Introduction
Concomitant strabismus is a common eye disease among
infants, children and adults, with a prevalence ranging from 2 to
6% (1,2). The prevalence of concomitant
exotropia is higher in Asian populations (3,4),
while concomitant esotropia is more likely to occur in Caucasians
(2). Affected patients may suffer
severe problems, such as binocular visual impairment, diplopia and
social stigma, and may require multiple corrective procedures
during their lifetime. However, the pathological mechanism of
concomitant strabismus remains unknown. Family studies have
demonstrated that genetic factors have a significant role in
concomitant strabismus development (5). Hu (6) reported that the incidence of
strabismus in first-, second- and third-degree relatives of 425
patients with exotropia was 9.0, 2.2 and 1.1%, respectively, and
the heritability of exotropia was 81.3%. In two other studies,
multivariate analyses revealed that children with a family history
were at a significantly higher risk of strabismus than children
without a family history (7,8).
Furthermore, a study of strabismus in twins concluded that 67.3% of
the strabismic phenotypes were concordant in 49 pairs of twins and
the concordance rate for monozygotic twins (82.4%) was higher than
that for multizygotic twins (47.6%) (9).
To date, only a few genes associated with
concomitant strabismus have been reported in families from multiple
countries. In 2003, Parikh et al (10) identified the first concomitant
strabismus locus, STBMS1 gene on chromosome 7p22.1 locus
based on a model of recessive inheritance in one esotropia
pedigree. Furthermore, it was suggested that MGST2 on
chromosome 4q28.3 and WNT2 on chromosome 7q31.2 were both
potential candidate genes for concomitant strabismus in 55 Japanese
pedigrees (11,12). However, these genes have only been
reported in concomitant esotropia or unspecified subtypes of
concomitant strabismus. There has been no exact gene reported to be
associated with concomitant exotropia.
Materials and methods
Whole-exome capture and
sequencing
Genomic DNA was extracted from peripheral blood and
exome capture was subsequently conducted using SureSelect Human All
Exon V6 (Agilent Technologies, Inc.). Exome sequencing was
performed on the Illumina Novaseq 6000 platform (Illumina, Inc.)
according to the manufacturer's instructions.
Whole-exome sequencing (WES) data
analyses
Fastq data were generated from raw sequencing files
using Illumina bcl2fastq software (Illumina, Inc.), the adapter was
removed and low-quality reads were removed. Subsequently,
Burrows-Wheeler Aligner was used to align fastq data to the Hs37d5
reference human genome, while marking of duplicate reads was
performed by sambamba tools (13)
to generate 150 base pair (bp) paired-end reads.
The sequencing reads were aligned to the human
reference genome hg19, single nucleotide variants (SNVs) and indels
were called using Samtools, and variants were filtered according to
the hard-filter criteria: i) Read depth, >4; ii) quality of
variant, >20; iii) root-mean-square mapping quality >30.
Variants were annotated with ANNOVAR (version 20180416) (14).
Identification of deleterious
variants
Possible deleterious genetic variants were required
to meet the following criteria: i) SNVs and indels are located at
exons or splicing sites. ii) Functional annotations are required to
be nonsynonymous SNVs, stop loss, stop gain, frameshift indels or
variants at splicing donor/recipient sites. iii) SIFT (15) and PolyPhen-2(16) annotations are required to be ‘.’ or
‘D’. iv) The variant allele frequency in East Asian Population of
the 1000G (17), ExAC (version
0.3) (18), gnomAD (version 2.1.1)
(19) and ChinaMAP (20) databases are all <0.01.
Based on the kinship of the affected family, the
likely inheritance pattern was tested for the possible pathogenetic
variants. For an autosomal dominant inheritance model, the proband
and the proband's parents should be heterozygous for the candidate
mutation; for an autosomal recessive inheritance model, both the
proband and one parent should be homozygous for the mutation and
the other parent should be heterozygous. The possibility of parents
carrying the same or different pathogenic genes was considered.
Gene prioritization based on relevant phenotypes was
performed using Phenolyzer (21).
CNV analysis
CNVs were called from the WES data using CoNIFER
(22) with the SVD threshold set
as 10. CNV calls located in the segment duplication regions
reported in the Database of Genomic Variants (DGV) (23) and those in the region of repeated
sequence annotated with RepeatMasker were excluded from further
analyses for being prone to be false positive. CNV calls located in
scattered repeating sequence or low complexity sequence are prone
to have alignment errors and were thus also excluded from further
analyses.
CNV calls were further filtered based on the
pathogenicity annotation. Those annotated as benign by
StringentLib, InclusiveLib (24)
and DGV GoldStandard (July 2015) were removed from analyses. CNVs
were annotated as being of high priority if they had 50% overlap
with those in the database of CNVD (25), which contains 212,277 CNV data
records related to human diseases.
The quality of the CNVs was inspected using
Integrative Genomics Viewer (version no. 2.11.0; software.broadinstitute.org/software/igv/home).
Quantitative (q)PCR validation of
CNVs
For CNVs that passed IGV inspection, qPCR validation
was performed. Genomic DNA (20 ng) was used in a final volume of 10
µl according to the recommended protocol provided by the
manufacturer using the SYBR® Premix Ex Taq™ II (Takara
Biotechnology Co., Ltd.). The optimal reaction conditions are 45
cycles of two-step amplification at 95˚C for 12 sec and 62˚C for 45
sec. The GAPDH gene was used as an internal control. Primer
sequences were as follows: STRCPl forward,
5'-AGCTCCAGCCATCTATCTGC-3' and reverse, 5'-GATCCTGCAGCTCGGTAGAC-3';
STRC forward, 5'-CCTGGGTCTCCTGCAAATAA-3' and reverse,
5'-GTGCAGATGTACGAGGGACA-3'; PCDHA6 forward,
5'-CGTGTACCTGATCATCGCCA-3' and reverse, 5'-AGGACAAGGTGAAAGGCTGG-3'.
