Introduction
Inherited epidermolysis bullosa (IEB) represents a
group of rare heterogeneous genetic dermatoses characterized by
mucocutaneous fragility and blister formation, often induced by
trivial trauma (1). Patients with
IEB can be affected mildly to severely, while in extreme cases, the
disease can be debilitating or mortal (1). Patients with severe EB may show the
involvement of not only the skin tissue but also any
epithelial-lined organ (2).
Recently, due to the emergence of EB-related
pathogenic genes and clinical subtypes after a previous
classification was revised in 2014(3), those practitioners studying IEB have
to understand it anew. According to the latest consensus report,
>30 subtypes of EB comprised four main types: EB simplex (EBS),
Junctional EB (JEB), Dystrophic EB (DEB) and the rarely occurring
Kindler EB (KEB), generally based on the level of skin cleavage
(4). Other disorders with
relatively minor skin blisters are also classified into separate
categories, including peeling skin disorders, erosive disorders,
hyperkeratotic disorders and connective tissue disorders with skin
fragility (4). As examples of
large cohorts, according to a long-term and large-sample
epidemiological survey (the National EB Registry of USA,
~1986-2002), IEB occurred at a rate of ~11.1 cases per million
individuals and 19.6 cases per million live births, with no
differences among the sexes or ethnic groups (5). EB, in general, or its specific
subtypes, has been reported to have a higher prevalence in some
ethnic groups, which may simply reflect improved case collection
and integrity of molecular diagnostics in these studies (6,7).
Among East Asians, the largest ethnic group, the incidence of IEB
has not been accurately reported, except for Japan (8). This limits the development of
research on the disease in this region.
To date, pathogenetic variations in at least 20
distinct genes encoding proteins influencing cellular integrity and
adhesion have been implicated in IEB (1). EBS is the predominant type accounting
for ~70% of the EB cases, which could be caused by mutations in
KRT5, KRT14, PLEC, KLHL24, DST,
EXPH5 (syn. SLAC2B), CD151 (syn.
TSPAN24), TGM5, PKP1, DSP and
JUP genes. JEB is associated with mutations in LAMA3,
LAMB3, LAMC2, COL17A1, ITGA6,
ITGB4 and ITGA3, while DEB and KEB are caused by
mutations in COL7A1 and FERMT1 (syn. KIND1),
respectively (4). An accurate
diagnosis is established based on multimodal methods consisting of
transmission electron microscopy (TEM), immuno-fluorescence antigen
mapping of the affected skin and DNA mutational analysis (9). Recently, the advancement in gene
sequencing techniques promises faster, cheaper and more
comprehensive diagnosis, facilitating the identification of new
genes and ultimately personalized treatments (10,11).
Precise molecular diagnosis, although not currently fully
functional, is essential to advance the understanding of disease to
provide a basis for the potential stratification and
prognostication, as well as a platform for tailored or stratified
management of disease, including genetic counseling and targeted
therapies (12-15).
The present study aimed to provide a definite
molecular diagnosis on six enrolled cases with suspected IEB. It
conducted a comprehensive survey of clinical and family history and
detected mutations using whole exome sequencing. The findings
confirmed the complexity of clinical and genetic characteristics
for IEB.
Materials and methods
Subjects
The six cases with apparent EB were recruited in the
Department of Dermatology, the First Hospital of Hebei Medical
University, Shijiazhuang, between January 2018 and December 2021.
The clinical evaluation was made by GZ via routine clinical
examination, family survey and TEM testing of the skin tissue
obtained by biopsy (only for Case 1). Genomic DNA was extracted
from the peripheral blood specimens of the patients and their
parents using the QIAamp DNA Midi kit (Qiagen GmbH) for further
testing.
Whole-exome sequencing (WES)
WES was used to detect the sequence variants in the
samples of the probands (16).
Briefly, the sequences of the target region were enriched using the
Agilent Sure Select Human Exon Sequence Capture kit (Agilent
Technologies, Inc.). The DNA libraries were tested using
quantitative PCR, where the size, distribution and concentration
were determined using an Agilent Bioanalyzer 2100 (Agilent
Technologies, Inc.). The DNA of ~150 bp paired-end reads was
sequenced using the NovaSeq6000 platform (Illumina, Inc.), taking
~300 pM of DNA per sample using the NovaSeq Reagent kit. Sequencing
raw reads (quality level of Q30%>90% and the criteria for
quality listed at https://www.illumina.com/science/technology/next-generation-sequencing/plan-experiments/quality-scores.html)
were aligned to the human reference genome (accession No.
hg19/GRCh37) using the Burrows-Wheeler Aligner tool
(bwa-0.7.17.tar.bz2) (17),
following which, duplicate PCR products were removed using the
program Picard v1.57 (https://github.com/broadinstitute/picard). Variant
calling was performed using the Verita Trekker® Variants
Detection system (v2.0; Berry Genomics Co., Ltd.) and Genome
Analysis Tool kit (https://software.broadinstitute.org/gatk/). Then,
variants were annotated and interpreted using the ANNOVAR (v2.0)
(18) and Enliven®
Variants Annotation Interpretation systems (Berry Genomics Co.,
Ltd.), according to the guidelines by ACMG (American College of
Medical Genetics and Genomics) (19). To assist in the interpretation of
variant pathogenicity, the present study referred to three
frequency databases, namely ExAC_EAS (http://exac.broadinstitute.org), gnomAD_exome_EAS
(http://gnomad.broadinstitute.org),
1000G_2015aug_eas (https://www.internationalgenome.org) and Human Gene
Mutation Database Pro v2019 (https://www.hgmd.cf.ac.uk/ac/index.php). The Revel
score (a combined method of pathogenicity prediction) (20) and pLI score (representing the
tolerance for truncating variants) were also employed.
