Introduction
Hereditary protein C deficiency (PCD) is engendered
by mutations in the PC gene (PROC) located on chromosome
2q14.3(1). PCD has been estimated
to affect 1 in 20,000 individuals who present with clinical
symptoms (2). The liver produces a
vitamin K-dependent serine protease, PC (1). Activated PC inhibits the activation
of coagulation factor X and II by suppressing coagulation factors
VIIIa and Va, respectively, thereby preventing coagulation. PCD is
a risk factor for thrombosis, with symptoms involving an
asymptomatic presentation, venous thromboembolism (VTE) and
life-threatening neonatal purpura fulminans (1). According to the abnormal quantity and
quality of PC, PCD is divided into two types. Type I is
characterized by the simultaneous decrease of PC antigen (PC:Ag)
and activity (PC:A). Type II is characterized by normal PC:Ag and
decreased PC:A. Type II deficiency is less common compared with
type I (3). In Asia, noncirrhotic
patients with portal vein thrombosis (PVT) secondary to PCD have
been rarely reported. The present study reported a patient with PVT
caused by type II PCD. Furthermore, the mutation of PROCc.152G>A
was observed in the patient of the present study, and according to
the current literature, there has been no previous report of this
mutation of this gene in China.
Case report
A 30-year-old male visited Guizhou Provincial
Orthopedics Hospital (Guiyang, China) in March 2019 due to
abdominal pain and recurring episodes of vomiting. The patient had
developed persistent distension pain in the left lower abdomen 20
days ago, with no radiating pain and certain relief in the knee
flexion position. Two days previously, the patient experienced
nonejective vomiting and vomiting immediately after eating;
however, no fever, diarrhea, bloody stool or other discomfort were
observed. When the patient was 21 years old, he had a filter for
deep vein thrombosis inserted in the right lower extremity.
Physical examination indicated deep tenderness in the left lower
abdomen with no rebound pain and muscle tension.
From the laboratory examination, the following
results were obtained: Increased leukocyte count
(15.14x109/l; normal range: 3.5-9.5x109/l)
with increased neutrophils (78.5%; normal range: 40-75%), increased
C-reactive protein (142.15 mg/l; normal range: 0-10 mg/l), positive
fecal occult blood test, increased fibrinogen degradation products
(55 µg/ml; normal range, 0-5 µg/ml) and D-dimer (11.01 µg/ml;
normal range, 0.00-1.00 µg/ml). Other laboratory tests indicated no
signs of renal, hepatic, pancreatic or cardiac disease. Since the
laboratory parameters indicated a hypercoagulable state and the
patient was a young male with no obvious predisposing factors for
recurrent vein thrombosis, exhaustive investigations were started.
The screening results related to hereditary anticoagulant protease
deficiency, as determined by chromogenic substrate, suggested
normal PC antigen values (115.2%; normal range, 70.0-140%) and
antithrombin III (101.5%; normal range, 75.0-125.0%), but
diminished PC activity (35%; normal range, 70.0-130.0%) and protein
S activity (69.5%; normal range, 75.0-130%). PC, PS and
antithrombin III were determined by chromogenic substrate.
Coagulation factor V and anticardiolipin antibodies were within the
normal range. The patient was considered to have type II hereditary
PCD based on decreased PC activity but normal PC antigen values. To
identify disease-causing gene variants, blood coagulation-related
genes (including 128 genes; Fig.