GAPDH forward, 5'-CACCCGCCCCAGTCTCTG-3' and reverse,
5'-AACTCAAAGGGCAGGAGTAAAGG-3'.
Each sample was repeated three times independently.
Changes in the expression of target genes were determined based on
the relative values of 2-ΔΔCq (26).
Plasmid construction and Sanger
sequencing
For PCR amplification, genomic DNA (100 ng) was used
in a final volume of 50 µl according to the recommended protocol
provided by the manufacturer of the 2X Phanta® Flash
Master Mix (Nanjing Vazyme Biotechnology Co., Ltd.). The reaction
conditions were as follows: 95˚C for 3 min, followed by 30 cycles
of 3-step PCR (95˚C for 15 sec, 56˚C for 15 sec and 72˚C for 1
min), 72˚C for 5 min and then hold at 4˚C. Oligonucleotide primers
for PCR reactions of variants were designed by the website Primer 3
(bioinfo.ut.ee/primer3-0.4.0/) and the
sequences are listed in Table SI.
For indel variants, after PCR amplification, the two alleles were
cloned into the pEASY-Blunt Zero Cloning vector, followed by
transforming into Trans1-T1 competent cells (storage at
-70˚C and incubation in LB medium containing ampicillin; Beijing
Transgen Biotechnology Co., Ltd.) using the pEASY-Blunt Zero
Cloning Kit (Beijing Transgen Biotechnology Co., Ltd.) according to
the manufacturer's protocol. After incubation overnight, certain
single clones were selected for sequencing.
Results
Three-generation family with
concomitant exotropia
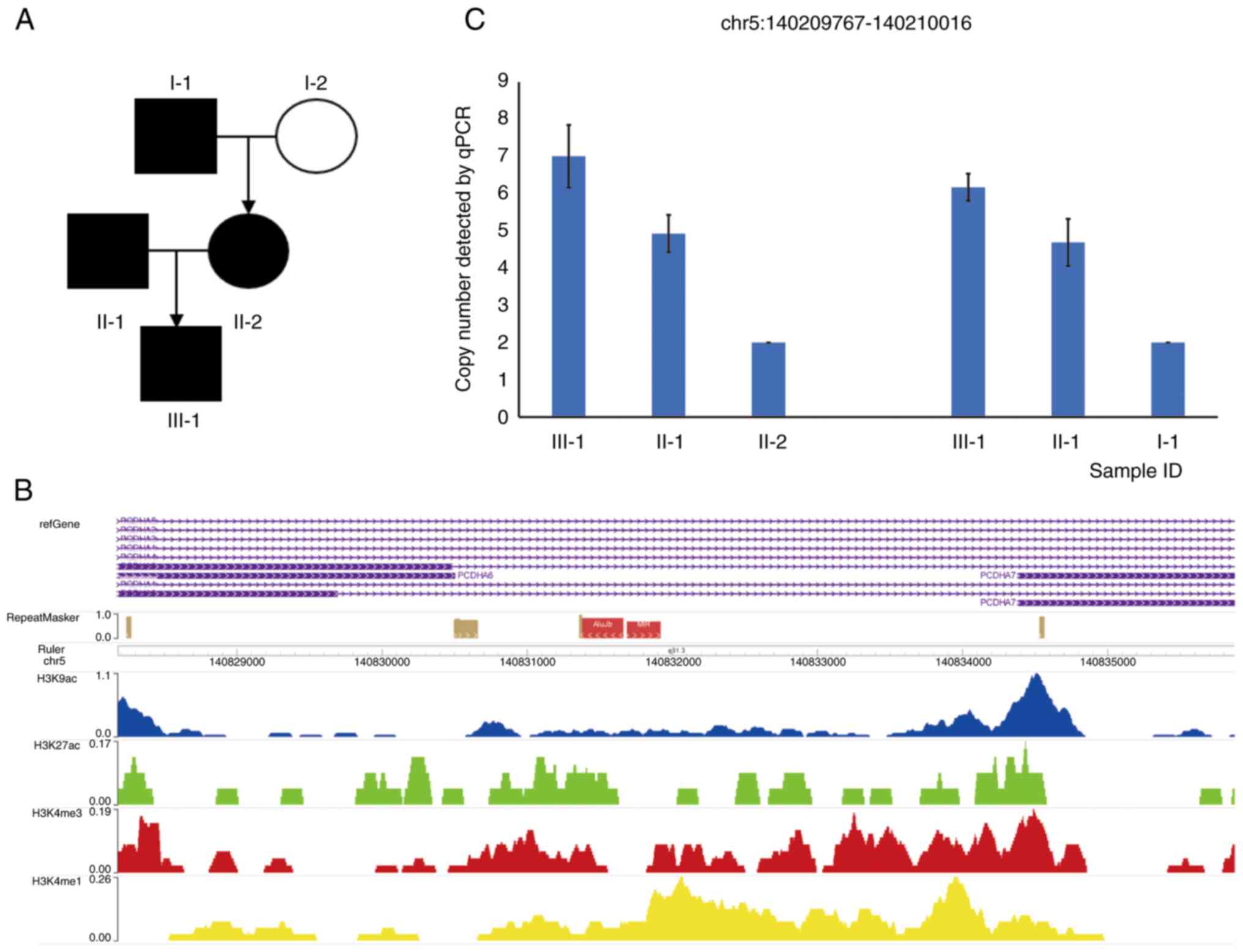
The present study recruited a Chinese family with
three generations and four members suffering from concomitant
exotropia (Fig. 1A). The proband,
an 11-year-old male, was found to have intermittent exotropia
during the physical examination in November 2020 at Tianjin Eye
Hospital, Tianjin, China. The patient had 35 PD of exotropia at
near distance and 20 PD at far distance. There were no refraction
errors with uncorrected visual acuities of 20/20 in both eyes. The
parents and maternal grandfather of the proband had concomitant
exotropia. The maternal grandmother had no presentation of
exotropia. The clinical manifestations of the children were more
severe than those of the parents. All subjects underwent cover
tests to check for strabismus conducted by a professional
ophthalmologist. As the exotropic phenotype appears in three
generations, it was speculated that the etiology of strabismus in
this family may have a strong genetic component. Genomic DNA from
all the individuals with concomitant exotropia in this family was
analyzed by WES (Fig. 1A).