Sanger sequencing
For validation, Sanger sequencing was performed on
potentially causative-specific variants using the 3730 DX Genetic
Analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.).
Analysis of conservatism
The evolutionary conservatism of all affected amino
acid (AA) residues by the corresponding missense variants was
analyzed using the online tool MEGA7 (http://www.megasoftware.net/previousVersions.php)
with default parameters.
Results
Clinical manifestations
All six patients included in this study showed
EB-like phenotypes shortly after birth, except for Case 4, who
began to develop multiple skin breakages and blister formation at
~8 years of age. The parents of this patient denied any family
history of genetic disease, except that the elder brother of this
patient also had an EB presentation. Specifically, the clinical
characteristics and family history of these cases were as
follows:
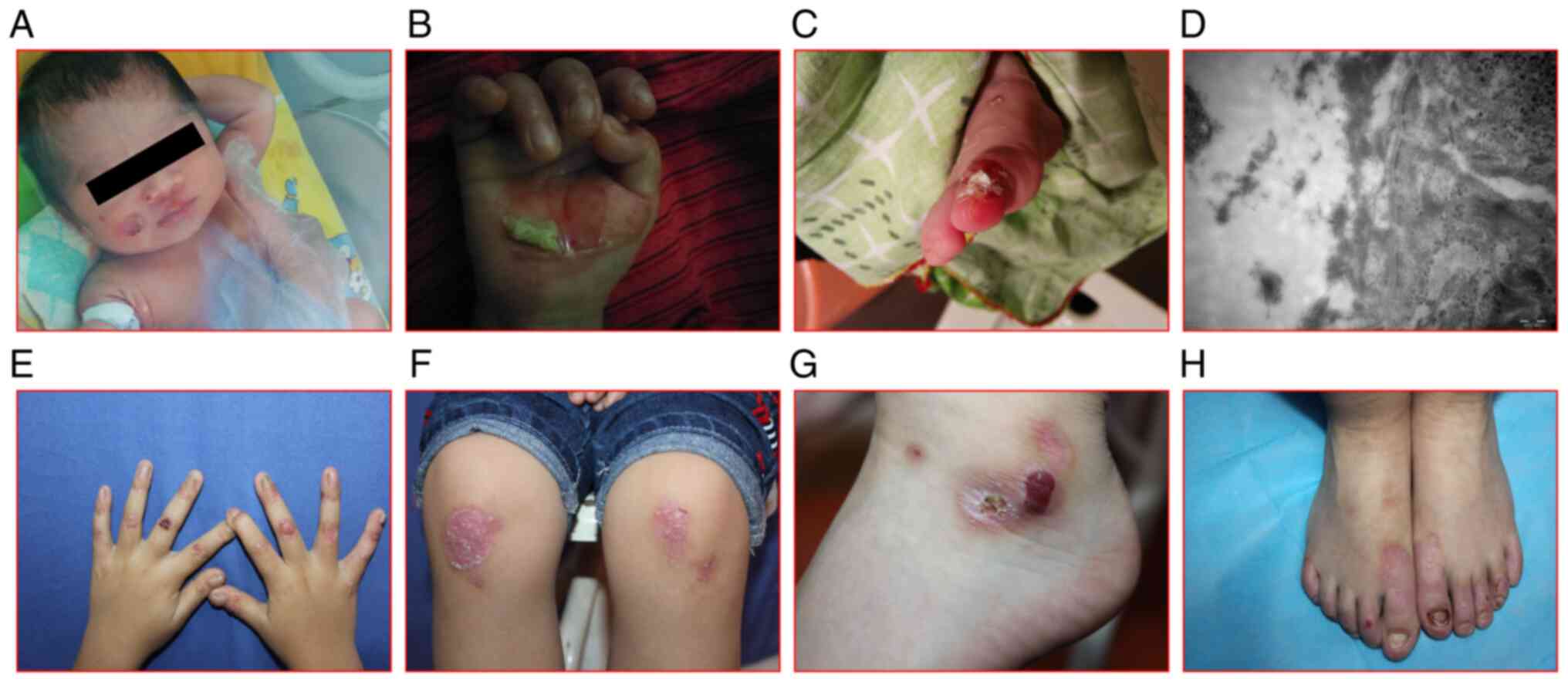
Case 1, a 4-month-old girl, initially exhibited
erosion and desquamation at both palms, soles and oral mucosa after
birth. Then, her entire body showed recurrent blisters, tatters and
scabs, part of which developed infections (Fig. 1A-C). TEM revealed a split epidermis
