiort O and Partsch CJ: Early
manifestation of calcinosis cutis in pseudohypoparathyroidism type
Ia associated with a novel mutation in the GNAS gene. Eur J
Endocrinol. 152:515–519. 2005.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Snanoudj S, Molin A, Colson C, Coudray N,
Paulien S, Mittre H, Gérard M, Schaefer E, Goldenberg A, Bacchetta
J, et al: Maternal transmission ratio distortion of GNAS
loss-of-function mutations. J Bone Miner Res. 35:913–919.
2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kodo K, Maeda H, Morimoto H, Wada M, Imura
T and Nakajima H: A case of pseudohypoparathyroidism type Ia with a
novel frameshift mutation in the GNAS gene: Early diagnosis of
osteoma cutis by skin biopsy. Clin Pediatr Endocrinol. 28:15–18.
2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Mantovani G and Elli FM: Inactivating
PTH/PTHrP signaling disorders. Front Horm Res. 51:147–159.
2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Del Monte P, Cuttica CM, Marugo A,
Foppiani L, Audenino D, Godowicz TT, Elli FM, Mantovani G and Di
Maria E: Unrecognized pseudohypoparathyroidism type 1A as a cause
of hypocalcemia and seizures in a 64-year-old woman. Case Rep
Endocrinol. 2019(8456239)2019.PubMed/NCBI View Article : Google Scholar
|