Mutations in PCDHA, COL3A1 (p.A1259T)
and NCOA7 (p.S165del) detected by WES analysis
WES is an effective and efficient method for
studying gene loci within the human genome. It has had great
success in the genetic research of numerous complex diseases. In
the present study, WES was used to identify a CNV duplication in
PCDHA, a c.3775G>A (p.A1259T) mutation in COL3A1
and a c.492CAGT>C (p.S165del) mutation in NCOA7
considered as the likely causative genes in a Chinese pedigree with
concomitant exotropia, including the proband, the proband's parents
and maternal grandfather.
Identification of variants in PCDHA
and COL3A1 (p.A1259T) carried by the proband and the proband's
father
After CNV calling, CNVs carried by the proband and
by the proband's father or mother were screened. Due to uncertainty
in CNV calling, like DGV, CNVs of different individuals whose scope
overlaps 70% or more were considered as shared CNVs (23). Based on genetic analysis and
functional annotation of the CNVs, one CNV at the PCDHA gene
cluster was found to be likely to be a pathogenic CNV carried by
the proband (chr5:1402077661-40215463) and the proband's father
(chr5:140207600-140216026). This CNV region is a duplication in the
exon region of genes PCDHA6 and PCDHA7, and intron
region of genes PCDHA1, PCDHA2, PCDHA3,
PCDHA4 and PCDHA5. This region contains histone
modification peaks of H3K4me1 and H3K4me3 indicative of active
promoter and enhancer in neuronal progenitor cultured cells and
different brain regions based on the Encode dataset (Fig. 1B). Previous studies indicated that
PCDHA gene clusters encoding neurocadherin-like cell
adhesion proteins have important roles in the establishment of
specific cell-cell connections in the brain (27). In the present study, four family
members were analyzed by qPCR to validate the presence of
PCDHA CNVs. The qPCR amplification region is at the center
of CNVs called by WES. The results suggested the increased copy
number of PCDHA in the proband and the proband's father
compared to that of the mother or the maternal grandfather
(Fig. 1C).
After variant filtering based on calling quality,
functional annotation and inheritance mode, it was indicated that
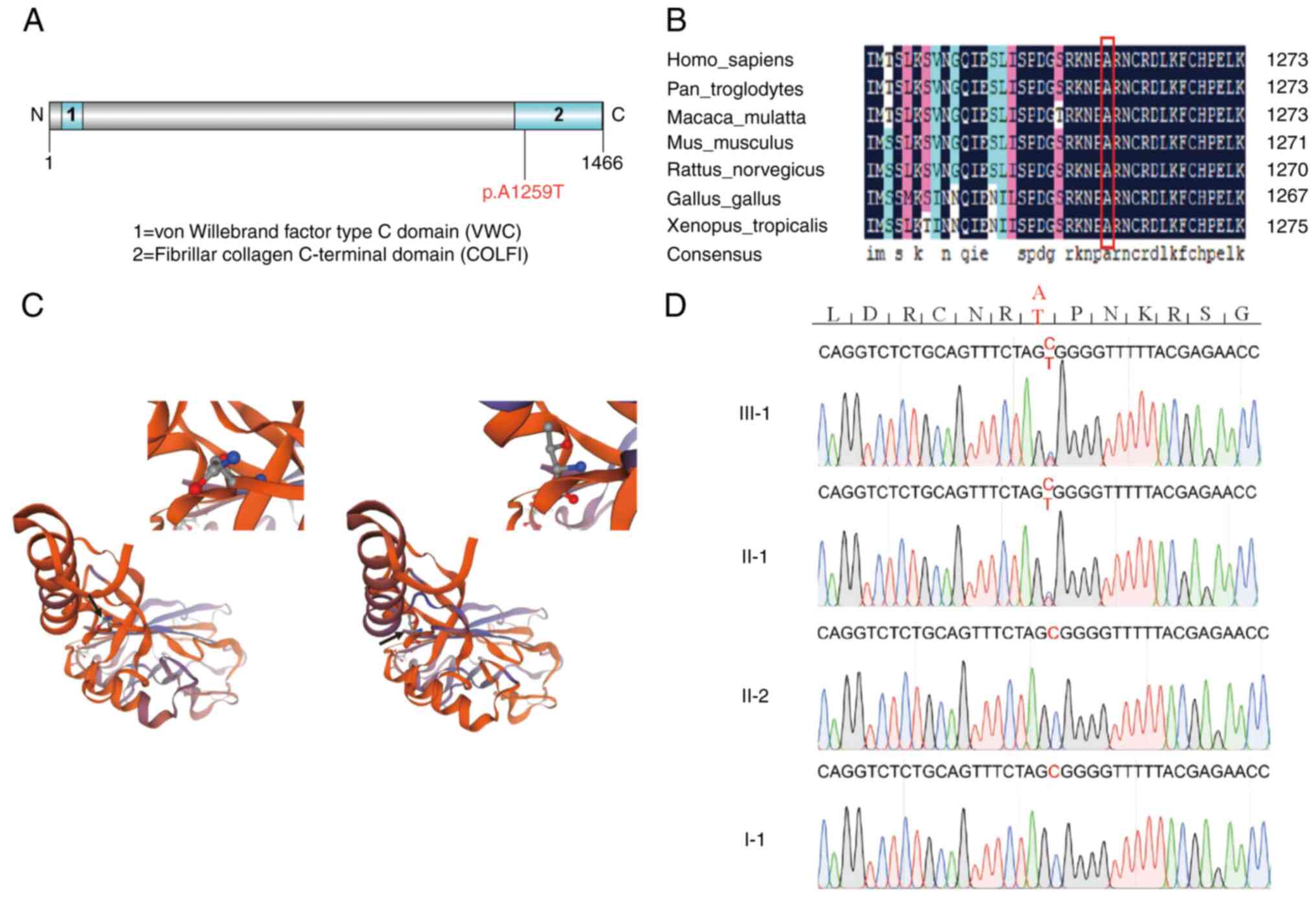
the proband and the proband's father carry a heterozygous mutation
in gene COL3A1 (chr2: 189873899, p.A1259T) which fits into a
dominant inheritance mode. There are no recessive SNVs passing all
the filtering criteria. The COL3A1 gene is the most relevant
gene according to Phenolyzer (21)
and the mutation is predicted to be ‘dangerous’ by SIFT, PolyPhen2
and PROVEAN_pred. The pro-alpha 1 chain of type III collagen