S1) were determined by next generation high-throughput
sequencing (MyGenostics Inc., Beijing, China). The results
indicated that a homozygous mutation in PC resulted in an amino
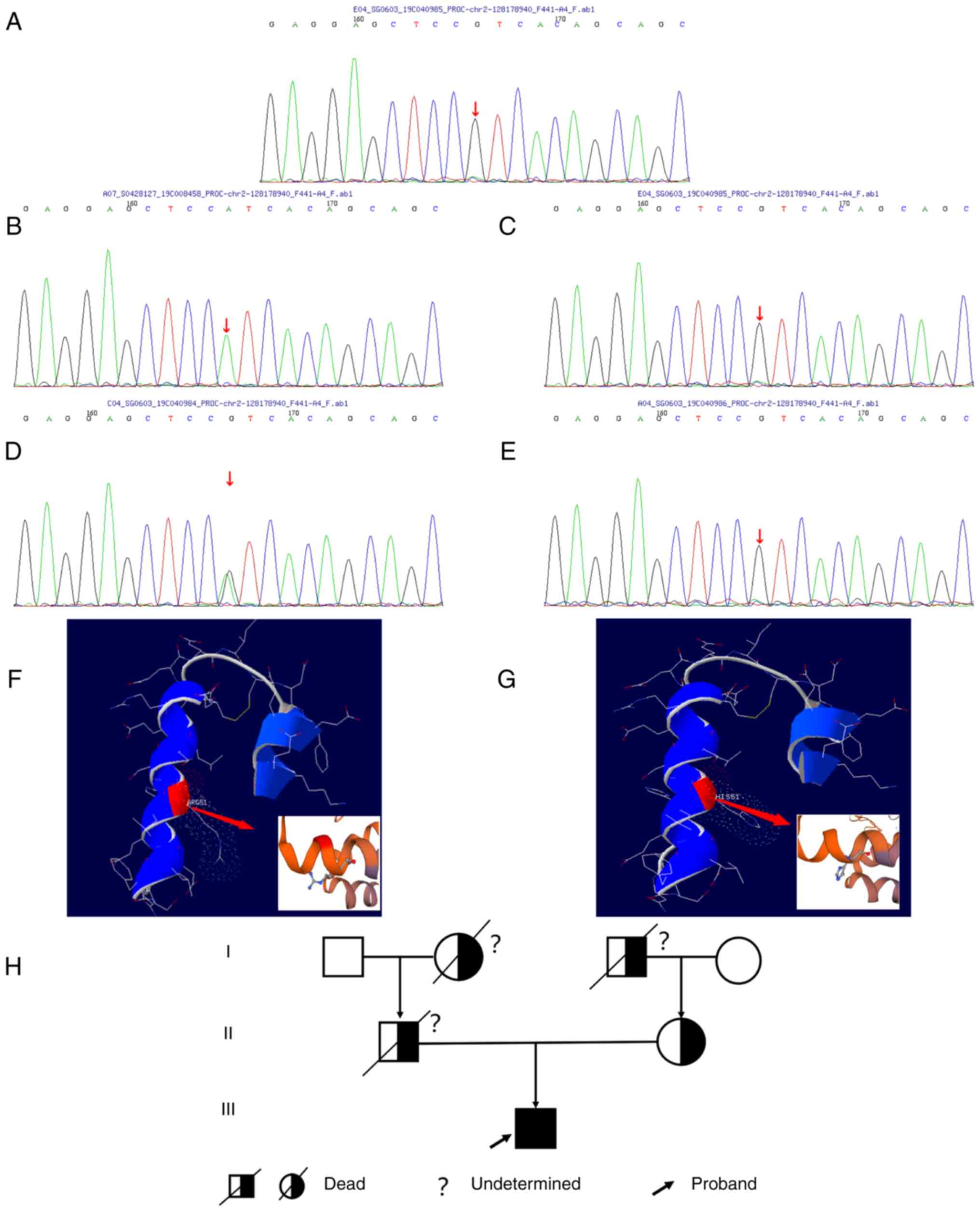
acid missense mutation (Fig. 1),
i.e., compared with the normal PC gene (Fig. 1A), the nucleotide site 152 of exon
3, which was located in the light chain of PC encoded by chromosome
2 and was mutated from guanine to adenine (Fig. 1B), resulting in the mutation of
amino acid 9 of PC light chain from arginine to histidine (Fig. 1F and G). Further analysis of the PC gene of the
mother, maternal grandmother and paternal grandfather revealed that
the mother's PC had a heterozygous mutation (Fig. 1D), whereas those of the maternal
grandmother and paternal grandfather did not (Fig. 1C-E). For various reasons, such as
the death of the patient's father in a car accident (the father had
no syndromes associated with thrombosis during his lifetime), only
the patient's mother, maternal grandmother and paternal grandfather
were genetically tested. However, the proband had a homozygous
mutation and the proband's mother had a heterozygous mutation. It
was thus inferred that the proband's father had a heterozygous
mutation as well. The patient's parents were carriers of pathogenic
genes and the genetic pattern was of autosomal recessive
inheritance (Fig. 1H).