(Fig. 1D). Case 2, a 3.5-year-old
boy who displayed mild EB phenotype presenting with localized
repeated skin breakages on the fingers, toes, knees and ankles
(Fig. 1E-H). In addition, the boy
showed autism-like features, such as difficulty in communication
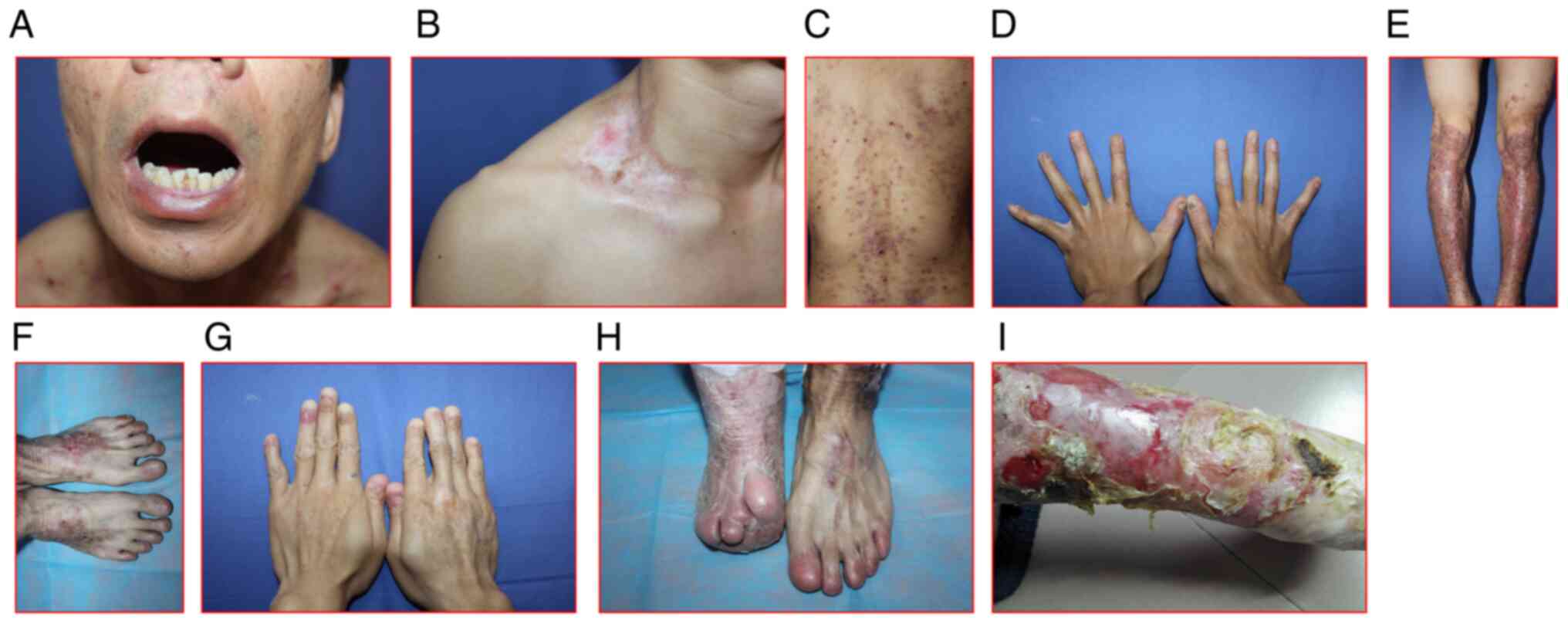
and concentration. Case 3, a 34-year-old male who, along with his
elder brother, presented with moderate to severe EB that was mainly
localized in the back, neck, elbows, lower extremities and fingers
(Fig. 2A-F for the patient; G-H
for the elder brother). The patient had progressive nail loss.
Also, his brother developed subcutaneous pustules and infections at
the shins, as well as truncated and fused fingers and toes as the
result of prolonged illness and poor care.
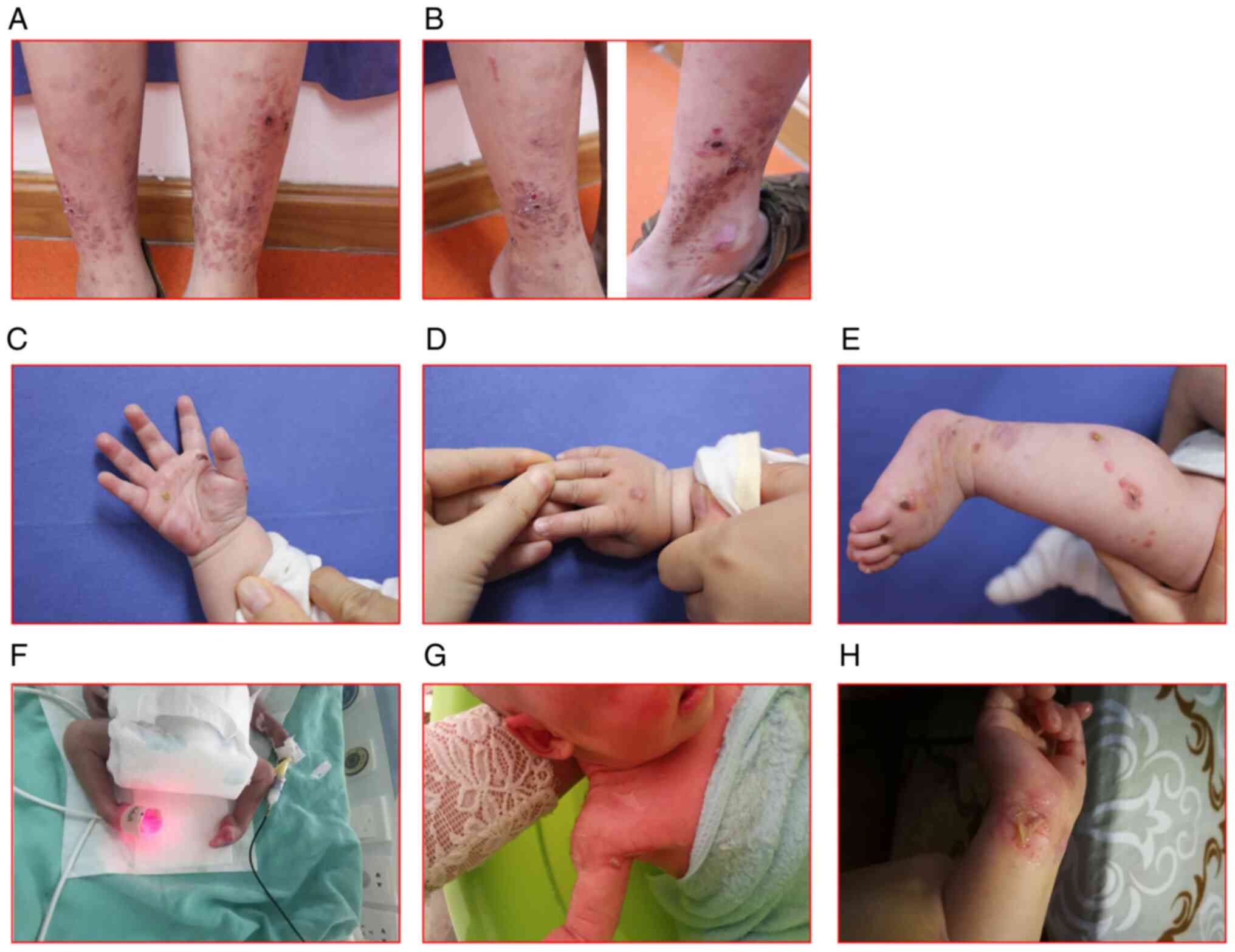
Case 4, a 13-year-old boy, who had been exhibiting
mild to moderate EB phenotype mainly with erythema blisters on the
distal extremities since the age of 8 years (Fig. 3A and B). Case 5, a 4-month-old girl, started
showing multiple skin lesions and blistering shortly after birth
(Fig. 3C-E). Case 6, a newborn
girl, showed multiple skin lesions and strephenopodia (Fig. 3F-H).