encoded by the COL3A1 gene is involved in regulating the
integrity of the pial basement membrane and cortical laminate in
the brain, which is critical for neuronal migration (28). The mutation site c.3775G>A
(p.A1259T) is located in the COLFI conserved domain (Fig. 2A) and the altered amino acid
residue is highly conserved across species (Fig. 2B). In addition, the
three-dimensional structure of the mutant COL3A1 protein is
predicted to be destabilized (Fig.
2C). Sanger sequencing validated the identification of the
COL3A1(p.A1259T) mutation (Fig.
2D).
Considering that concomitant strabismus is an eye
disease related to neural development, the mutation in
COL3A1 (p.A1259T) and large CNV covering gene cluster
PCDHA1-7 are likely to contribute to its pathogenesis. The
known functions of these genes are summarized in Table SII. Knockout mice of PCDHA
or COL3A1 exhibited defects in brain morphology or muscle
morphology (Table SIII).
Identification of variant in NCOA7 (p.
S165Δ) carried by the proband and the proband's mother
Similar variant and CNV analyses on the proband's
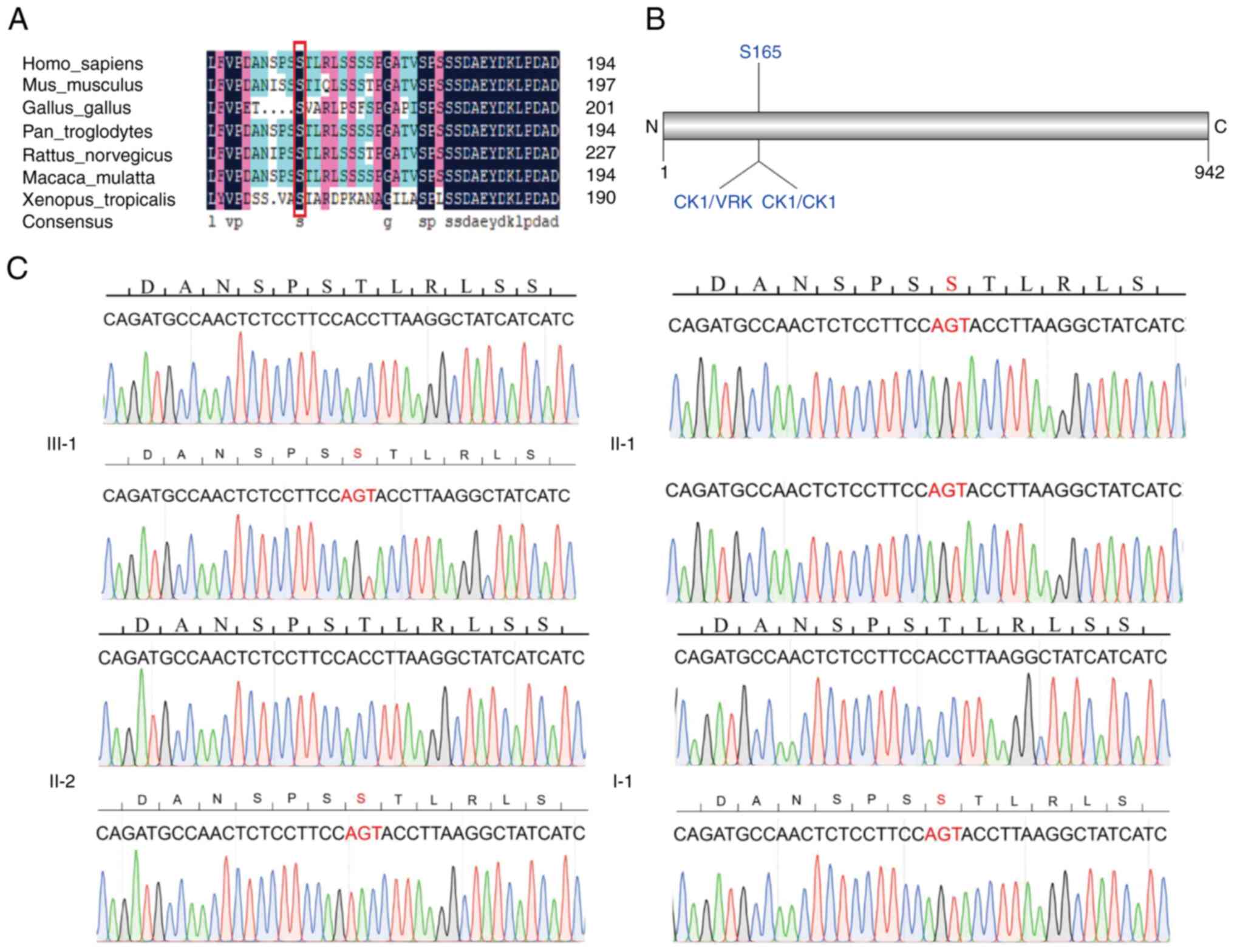
maternal side yielded the identification of a three-bp in-frame
deletion in the gene NCOA7. The NCOA7 gene encodes an
important V-ATPase regulatory protein in the brain that modulates
lysosomal function, neuronal connectivity and behavior. This indel
results in the deletion of S165, which is highly conserved across
species (Fig. 3A) and S165 is
predicted to be a phosphorylation site of serine/threonine kinase
vaccina related kinase (VRK) of the casein kinase 1 (CK1) family
(Fig. 3B). The details of the
candidate causative variant are presented in Table I. PCR amplification of the 563 bp
genomic region surrounding NCOA7(s165Δ) was performed and
the PCR products were cloned into the pEASY-Blunt Zero
Cloning vector. Sanger sequencing of individual clones confirmed
the presence of the heterozygous NCOA7(s165Δ) mutation
(Fig. 3C). The functions of the
NCOA7 gene are summarized in Table SII. Previous research indicated
that NCOA7del/del mice displayed abnormal brain
and synapse morphology (Table
SIII).
 | Table IDetailed information of the variants
COL3A1 (p.A1259T) and NCOA7 (p. S165Δ). |
Table I
Detailed information of the variants
COL3A1 (p.A1259T) and NCOA7 (p. S165Δ).
| Carrier | Chr | Pos (GRCh37) | Ref | Alt | Gene | AD | ChinaMap | ExAC | GnomAD | 1000G | SIFT | Polyphen2 | PROVEAN_pred |
|---|
| III-1 and II-1 | 2 | 189873899 | G | A | COL3A1 | III-1: 45,55; II-1:
34,32 | 0.000756 | 0.0012 | 0.0012 | - | D | D | D |
| III-1, II-2 and
I-1 | 6 | 126202268 | CAGT | C | NCOA7 | III-1: 49,42; II-2:
15,14; I-1: 43,32 | 0.00298 | 0.0027 | 0.0027 | - | - | - | - |