In terms of imaging examinations, abdominal
computerized tomography (CT) revealed incomplete small intestinal
obstruction and proximal intestinal wall thickening, considering
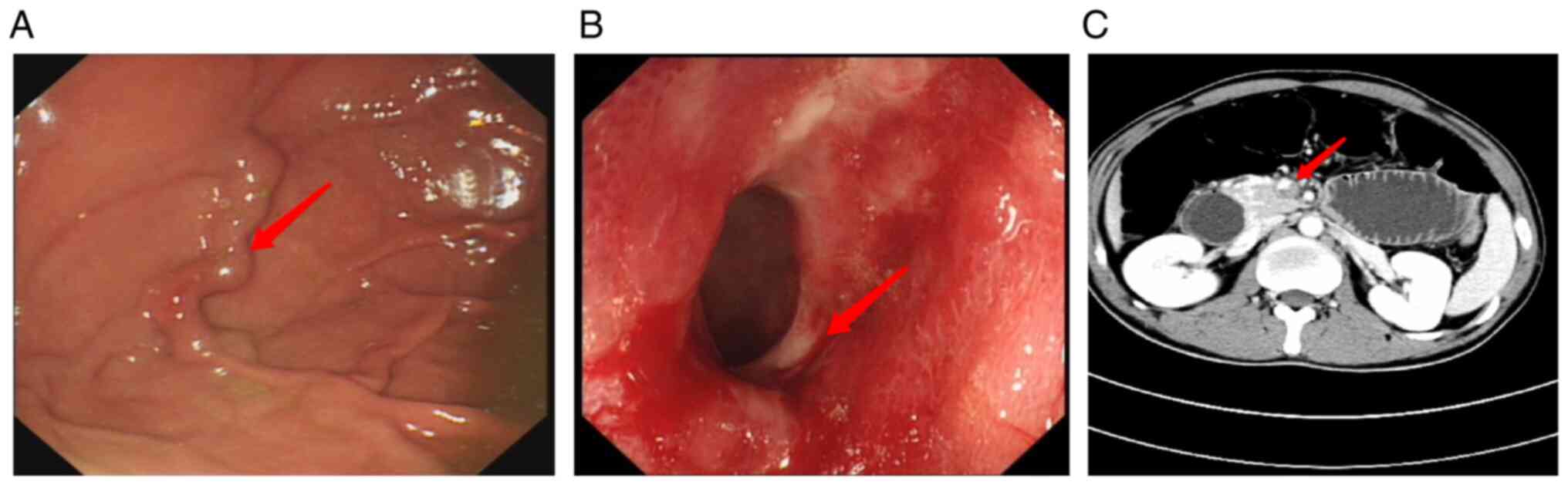
the possibility of ischemia. During gastroscopy, a varicose vein in
the fundus of the stomach was observed (Fig. 2A). Multiple ulcers and diverticula
in the sigmoid colon, as well as erosive small ulcers throughout
the colon, were discovered during a colonoscopy (Fig. 2B). A colonic pathological
examination revealed moderate chronic mucous membrane inflammation
with erosion and lamina propria hemorrhage. Since the patient's
laboratory examination indicated a hypercoagulable state and
colonoscopy suggested multiple erosion and ulcer formation of the
whole colon, abdominal angiography was performed to identify
ischemic bowel disease. Abdominal angiography revealed thrombosis
of the portal vein and its branches (Fig. 2C); thus, it was speculated that
ischemic bowel disease was the cause of the patient's abdominal
pain.