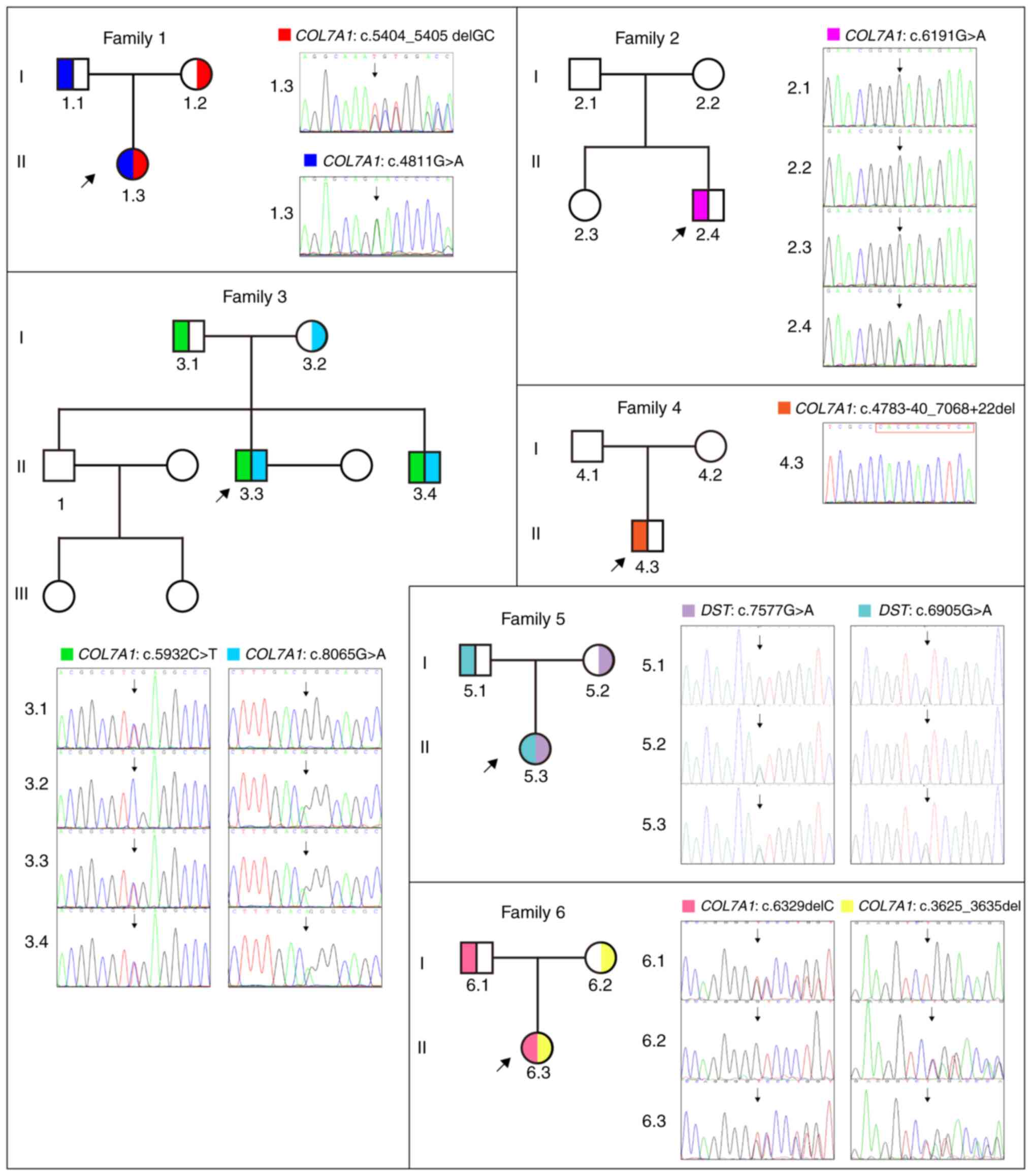
Genetic findings
All six cases showed positive results for WES
detection, which was also confirmed by Sanger sequencing. A total
of 10 variants distributed in COL7A1 and DST genes
were detected. The detailed information of all variants is
presented in Table I, while the
pattern shown by the members of each family is presented in
Table I and Fig. 4.
 | Table IInformation of the genetic variants
identified in this study. |
Table I
Information of the genetic variants
identified in this study.