Discussion
Concomitant exotropia is an eye disease with genetic
heterogeneity. In the present study, WES was performed on a
three-generation Chinese strabismus pedigree involving an affected
male pediatric patient, as well as the patient's parents and
maternal grandfather. A CNV involving PCDHA1-7 and a
heterozygous mutation c.3775G>A (p.A1259T) in the COL3A1
gene were identified in the proband and the proband's father.
Furthermore, a deletion of one amino acid (S165) in NCOA7 was
detected in family members over three generations, including the
proband as well as the proband's mother and maternal
grandfather.
Both the visual and oculomotor systems are comprised
of neural networks that link numerous structures from the eyes to
the brain. All of these structures must be functionally coordinated
and work properly in order to achieve normal visual function,
including normal binocular vision with a normal alignment of the
eyes. The cause of strabismus is generally an abnormal or immature
development of the neural networks caused by genetic or acquired
factors. For instance, a primary lack of innervation of extraocular
muscle from deficient, absent, or misguided cranial nerves causes
various forms of complex incomitant strabismus (29-32).
Certain gene mutations have been identified for these special types
of strabismus, such as KIF21A mutations for congenital
fibrosis of the extraocular muscles type 1 (CFEOM 1), PHOX2A
mutations for CFEOM 2, TUBB3 mutations for CFEOM 3,
CHN1 mutations for Duane retraction syndrome type 2 and
ROBO3 mutations for horizontal gaze palsy with progressive
scoliosis (33), all of which are
now known as congenital cranial dysinnervation disorders. However,
the pathogenesis of concomitant strabismus is much more complex. To
date, it has not been attributed to a specific gene. Previous
studies support a hypothesis that any insult or injury during the
normal processes of neurogenesis, neuronal migration, axonal
growth, synaptogenesis and myelination may potentially lead to
strabismus (34). For instance,
when cutting the cortical-cortical connections of cats, they
rapidly exhibit misaligned eyes and strabismus (35,36).
Strabismus may also be caused by abnormal inputs from cortical
structures, such as the frontal eye field, supplementary eye field
and parietal eye field, all of which have a critical role in
controlling eye movements (34).
The gray matter volume of the cortical areas of the eyes of
patients with strabismus is always abnormal, either larger or
smaller, in neuroimaging studies (37). It remains unknown, however, whether
genetic influences have a substantial role in all these processes
of brain development (38).