The patient was diagnosed with type II hereditary
PCD with PVT and ischemic bowel disease. Anticoagulant therapy was
provided after definite diagnosis (low-molecular-weight heparin,
5,000 U every 12 h for 10 days, which was then changed to oral
warfarin 3 mg/day). After 15 days, the patient's blood coagulation
function was restored to normal and the symptoms were substantially
relieved. The patient was discharged from the hospital with
instructions to take warfarin 3 mg/day and to keep the
international normalized ratio (INR) at 2-3. However, the patient
did not take his medication as prescribed on a regular basis and
did not monitor his blood coagulation function. Over 20 days after
discharge, the patient went to a local hospital for oliguria and
was diagnosed with ‘acute renal failure’ (the specific diagnosis
and treatment were unknown), and was discharged after treatment.
Two months later, the patient had sudden convulsions and loss of
consciousness at home and died after ~1 h.
Discussion
Hereditary PCD comprises mostly autosomal dominant
and heterozygous mutations, whereas certain cases feature autosomal
recessive, homozygous or compound heterozygous mutation (4). These patients usually have severe
PCD, as well as early-onset and severe clinical symptoms, such as
skin purpura fulminans, pulmonary embolism and disseminated
intravascular coagulation (4).
Severe PCD (homozygous or compound heterozygous form) is rare with
an incidence of 1 in 500,000-1 in 750,000 births (5). PROC mutations include missense
mutations, frameshift mutations, nonsense mutations and splice site
abnormalities, with missense mutations being the most common. To
date, 391 different PROC gene mutations have been reported
worldwide, most of which are from Western populations and a smaller
number from Asian populations (6-9).
The most common gene mutations in the Chinese population are
PROCc.565C>T and PROCc.10230C>T (10,11).
PROCc.152G>A was observed in the patient of the present study.
As per the current literature, there has been no report regarding
the mutation of this gene in China. However, the mutation is
associated with PCD, as reported by a study from Italy (11).
PVT unrelated to solid malignancy is common in
patients with cirrhosis; however, it is less frequently observed in
patients without cirrhosis. Failure to detect and treat thromboses
may lead to mesenteric ischemia, chronic cavernous transformation
and portal hypertension complications (12). Previously reported cases of PVT
caused by PCD in the literature are summarized and compared in
Table I. PVT induced by PCD
exhibits a chronic onset and cavernous transformation of the portal
vein; i.e., development or dilatation of small vessels around the
main trunk of the portal vein (13,14).
Anticoagulant therapy is currently considered the gold standard to
achieve portal vein recanalization. The European Association for
the Study of the Liver published a guideline in 2015 and
recommended initial treatment with low-molecular-weight heparin
targeting a level of 0.5-0.8 IU/ml, which is based on a grade A1
level of evidence (15). Oral
vitamin K antagonists are used for long-term anticoagulant
treatment targeting an INR of 2-3, based on a grade B1 level of
evidence (15). Anticoagulant
therapy achieved remarkable results in the reported cases (Table I) as well as the present case.
 | Table ICases of PVT caused by hereditary PC
deficiency reported in the literature. |
Table I
Cases of PVT caused by hereditary PC
deficiency reported in the literature.