| Case No. | Carrier ID | Genea | DNA variation | Protein
variation | Variation
frequencies in 3 databasesb | HGMD
ratingc | CADD_ PHREAD
scored | Revel
scoree | Pathogenicity
rating (evidences)f |
|---|
| 1 | 1.1;1.2 | COL7A1 |
c.5404_5405delGC |
p.Ala1802Trpfs*69 | -; -; - | DM | / | / | LP (pvs1+pm2) |
| | 1.1;1.3 | COL7A1 | c.4811G>A | p.Gly1604Glu | -; -; - | DM | 27.6 | 0.936 | VUS
(pm2+pm5+pp3) |
| 2 | 2.4 | COL7A1 | c.6191G>A | p.Gly2064Glu | -; -; - | DM | 28.4 | 0.982 | LP
(pm2+pm5_strong+pp3) |
| 3 | 3.1;3;3;3.4 | COL7A1 | c.5932C>T | p. Arg1978* | -; -; - | DM | 35 | / | LP (pvs1+pm2) |
| | 3.2;3.3;3.4 | COL7A1 | c.8065G>A | p.Gly2689Arg | -; -; 0.000054 | DM | 31 | 0.982 | LP
(pm2+ps1+pp3) |
| 4 | 4.3 | COL7A1 |
c.4783-40_7068+22del | / | -; -; - | / | / | / | P (one copy loss of
2 exons) |
| 5 | 5.1;5.3 | DST | c.7577G>A | p.Ser2526Asn | -; 0.000356;
0.000390 | / | 19.47 | 0.039 | VUS (pm2+bp4) |
| | 5.2;5.3 | DST | c.6905G>A | p.Arg2302His | -; 0.000204;
0.000250 | / | 23.8 | 0.267 | VUS (pm2) |
| 6 | 6.1;6.3 | COL7A1 | c.6329delC | p.P2110Lfs*96 | -; -; - | / | / | / | LP (pvs1+pm2) |
| | 6.2;6.3 | COL7A1 | c.3625_3635del | p.S1209Lfs*6 | -; -; - | DM | 32 | / | P
(pvs1+pm2+ps4_supporting) |
Specifically, Case 1 harbored a compound
heterozygous variation in COL7A1 that consisted of two
variants, c.5404_5405 delGC (p.Ala1802Trpfs*69) and c.4811G>A
(p.Gly1604Glu), which were inherited from her parents (Fig. 4: Family 1). Case 2 had a de
novo heterozygous missense variation, namely COL7A1:
c.6191G>A (p.Gly2064Glu; Fig.
4: Family 2). Case 3 carried a compound heterozygous
COL7A1 variation consisting of c.5932C>T (p. Arg1978*)
and c.8065G>A (p.Gly2689Arg) (Fig.
4: Family 3). Case 4 had a de novo heterozygous
intergenic deletion, COL7A1: c.4783-40_7068+22del (Fig. 4: Family 4). Case 5 carried a
compound heterozygous variation in the DST gene, consisting
of c.7577G>A (p.Ser2526Asn) and c.6905G>A (p.Arg2302His)
(Fig. 4: Family 5). Case 6 carried
a compound heterozygous COL7A1 variation, consisting of
c.6329delC (p.P2110Lfs*96) and c.3625_3635del (p.S1209Lfs*6)
variants (Fig. 4: Family 6). Among
these variants, four, namely COL7A1: c.4783-40_7068+22del,
DST: c.7577G>A (p.Ser2526Asn), DST: c.6905G>A
(p.Arg2302His) and COL7A1: c.6329delC (p.P2110Lfs*96), were
newly identified.
Regarding Case 2, WES also revealed two suspected
variations that might contribute to the autistic symptoms of the
patients. One variation was a compound heterozygous variation in
the LFNG (NM_001166355) gene, consisting of c.139_142del
(p.Asp55Serfs*141) and c.142_143insGATG (p.Glu56Glyfs*2), which
were inherited from the patient's parents. Another variation was a
de novo missense variation, namely SCN9A (NM_002977):
c.554G>A (p.Arg185His). These two variations and their carrying
status are presented in Fig.
S1.
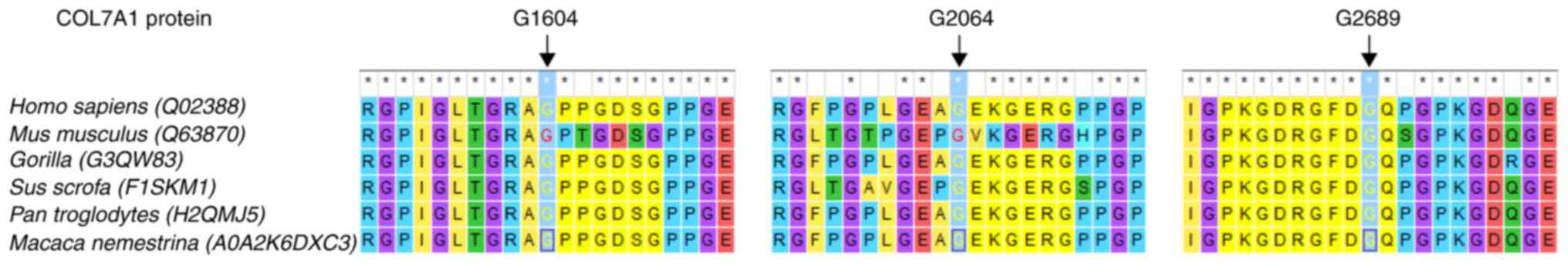
The conservatism of the amino acid
residues affected by missense variations
A total of five missense variants, namely
COL7A1:c.4811G>A (p.Gly1604Glu), COL7A1:
c.6191G>A (p.Gly2064Glu), COL7A1: c.8065G>A
(p.Gly2689Arg), DST: c.7577G>A (p.Ser2526Asn) and
DST: c.6905G>A (p.Arg2302His), were detected in the
present study. The homologous sequences of the DST protein have
been resolved in only a few species, so the nature of conservatism
of the two amino acids (Ser2526 and Arg2302) in it were not
analyzed. The MEGA7 analysis demonstrated that the AA residues
affected by the three variants in COL7A1 remained highly conserved
among multiple species (Fig.