In the present study, a heredity analysis of a
Chinese strabismus family was performed and PCDHA mutations
that may cause genetic susceptibility to strabismus were
discovered. The protocadherin alpha gene cluster is one of three
related clusters connected in tandem on chromosome 5. The alpha
gene cluster is made up of 15 cadherin superfamily genes, including
13 highly similar and 2 more distantly related coding sequences.
The most likely function of the protocadherin alpha gene encoding
neurocadherin-like cell adhesion proteins is to participate in the
establishment of specific cell-cell connections in the brain
(27). Compared with wild-type
mice, a mouse mutant (PCDHAΔCR/ΔCR) exhibited
abnormal serotonergic fibers in the cerebral cortex, hippocampus,
basal ganglia and thalamus. Serotonergic fibers gather around the
dorsal lateral geniculate nucleus and the medial geniculate nucleus
but are scarce in the central regions of these nuclei (Table SIII) (39). In PCDHA knockout mice, huge
aggregates, formed by the terminals of retinal ganglion cells
projecting to the dorsal lateral geniculate nucleus, contribute to
vision loss (40).
In addition, a COL3A1 mutation was discovered
in the present study. The COL3A1 gene is located on
chromosome 2q32.2 and encodes the pro-alpha 1 chain of type III
collagen, which is used as a ligand for the adhesion receptor GPR56
(ADGRG1). This interaction regulates the integrity of the pial
basement membrane and cortical laminate in the brain, which is
critical for neuronal migration (28). COL3A1 is related to
Ehlers-Danlos syndrome (EDS), vascular type and polymicrogyria with
or without vascular-type EDS. Col3a1-/- mice have
cobblestone-like cortical malformations with breakdown of the pial
basement membrane and marginal zone heterotopias. There was also
neuronal overmigration and radial glial detachment (41) (Table
SIII). Research has indicated that collagen III has a critical
role in the development of the brain (41). In the results of the present study,
the missense mutation c.3775G>A caused the changed amino acid
residue (p.A1259T) located in the conserved COLFI superfamily
domain of COL3A1.
A novel NCOA7 variant was also discovered in
the present study. The NCOA7 gene, mapped to chromosome
6q22.33, contains 15 exons and spans ~150 kb of genomic DNA. The
protein containing 942 amino acids encoded by NCOA7 is also
known as ERAP140(42). In
neuroblastoma-derived RTBM1 cells, the expression level of
ERAP140/NBLA10993 was increased at the mRNA level during the
process of neuronal differentiation mediated by all-trans retinoic
acid (43). NCOA7 is an important
V-ATPase regulatory protein in the brain that modulates lysosomal
function, neuronal connectivity and behavior.
NCOA7del/del mice exhibit a larger number of
proximal neurites on cortical neuronal processes, a reduced number
of calbindin (CB)-positive interneurons in the somatosensory and
visual cortex, and reduced inhibitory contacts on cortical and
somatosensory cortex neurons (44)
(Table SIII). In the case of the
present study, the deletion of three adjacent nucleotides resulted
in the loss of serine, which is a highly conserved amino acid
throughout evolution. Besides, this amino acid site is located in
the CK1/VRK and CK1/CK1 kinase-specific phosphorylation site. These
findings support that the novel NCOA7 variants, as a
possible pathogenic mutation, may have a potential role in the
pathogenesis of strabismus.
In conclusion, the present results indicated that a
CNV duplication variant in PCDHA, c.3775G>A (p.A1259T)
mutation in COL3A1 and c.492CAGT>C (p.S165del) mutation
in NCOA7 may have contributed to the susceptibility to
concomitant exotropia in the Chinese family examined by an additive
effect and suggested that aberrant cortical neuronal development
may contribute to the origin of concomitant strabismus.
Supplementary Material
PCR primers for Sanger
sequencing.
Mutant genes identified in the present
study.
Phenotypes of candidate gene knockout
mice.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by the National Natural Science
Foundation of China (grant nos. 81770956 and 81371049), the Science
Fund for Distinguished Young Scholars of Tianjin (grant no.
17JCJQJC46000), Project of Tianjin 131 Innovative Talent Team
(grant no. 201936), Jinmen Medical Talent Project of Tianjin, the
Science and Technology Planning Project of Tianjin (grant no.
21JCYBJC00780) and Tianjin Key Medical Discipline (Specialty)
Construction Project (grant no. TJYXZDXK-016A).
Availability of data and materials
The datasets analyzed during the current study are
not publicly available due to privacy or ethical restrictions but
are available from the corresponding author on reasonable
request.
Authors' contributions
JXL collected the clinical samples, performed the
experiments, analyzed and interpreted the data and wrote the
manuscript. YM and WZ analyzed and interpreted the data and edited
the manuscript. WL and LW collected the clinical samples and
performed the experiments. SM analyzed the data. JL and XS
conceptualized and designed the study, reviewed the manuscript and
confirmed the authenticity of all the raw data. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
The present study followed the Declaration of
Helsinki. Written informed consent regarding genetic testing was
provided by all participants or the legal guardian. The study was
approved by the Ethics Committee of Tianjin Eye Hospital (Tianjin,
China; no. 202015).