| Author, year | N | Age/sex | PVT or MVT | Symptoms | PC values/PC
activity, % | Treatments | Survival status at
last follow-up | (Refs.) |
|---|
| Momoi, 2003 | 1 | 39/M | MVT | Abdominal
pain/melena | 58/44 |
Anticoagulant/surgical | Alive (12
months) | (13) |
| Mitani, 1999 | 1 | 83/M | MVT | Abdominal
pain/vomiting | 45/45 |
Anticoagulant/surgical | Alive (16
months) | (22) |
| Yates, 1991 | 2 | 27/F | MVT | Suprapubic
pain/urinary symptoms | 46/- |
Anticoagulant/surgical | Alive (-) | (23) |
| | | 24/M | MVT | Abdominal
pain/vomiting | 36/- |
Anticoagulant/surgical | Alive (-) | |
| Hsu, 2015 | 1 | 47/M | MVT | Melaena | 33.6/- | Anticoagulant | Alive (17
months) | (24) |
| Matsushita, 2000 | 1 | 64/M | MVT | Leg
edema/hypoproteinemia | 40/37 |
Anticoagulant/activated PC
concentrate | Alive (18
months) | (25) |
| Valla, 1988 | 1 | 45/M | PVT | Abdominal
pain/melaena | 37/45 | - | - | (26) |
| Rodríguez-Leal,
2014 | 1 | 63/M | PVT | Abdominal
pain/nausea/diarrhea | 39/54 | Anticoagulant | Alive (36
months) | (14) |
| Yang, 1999 | 2 | 25/M | PVT | Abdominal
pain/fever | 55/- | Anticoagulant | Alive (60
months) | (27) |
| | | 31/M | PVT/MVT | Oesophageal variceal
bleeding | -/- |
Anticoagulant/surgical | Alive (36
months) | |
| Orozco, 1988 | 2 | 27/M | PVT/MVT | Upper
gastrointestinal hemorrhage | 10% of normal/- |
Anticoagulant/surgical | Alive (24
months) | (28) |
| | | 55/M | PVT/MVT | Upper
gastrointestinal hemorrhage | 10% of normal/- |
Anticoagulant/surgical | Alive (2 months) | |
| Choi, 2011 | 1 | 66/M | PVT | Fever/stomach
ache/nausea | 41/- | Anticoagulant | Alive (6
months) | (29) |
Review of the medical history of the case of the
present study indicated that the patient experienced multiple
venous thromboses, including deep venous thrombosis of the right
lower limb at the age of 21, and during hospitalization, thrombosis
of the portal vein and its branches was revealed by abdominal
arteriovenous CT imaging. The diagnosis of acute renal failure from
the local hospital after discharge may be attributed to renal vein
thrombosis. Hereditary PCD may increase the risk of thrombosis
(1). In addition to deep venous
thrombosis, cerebral infarction has been reported (16-18).
In the present case, sudden convulsions and loss of consciousness
at the time of death may have resulted from cerebral infarction
caused by PCD. A previous study demonstrated that in heterozygous
individuals, thrombotic episodes occur at the age of 30-40 years
and is rare prior to the age of 20 years (19). In the patient of the present study,
the age at onset of the first thrombotic episode was 21 years,
which is the average age of the first onset of VTE in homozygous
individuals and was earlier than that in heterozygous individuals
(4).
Replacement of PC and the use of oral anticoagulant
therapy to treat and prevent thrombosis are the currently applied
treatments for severe PCD (1).
Long-term anticoagulant therapy is critical for patients with PCD
(20). The anticoagulant treatment
of the patient of the present study achieved remarkable results
(blood coagulation function returned to normal and abdominal pain
was significantly reduced). Of note, warfarin-induced skin necrosis
is a serious potential complication of PCD that occurs in adult
patients (21), generally on the
third or fourth day after starting warfarin. Pain, bruising and
redness in the affected area are the first symptoms, and lesions
may progress to a well-demarcated, inflamed lesion, and eventually
to skin necrosis (21). PC
replacement therapy has been indicated to be an effective treatment
for patients with warfarin-induced skin necrosis, in association
with low-molecular-weight heparin (5).
In conclusion, PCD is a predisposing cause of PVT,
particularly in patients with noncirrhotic, thrombosis-related
portal hypertension. Clinicians should focus on the differential
diagnosis of unknown vascular diseases, particularly in younger
patients. When routine tests fail to determine the cause of
thromboembolic disease, additional examinations of hereditary
anticoagulant protease deficiency and genetic testing are
necessary. According to our successful experience with the present
case, long-term anticoagulant therapy is a crucial treatment step
for patients with PCD. However, care should be taken to avoid
warfarin-induced skin necrosis during treatment.