5).
Discussion
IEB comprises various conditions with overlapping
skin and epidermal-link phenotypes, each with unique
characteristics (1,21-23).
Clinically, it is difficult to accurately diagnose the subtype of
IEB, especially in newborns. However, an accurate diagnosis is
vital for prognostics, genetic counseling and patient management
(12,24). The present study used WES to
directly detect the causative gene of EB in six Chinese families
and found disease-associated variants in the known EB genes of
COL7A1 and DST. The distribution of the subtypes of
EB is different in different countries. Worldwide variations in the
population and level of immigration (ethnic background,
consanguineous marriages and spectrum of mutations) may affect the
epidemiology and distribution of the subtypes of EB per region
(1). Research indicates that the
recurrent mutations R578X, 7786delG and R2814X in COL7A1
seem to be exclusive to a specific ethnic group, the British
population; in addition, the mutations 5818delC, 6573+1G->C and
E2857X are present only in individuals of Japanese ethnic origin
(8). However, due to the limited
sample size, a larger screening effort is necessary for us to
further clarify whether there are ethnic difference between Asian
patients and non-Asian patients (1,25).
Dystrophic EB (DEB, MIM #131750 and #226600) is
characterized by the cleavage of the upper dermis (22). DEB arose from the COL7A1
(MIM *120120) mutations that resulted in mutant type VII collagen
and disrupted anchoring fibrils (1,22).
In the present study, DEB accounted for the majority (five) of the
six cases, which was not consistent with the situation in other
studies, in which EBS had the highest incidence. This may be
attributed to the small sample size of the present study or
differences in our ability and standard of clinical identification
(5,15,26).
Among the five subjects with DEB, two (Case 2 and 4) had de novo
COL7A1 variations and conformed to the autosomal dominant
pattern, while three (Case 1, 3 and 6) carried compound
heterozygous variations in COL7A1 and conformed to the
autosomal recessive pattern. Mutations of COL7A1 were linked
to ADDEB in 1991 by Ryynänen et al (27) and ARDEB in 1993 by Christiano et
al (28). Until now, ~1,000
variants of COL7A1 have been related to DEB (http://www.col7a1-database.info; http://www.hgmd.cf.ac.uk/ac/index.php)
(29). Generally, the symptoms of
ARDEB are more severe, including skin fragility that is manifested
by blistering with minimal trauma that heals with milia and
scarring, while in ADDEB, blistering is often mild and limited to
the hands, feet, knees and elbows, although it heals with scarring
(22). The clinical phenotypes of
our five DEB cases were consistent with this pattern; thus, Cases
1, 3 and 6 showed more severe and widespread symptoms. However, it
was also observed that the brothers in Case 3 showed some
difference in phenotypic severity, which suggests that there may be
other factors regulating DEB phenotypes, which need further
clarification (30).
To the best of the authors' knowledge, the patient
in Case 2 was the first known case in which both DEB and autism
were involved. Intriguingly, mutations in two genes possibly
contributed to the patient's autism-like phenotype. The
SCN9A gene (MIM *603415), which encodes a voltage-gated
sodium channel that is enriched in the nociceptive and sympathetic
neurons of the peripheral nervous system, is involved in a group of
nociception-related neuropathies (31,32).
Another gene LFNG (MIM *602576), is the causative gene for
the autosomal recessive spondylocostal dysostosis 3, a rare
skeletal dysplasia (33).
Generally, the existing evidence is insufficient to support our
diagnosis of these two gene variations and further functional
studies are required.
The affected child in Case 5 was a patient of EBS,
the pathogenic variation of which possesses a compound heterozygous
variation of the DST gene (MIM *113810), a rare non-keratin
cause for EBS. So far, only 10 variants of DST, almost all
of which are truncating variants, have demonstrated clear
associations with EBS (http://www.hgmd.cf.ac.uk/ac/index.php) and lack
universal distribution across ethnic groups (34-36).
Moreover, the function of the DST gene in human diseases has
been under-studied. Based on the available evidence, the present
study could only identify two variants as variant with unknown
significance at the genetic level. The findings of the present
study may contribute to the expansion of the mutation spectrum for
this disease in the Chinese population, although further studies,
including in situ electron microscopy, immunofluorescence
assays and possibly functional experiments, are needed to clarify
the pathogenicity of the novel variations. In addition, in
silico structural analysis would contribute to elucidating the
pathogenicity of these missense variants. The cross-species
conservatism nature of the amino acid residues affected by the
three missense variants in COL7A1 supports their
pathogenicity. It also demonstrates that in silico methods
play an increasingly important role in the analysis of rare disease
mutations (37).
The findings of the present study may also have some
implications for the recent insights into the pathogenesis of IEB
and the emerging potential for new therapies. For example, a recent
study on applied Adenine Base Editors to correct the pathogenic
mutation of COL7A1 or to bypass a premature stop codon in
fibroblasts of patients produced encouraging results (38). Another study identified several
molecules that effectively increased the expression of type 7
collagen in keratinocytes, showing some therapeutic promise
(39). The enzymatic modification
of structural proteins by non-structural proteins such as PLOD3,
USB1, EXPH5 and KLHL24 may be an important supplement to the
pathogenesis of IEB (40).
In conclusion, the findings of the present study
established the genetic diagnosis of six IEB cases, expanded the
mutation spectrum of the related genes and diseases and provided a
solid basis for further analysis of the disease prognosis,
treatment design and reproductive guidance for the affected
families.