Patient consent for publication
The subjects or the legal guardian provided written
consent for the publication of their data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang Y, Zhao A, Zhang X, Huang D, Zhu H,
Sun Q, Yu J, Chen J, Zhao X, Li R, et al: Prevalence of strabismus
among preschool children in eastern China and comparison at a
5-year interval: A population-based cross-sectional study. BMJ
Open. 11(e055112)2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Fieß A, Elflein HM, Urschitz MS, Pesudovs
K, Münzel T, Wild PS, Michal M, Lackner KJ, Pfeiffer N, Nickels S
and Schuster AK: Prevalence of strabismus and its impact on
vision-related quality of life: Results from the German
population-based Gutenberg health study. Ophthalmology.
127:1113–1122. 2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Matsuo T and Matsuo C: The prevalence of
strabismus and amblyopia in Japanese elementary school children.
Ophthalmic Epidemiol. 12:31–36. 2009.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Chia A, Dirani M, Chan YH, Gazzard G, Eong
KG, Selvaraj P, Ling Y, Quah BL, Young TL, Mitchell P, et al:
Prevalence of amblyopia and strabismus in young singaporean chinese
children. Invest Ophthalmol Vis Sci. 51:3411–3417. 2010.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Maconachie GD, Gottlob I and McLean RJ:
Risk factors and genetics in common comitant strabismus: A
systematic review of the literature. JAMA Ophthalmol.
131:1179–1186. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Hu DN: Prevalence and mode of inheritance
of major genetic eye diseases in China. J Med Genet. 24:584–588.
1987.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Mohney BG, Erie JC, Hodge DO and Jacobsen
SJ: Congenital esotropia in Olmsted county, Minnesota.
Ophthalmology. 105:846–850. 1998.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Pennefather PM, Clarke MP, Strong NP,
Cottrell DG, Dutton J and Tin W: Risk factors for strabismus in
children born before 32 weeks' gestation. Br J Ophthalmol.
83:514–518. 1999.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Matsuo T, Hayashi M, Fujiwara H, Yamane T
and Ohtsuki H: Concordance of strabismic phenotypes in monozygotic
versus multizygotic twins and other multiple births. Jpn J
Ophthalmol. 46:59–64. 2002.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Parikh V, Shugart YY, Doheny KF, Zhang J,
Li L, Williams J, Hayden D, Craig B, Capo H, Chamblee D, et al: A
strabismus susceptibility locus on chromosome 7p. Proc Natl Acad
Sci USA. 100:12283–12288. 2003.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Shaaban S, Matsuo T, Fujiwara H, Itoshima
E, Furuse T, Hasebe S, Zhang Q, Ott J and Ohtsuki H: Chromosomes
4q28.3 and 7q31.2 as new susceptibility loci for comitant
strabismus. Invest Ophthalmol Vis Sci. 50:654–661. 2009.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhang J and Matsuo T: MGST2 and WNT2 are
candidate genes for comitant strabismus susceptibility in Japanese
patients. PeerJ. 5(e3935)2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Tarasov A, Vilella AJ, Cuppen E, Nijman IJ
and Prins P: Sambamba: Fast processing of NGS alignment formats.
Bioinformatics. 31:2032–2034. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38(e164)2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Sim N, Kumar P, Hu J, Henikoff S,
Schneider G and Ng PC: SIFT web server: Predicting effects of amino
acid substitutions on proteins. Nucleic Acids Res. 40:W452–W457.
2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010.PubMed/NCBI View Article : Google Scholar
|
|
17
|
1000 Genomes Project Consortium. Auton A,
Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL,
McCarthy S, McVean GA and Abecasis GR: A global reference for human
genetic variation. Nature. 526:68–74. 2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Lek M, Karczewski KJ, Minikel EV, Samocha
KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ,
Cummings BB, et al: Analysis of protein-coding genetic variation in
60,706 humans. Nature. 536:285–291. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Karczewski KJ, Francioli LC, Tiao G,
Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A,
Birnbaum DP, et al: The mutational constraint spectrum quantified
from variation in 141,456 humans. Nature. 581:434–443.
2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Cao Y, Li L, Xu M, Feng Z, Sun X, Lu J, Xu
Y, Du P, Wang T, Hu R, et al: The ChinaMAP analytics of deep whole
genome sequences in 10,588 individuals. Cell Res. 30:717–731.
2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Yang H, Robinson PN and Wang K:
Phenolyzer: Phenotype-based prioritization of candidate genes for
human diseases. Nat Methods. 12:841–843. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Krumm N, Sudmant PH, Ko A, O'Roak BJ,
Malig M and Coe BP: NHLBI Exome Sequencing Project. Quinlan AR,
Nickerson DA and Eichler EE: Copy number variation detection and
genotyping from exome sequence data. Genome Res. 22:1525–1532.
2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
MacDonald JR, Ziman R, Yuen RKC, Feuk L
and Scherer SW: The database of genomic variants: A curated
collection of structural variation in the human genome. Nucleic
Acids Res. 42:D986–D992. 2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zarrei M, MacDonald JR, Merico D and
Scherer SW: A copy number variation map of the human genome. Nat
Rev Genet. 16:172–183. 2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Qiu F, Xu Y, Li K, Li Z, Liu Y, DuanMu H,
Zhang S, Li Z, Chang Z, Zhou Y, et al: CNVD: Text mining-based copy
number variation in disease database. Hum Mutat. 33:E2375–E2381.