Supplementary Material
List of the 128 blood
coagulation-related genes tested in the proband.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
CZ, TL and JZ designed the experiments. LLo and LLi
collected and evaluated the clinical data. CZ drafted the
manuscript. CZ, TL and JZ confirm the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
The patient provided written informed consent for
the treatment, interventions, images, data collection and
submission of this article for publication prior to his death.
Furthermore, the patient's relatives (mother, grandmother,
grandfather) provided written informed consent for the publication
of this case report and all accompanying results of genetic
testing.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dinarvand P and Moser KA: Protein C
deficiency. Arch Pathol Lab Med. 143:1281–1285. 2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Dahlbäck B: The protein C anticoagulant
system: Inherited defects as basis for venous thrombosis. Thromb
Res. 77:1–43. 1995.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Kottke-Marchant K and Comp P: Laboratory
issues in diagnosing abnormalities of protein C, thrombomodulin,
and endothelial cell protein C receptor. Arch Pathol Lab Med.
126:1337–1348. 2002.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Pescatore SL: Clinical management of
protein C deficiency. Expert Opin Pharmacother. 2:431–439.
2001.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Goldenberg NA and Manco-Johnson MJ:
Protein C deficiency. Haemophilia. 14:1214–1221. 2008.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Gandrille S, Greengard JS, Alhenc-Gelas M,
Juhan-Vague I, Abgrall JF, Jude B, Griffin JH and Aiach M:
Incidence of activated protein C resistance caused by the ARG 506
GLN mutation in factor V in 113 unrelated symptomatic protein
C-deficient patients. The French network on the behalf of INSERM.
Blood. 86:219–224. 1995.PubMed/NCBI
|
|
7
|
Miyata T, Sakata T, Yasumuro Y, Okamura T,
Katsumi A, Saito H, Abe T, Shirahata A, Sakai M and Kato H: Genetic
analysis of protein C deficiency in nineteen Japanese families:
Five recurrent defects can explain half of the deficiencies. Thromb
Res. 92:181–187. 1998.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Reitsma PH, Poort SR, Allaart CF, Briët E
and Bertina RM: The spectrum of genetic defects in a panel of 40
Dutch families with symptomatic protein C deficiency type I:
Heterogeneity and founder effects. Blood. 78:890–894.
1991.PubMed/NCBI
|
|
9
|
Shen MC, Lin JS and Tsay W: High
prevalence of antithrombin III, protein C and protein S deficiency,
but no factor V Leiden mutation in venous thrombophilic Chinese
patients in Taiwan. Thromb Res. 87:377–385. 1997.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ding Q, Shen W, Ye X, Wu Y, Wang X and
Wang H: Clinical and genetic features of protein C deficiency in 23
unrelated Chinese patients. Blood Cells Mol Dis. 50:53–58.
2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Faioni EM, Hermida J, Rovida E, Razzari C,
Asti D, Zeinali S and Mannucci PM: Type II protein C deficiency:
Identification and molecular modelling of two natural mutants with
low anticoagulant and normal amidolytic activity. Br J Haematol.
108:265–271. 2000.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Intagliata NM, Caldwell SH and Tripodi A:
Diagnosis, development, and treatment of portal vein thrombosis in
patients with and without cirrhosis. Gastroenterology.
156:1582–1599.e1. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Momoi A, Komura Y, Kumon I, Tamai M,
Tarumi Y, Matsubara J, Miyauchi K, Yamanouchi J and Hato T:
Mesenteric venous thrombosis in hereditary protein C deficiency
with the mutation at Arg169 (CGG-TGG). Intern Med. 42:110–116.
2003.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Rodríguez-Leal GA, Morán S, Corona-Cedillo
R and Brom-Valladares R: Portal vein thrombosis with protein C-S
deficiency in a non-cirrhotic patient. World J Hepatol. 6:532–537.