Supplementary Material
The variations in the SCN9A and
LFNG genes that were harbored by the affected child in Case
2.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Project Funded
by China Postdoctoral Science Foundation (grant no.
2022T150445).
Availability of data and materials
The datasets generated or analyzed during the
current study are available in the figshare repository, https://doi.org/10.6084/m9.figshare.20979547.v1.
Whole-exome sequencing data are not publicly available due to
patient privacy, but are available from the corresponding author on
reasonable request.
Authors' contributions
GQZ designed this study. YY and LXZ recruited the
case and did the clinical examination. KY, YY and KQ performed the
genetic and in silico studies. YY, KQ and LXZ analyzed the
experimental data and composed all figures and tables. GQZ wrote
this manuscript. YY and GQZ confirm the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
This research was approved by the Ethics Committee
of the First Hospital of Hebei Medical University (approval no.
HMU-FH-2020-02). Informed consent was signed by all the
participants or their guardians for participation in this study.
All procedures performed in the present study were following the
Declaration of Helsinki 1964 and its later amendments or comparable
ethical standards.
Patient consent for publication
Informed consent was signed by all the participants
or their guardians for the images of this manuscript to be
published.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bardhan A, Bruckner-Tuderman L, Chapple
ILC, Fine JD, Harper N, Has C, Magin TM, Marinkovich MP, Marshall
JF, McGrath JA, et al: Epidermolysis bullosa. Nat Rev Dis Primers.
6(78)2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Prodinger C, Reichelt J, Bauer JW and
Laimer M: Epidermolysis bullosa: Advances in research and
treatment. Exp Dermatol. 28:1176–1189. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Fine JD, Bruckner-Tuderman L, Eady RA,
Bauer EA, Bauer JW, Has C, Heagerty A, Hintner H, Hovnanian A,
Jonkman MF, et al: Inherited epidermolysis bullosa: Updated
recommendations on diagnosis and classification. J Am Acad
Dermatol. 70:1103–1126. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Has C, Bauer JW, Bodemer C, Bolling MC,
Bruckner-Tuderman L, Diem A, Fine JD, Heagerty A, Hovnanian A,
Marinkovich MP, et al: Consensus reclassification of inherited
epidermolysis bullosa and other disorders with skin fragility. Br J
Dermatol. 183:614–627. 2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Fine JD: Epidemiology of inherited
epidermolysis bullosa based on incidence and prevalence estimates
from the national epidermolysis bullosa registry. JAMA Dermatol.
152:1231–128. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Horn HM, Priestley GC, Eady RA and Tidman
MJ: The prevalence of epidermolysis bullosa in Scotland. Br J
Dermatol. 136:560–564. 1997.PubMed/NCBI
|
|
7
|
Vahlquist A and Tasanen K: Epidermolysis
bullosa care in Scandinavia. Dermatol Clin. 28:425–427.
2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Shinkuma S, Natsuga K, Nishie W and
Shimizu H: Epidermolysis bullosa in Japan. Dermatol Clin.
28:431–432. 2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Has C, Liu L, Bolling MC, Charlesworth AV,
El Hachem M, Escámez MJ, Fuentes I, Büchel S, Hiremagalore R,
Pohla-Gubo G, et al: Clinical practice guidelines for laboratory
diagnosis of epidermolysis bullosa. Br J Dermatol. 182:574–592.
2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Alharthi R, Alnahdi MA, Alharthi A,
Almutairi S, Al-Khenaizan S and AlBalwi MA: Genetic profile of
epidermolysis bullosa cases in king Abdulaziz medical City, Riyadh,
Saudi Arabia. Front Genet. 12(753229)2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ma THT, Luong TLA, Hoang TL, Nguyen TTH,
Vu TH, Tran VK, Nguyen DB, Trieu TS, Nguyen HH, Nong VH and Nguyen
DT: Novel and very rare causative variants in the COL7A1 gene of
Vietnamese patients with recessive dystrophic epidermolysis bullosa
revealed by whole-exome sequencing. Mol Genet Genomic Med.
9(e1748)2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Khan FF, Khan N, Rehman S, Ejaz A, Ali U,
Erfan M, Ahmed ZM and Naeem M: Identification and computational
analysis of novel pathogenic variants in pakistani families with
diverse epidermolysis bullosa phenotypes. Biomolecules.
11(620)2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Mayr E, Ablinger M, Lettner T, Murauer EM,
Guttmann-Gruber C, Piñón Hofbauer J, Hainzl S, Kaiser M, Klausegger
A, Bauer JW, et al: 5'RNA Trans-splicing repair of COL7A1 mutant
transcripts in epidermolysis bullosa. Int J Mol Sci.
23(1732)2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Subramaniam KS, Antoniou MN, McGrath JA
and Lwin SM: The potential of gene therapy for recessive dystrophic
epidermolysis bullosa. Br J Dermatol. 186:609–619. 2022.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Lucky AW, Dagaonkar N, Lammers K, Husami
A, Kissell D and Zhang K: A comprehensive next-generation
sequencing assay for the diagnosis of epidermolysis bullosa.
Pediatr Dermatol. 35:188–197. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhang J, Li YZ, Chen WQ, Yuan JY, Li Q,
Meng YX and Feng S: Genome sequencing identified a novel exonic
microdeletion in the RUNX2 gene that causes cleidocranial
dysplasia. Clin Chim Acta. 528:6–12. 2022.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Wang K LM and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from next-generation
sequencing data. Nucleic Acids Res. 38(e164)2010.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: a joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Ioannidis NM, Rothstein JH, Pejaver V,
Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E,
Karyadi D, et al: REVEL: An ensemble method for predicting the
pathogenicity of rare missense variants. Am J Hum Genet.