2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta C(T)) method. Methods. 25:402–408. 2001.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Yagi T and Takeichi M: Cadherin
superfamily genes: Functions, genomic organization, and neurologic
diversity. Genes Dev. 14:1169–1180. 2000.PubMed/NCBI
|
|
28
|
Vandervore L, Stouffs K, Tanyalçin I,
Vanderhasselt T, Roelens F, Holder-Espinasse M, Jørgensen A, Pepin
MG, Petit F, Van Kien PK, et al: Bi-allelic variants in COL3A1
encoding the ligand to GPR56 are associated with cobblestone-like
cortical malformation, white matter changes and cerebellar cysts. J
Med Genet. 54:432–440. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Graeber CP, Hunter DG and Engle EC: The
genetic basis of incomitant strabismus: Consolidation of the
current knowledge of the genetic foundations of disease. Semin
Ophthalmol. 28:427–437. 2013.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Engle EC: The genetic basis of complex
strabismus. Pediatr Res. 59:343–348. 2006.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Marillat V, Sabatier C, Failli V,
Matsunaga E, Sotelo C, Tessier-Lavigne M and Chédotal A: The slit
receptor Rig-1/Robo3 controls midline crossing by hindbrain
precerebellar neurons and axons. Neuron. 43:69–79. 2004.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Seeger M, Tear G, Ferres-Marco D and
Goodman CS: Mutations affecting growth cone guidance in Drosophila:
Genes necessary for guidance toward or away from the midline.
Neuron. 10:409–426. 1993.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ye XC, Pegado V, Patel MS and Wasserman
WW: Strabismus genetics across a spectrum of eye misalignment
disorders. Clin Genet. 86:103–111. 2014.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Quoc EB and Milleret C: Origins of
strabismus and loss of binocular vision. Front Integr Neurosci.
8(71)2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Payne BR, Berman N and Murphy EH: A
quantitative assessment of eye alignment in cats after corpus
callosum transection. Exp Brain Res. 43:371–376. 1981.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Elberger AJ and Hirsch HV: Divergent
strabismus following neonatal callosal section is due to a failure
of convergence. Brain Res. 239:275–278. 1982.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Chan S, Tang K, Lam K, Chan L, Mendola JD
and Kwong KK: Neuroanatomy of adult strabismus: A voxel-based
morphometric analysis of magnetic resonance structural scans.
Neuroimage. 22:986–994. 2004.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Buzsáki G, Logothetis N and Singer W:
Scaling brain size, keeping timing: Evolutionary preservation of
brain rhythms. Neuron. 80:751–764. 2013.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Katori S, Hamada S, Noguchi Y, Fukuda E,
Yamamoto T, Yamamoto H, Hasegawa S and Yagi T: Protocadherin-family
is required for serotonergic projections to appropriately innervate
target brain areas. J Neurosci. 29:9137–9147. 2009.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Meguro R, Hishida R, Tsukano H, Yoshitake
K, Imamura R, Tohmi M, Kitsukawa T, Hirabayashi T, Yagi T,
Takebayashi H and Shibuki K: Impaired clustered protocadherin-α
leads to aggregated retinogeniculate terminals and impaired visual
acuity in mice. J Neurochem. 133:66–72. 2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Jeong SJ, Li S, Luo R, Strokes N and Piao
X: Loss of Col3a1, the gene for Ehlers-Danlos syndrome type IV,
results in neocortical dyslamination. PLoS One.
7(e29767)2012.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Shao W, Halachmi S and Brown M: ERAP140, a
conserved tissue-specific nuclear receptor coactivator. Mol Cell
Biol. 22:3358–3372. 2002.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Arai H, Ozaki T, Niizuma H, Nakamura Y,
Ohira M, Takano K, Matsumoto M and Nakagawara A: ERAP140/Nbla10993
is a novel favorable prognostic indicator for neuroblastoma induced
in response to retinoic acid. Oncol Rep. 19:1381–1388.
2008.PubMed/NCBI
|
|
44
|
Castroflorio E, den Hoed J, Svistunova D,
Finelli MJ, Cebrian-Serrano A, Corrochano S, Bassett AR, Davies B
and Oliver PL: The Ncoa7 locus regulates V-ATPase formation and
function, neurodevelopment and behaviour. Cell Mol Life Sci.
78:3503–3524. 2021.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Smith LT, Schwarze U, Goldstein J and
Byers PH: Mutations in the COL3A1 gene result in the Ehlers-Danlos
syndrome type IV and alterations in the size and distribution of
the major collagen fibrils of the dermis. J Invest Dermatol.
108:241–247. 1997.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Fukuda E, Hamada S, Hasegawa S, Katori S,
Sanbo M, Miyakawa T, Yamamoto T, Yamamoto H, Hirabayashi T and Yagi
T: Down-regulation of protocadherin-alpha A isoforms in mice
changes contextual fear conditioning and spatial working memory.
Eur J Neurosci. 28:1362–1376. 2008.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Hasegawa S, Hamada S, Kumode Y, Esumi S,
Katori S, Fukuda E, Uchiyama Y, Hirabayashi T, Mombaerts P and Yagi
T: The protocadherin-alpha family is involved in axonal coalescence
of olfactory sensory neurons into glomeruli of the olfactory bulb
in mouse. Mol Cell Neurosci. 38:66–79. 2008.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Liu X, Wu H, Byrne M, Krane S and Jaenisch
R: Type III collagen is crucial for collagen I fibrillogenesis and
for normal cardiovascular development. Proc Natl Acad Sci USA.
94:1852–1856. 1997.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Smith LB, Hadoke PW, Dyer E, Denvir MA,
Brownstein D, Miller E, Nelson N, Wells S, Cheeseman M and
Greenfield A: Haploinsufficiency of the murine Col3a1 locus causes
aortic dissection: A novel model of the vascular type of
Ehlers-Danlos syndrome. Cardiovasc Res. 90:182–190. 2011.PubMed/NCBI View Article : Google Scholar
|