2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
European Association for the Study of the
Liver. Electronic address: simpleeasloffice@easloffice.eu.
EASL clinical practice guidelines: Vascular diseases of the liver.
J Hepatol. 64:179–202. 2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li P and Qin C: Recurrent cerebellar
infarction associated with hereditary heterozygous protein C
deficiency in a 35-year-old woman: A case report and genetic study
on the pedigree. Exp Ther Med. 16:2677–2681. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Majid Z, Tahir F, Ahmed J, Bin Arif T and
Haq A: Protein C deficiency as a risk factor for stroke in young
adults: A review. Cureus. 12(e7472)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Zhu H, Liu H and Liu J: Pathogenic
variants of PROC gene caused type II activity deficiency in a
Chinese family: A case report. Medicine (Baltimore).
100(e25160)2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Lensen RP, Rosendaal FR, Koster T, Allaart
CF, de Ronde H, Vandenbroucke JP, Reitsma PH and Bertina RM:
Apparent different thrombotic tendency in patients with factor V
Leiden and protein C deficiency due to selection of patients.
Blood. 88:4205–4208. 1996.PubMed/NCBI
|
|
20
|
De Stefano V, Mastrangelo S, Schwarz HP,
Pola P, Flore R, Bizzi B and Leone G: Replacement therapy with a
purified protein C concentrate during initiation of oral
anticoagulation in severe protein C congenital deficiency. Thromb
Haemost. 70:247–249. 1993.PubMed/NCBI
|
|
21
|
McGehee WG, Klotz TA, Epstein DJ and
Rapaport SI: Coumarin necrosis associated with hereditary protein C
deficiency. Ann Intern Med. 101:59–60. 1984.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Mitani M, Kuwabara Y, Kawamura H, Sato A,
Hattori K and Fujii Y: Mesenteric venous thrombosis associated with
protein C deficiency. J Gastroenterol. 34:387–389. 1999.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Yates P, Cumber PM, Sanderson S and
Harrison BJ: Mesenteric venous thrombosis due to protein C
deficiency. Clin Lab Haematol. 13:137–139. 1991.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Hsu WF, Tsang YM, Teng CJ and Chung CS:
Protein C deficiency related obscure gastrointestinal bleeding
treated by enteroscopy and anticoagulant therapy. World J
Gastroenterol. 21:1024–1027. 2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Matsushita I, Hanai H, Sato Y, Arai H,
Iida T, Hosoda Y, Kaneko E, Yasumi K and Sugimura H: Protein-losing
enteropathy caused by mesenteric venous thrombosis with protein C
deficiency. J Clin Gastroenterol. 30:94–97. 2000.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Valla D, Denninger MH, Delvigne JM, Rueff
B and Benhamou JP: Portal vein thrombosis with ruptured oesophageal
varices as presenting manifestation of hereditary protein C
deficiency. Gut. 29:856–859. 1988.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Yang YY, Chan CC, Wang SS, Chiu CF, Hsu
HC, Chiang JH, Tasy SH, Chang FY and Lee SD: Case report: Portal
vein thrombosis associated with hereditary protein C deficiency: A
report of two cases. J Gastroenterol Hepatol. 14:1119–1123.
1999.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Orozco H, Guraieb E, Takahashi T,
Garcia-Tsao G, Hurtado R, Anaya R, Ruiz-Arguelles G,
Hernandez-Ortiz J, Casillas MA and Guevara L: Deficiency of protein
C in patients with portal vein thrombosis. Hepatology. 8:1110–1111.
1998.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Choi BK, Yang SH, Suh KH, Hwang JA, Lee
MH, Si WK and Kim JH: A case of portal vein thrombosis by protein C
and s deficiency completely recanalized by anticoagulation therapy.
Chonnam Med J. 47:185–188. 2011.PubMed/NCBI View Article : Google Scholar
|