99:877–885. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Pfendner EG and Lucky AW: Junctional
Epidermolysis Bullosa. In: GeneReviews® [Internet]. Adam
MP, Ardinger HH and Pagon RA (eds). University of Washington,
Seattle, WA, 1993-2022.
|
|
22
|
Pfendner EG and Lucky AW: Dystrophic
Epidermolysis Bullosa. In: GeneReviews® [Internet]. Adam
MP, Ardinger HH and Pagon RA (eds). University of Washington,
Seattle, WA, 1993-2021.
|
|
23
|
Youssefia L, Vahidnezhad H and Uitto J:
Kindler Syndrome. In: GeneReviews® [Internet]. Adam MP,
Ardinger HH and Pagon RA (eds). University of Washington, Seattle,
WA, 1993-2022.
|
|
24
|
Sybert VP: Genetic counseling in
epidermolysis bullosa. Dermatol Clin. 28:239–243. 2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Mariath LM, Santin JT, Schuler-Faccini L
and Kiszewski AE: Inherited epidermolysis bullosa: Update on the
clinical and genetic aspects. An Bras Dermatol. 95:551–569.
2010.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Tenedini E, Artuso L, Bernardis I, Artusi
V, Percesepe A, De Rosa L, Contin R, Manfredini R, Pellacani G,
Giannetti A, et al: Amplicon-based next-generation sequencing: An
effective approach for the molecular diagnosis of epidermolysis
bullosa. Br J Dermatol. 173:731–738. 2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Ryynänen M, Knowlton RG, Parente MG, Chung
LC, Chu ML and Uitto J: Human type VII collagen: Genetic linkage of
the gene (COL7A1) on chromosome 3 to dominant dystrophic
epidermolysis bullosa. Am J Hum Genet. 49:797–803. 2015.PubMed/NCBI
|
|
28
|
Christiano AM, Greenspan DS, Hoffman GG,
Zhang X, Tamai Y, Lin AN, Dietz HC, Hovnanian A and Uitto J: A
missense mutation in type VII collagen in two affected siblings
with recessive dystrophic epidermolysis bullosa. Nat Genet.
4:62–66. 1993.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wertheim-Tysarowska K,
Sobczynska-Tomaszewska A, Kowalewski C, Skroński M, Swięćkowski G,
Kutkowska-Kaźmierczak A, Woźniak K and Bal J: The COL7A1 mutation
database. Hum Mutat. 33:327–331. 2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Tartaglia G, Cao Q, Padron ZM and South
AP: Impaired wound healing, fibrosis, and cancer: The paradigm of
recessive dystrophic epidermolysis bullosa. Int J Mol Sci.
22(5104)2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
McDermott LA, Weir GA, Themistocleous AC,
Segerdahl AR, Blesneac I, Baskozos G, Clark AJ, Millar V, Peck LJ,
Ebner D, et al: Defining the functional role of NaV1.7
in human nociception. Neuron. 101:905–919.e8. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Faber CG, Hoeijmakers JG, Ahn HS, Cheng X,
Han C, Choi JS, Estacion M, Lauria G, Vanhoutte EK, Gerrits MM, et
al: Gain of function Nanu1.7 mutations in idiopathic small fiber
neuropathy. Ann Neurol. 71:26–39. 2012.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Sparrow DB, Chapman G, Wouters MA,
Whittock NV, Ellard S, Fatkin D, Turnpenny PD, Kusumi K, Sillence D
and Dunwoodie SL: Mutation of the LUNATIC FRINGE gene in humans
causes spondylocostal dysostosis with a severe vertebral phenotype.
Am J Hum Genet. 78:28–37. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
34
|
Ganani D, Malovitski K, Sarig O, Gat A,
Sprecher E and Samuelov L: Epidermolysis bullosa simplex due to
bi-allelic DST mutations: Case series and review of the literature.
Pediatr Dermatol. 38:436–441. 2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Groves RW, Liu L, Dopping-Hepenstal PJ,
Markus HS, Lovell PA, Ozoemena L, Lai-Cheong JE, Gawler J, Owaribe
K, Hashimoto T, et al: A homozygous nonsense mutation within the
dystonin gene coding for the coiled-coil domain of the epithelial
isoform of BPAG1 underlies a new subtype of autosomal recessive
epidermolysis bullosa simplex. J Invest Dermatol. 130:1551–1557.
2010.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Liu L, Dopping-Hepenstal PJ, Lovell PA,
Michael M, Horn H, Fong K, Lai-Cheong JE, Mellerio JE, Parsons M
and McGrath JA: Autosomal recessive epidermolysis bullosa simplex
due to loss of BPAG1-e expression. J Invest Dermatol. 132:742–744.
2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Yang K, Xu YC, Hu HY, Li YZ, Li Q, Luan
YY, Liu Y, Sun YQ, Feng ZK, Yan YS and Yin CH: Investigation of a
Novel NTRK1 variation causing congenital insensitivity to pain with
anhidrosis. Front Genet. 12(763467)2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Hong SA, Kim SE, Lee AY, Hwang GH, Kim JH,
Iwata H, Kim SC, Bae S and Lee SE: Therapeutic base editing and
prime editing of COL7A1 mutations in recessive dystrophic
epidermolysis bullosa. Mol Ther. 30:2664–2679. 2022.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Thompson EL, Pickett-Leonard M, Riddle MJ,
Chen W, Albert FW and Tolar J: Genes and compounds that increase
type VII collagen expression as potential treatments for dystrophic
epidermolysis bullosa. Exp Dermatol. 31:1065–1075. 2022.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Harvey N, Youssefian L, Saeidian AH,
Vahidnezhad H and Uitto J: Pathomechanisms of epidermolysis
bullosa: Beyond structural proteins. Matrix Biol. 110:91–105.
2022.PubMed/NCBI View Article : Google Scholar
|