Introduction
Vitamin D exists as two main forms in the body:
Vitamin D2 (ergocalciferol) and vitamin D3 (cholecalciferol)
(1). Vitamin D2 is derived from
the diet, such as ryegrass, beef and lamb. Whilst vitamin D3 is
formed from the exposure of 7-dehydrocholesterol in the skin to
ultraviolet B (280-310 nm) light. Vitamin D is metabolized and
converted into its active form 1,25-dihydroxy-vitamin D3
[1,25(OH)2D] by several enzymes, mainly microsomal
CYP2R1 in the liver and 25-OH vitamin D-1α-hydroxylase in kidneys,
which promotes calcium absorption from the gut into the bloodstream
and regulates serum calcium concentration maintained at 2.25 to
2.75 mmol/l. Deficiencies in vitamin D metabolism or action can
cause hypocalcemia and high serum parathyroid hormone (PTH) levels
(2). These can adversely affect
the growth and development of the human musculoskeletal and nervous
systems, eventually resulting in vitamin D-dependent rickets (VDDR)
(3).
Cytochrome P450 family 27 subfamily B member 1
(CYP27B1) mutations may cause deficiencies in 1α-hydroxylase
activity, resulting in a vitamin D deficiency and causes a rare
autosomal recessive disorder known as VDDR type 1A (VDDR-1A)
(4). The clinical features of
VDDR-1A during childhood include muscle weakness, growth disorders,
joint pain, genu valgum, seizures (5), symptoms of rickets and increased
susceptibility to bone fractures (6). In adulthood, the main characteristic
of VDDR-1A is osteomalacia (7).
The laboratory findings of this condition typically reveal
hypocalcemia, high serum PTH concentrations, elevated serum
alkaline phosphatase levels, low serum 1,25(OH)2D levels
and normal or elevated serum 25-hydroxyvitamin D (25OH-D)
concentrations (8). The present
report describes the case of a patient with VDDR-1A caused by a
novel mutation in the CYP27B1 gene, which was diagnosed in
adulthood.
Case report
A male 39-year-old patient presented to the
Department of Endocrinology, Affiliated Hangzhou First People's
Hospital, Zhejiang University School of Medicine (Hangzhou, China)
and asked to be hospitalized due to his short stature in September,
2021. He had a 37-year history of rickets induced by calcium
deficiency, taking calcium carbonate orally at 600 mg each time for
~5 times per week, and vitamin D3 orally at 400 IU each time for 4
times per week. A physical examination revealed bilateral
deformities of the lower extremities, with an X-shaped lower left
limb and O-shaped lower right limb (height, 148.0 cm). His thorax
exhibited pectus carinatum and a rib flare. His forehead was
slightly raised. Laboratory tests indicated elevated serum PTH
[electrochemiluminescence immunoassay (ECLIA) using COBAS e 601;
cat. no. 11972103122; Roche Diagnostics GmbH] concentrations (115
pg/ml; normal range, 15-65 pg/ml) coupled with low serum 25OH-D
(ECLIA using COBAS e 601; cat. no. 05894913190; Roche Diagnostics
GmbH) (18.31 µg/l; normal range, 20-70 µg/l), serum calcium
(ASAIII, 11111371701, DiaSys Diagnostic System Co. Ltd., Beckman
coulter AU680) (1.67 mmol/l; normal range, 2.0-2.6 mmol/l) and 24-h
urine calcium (ASAIII, 11111371701, DiaSys Diagnostic Systems GmbH;
Beckman Coulter, Inc. AU680) (0.56 mmol/day; normal range, 1.0-7.5
mmol/day) concentrations. Additional radiographic examinations
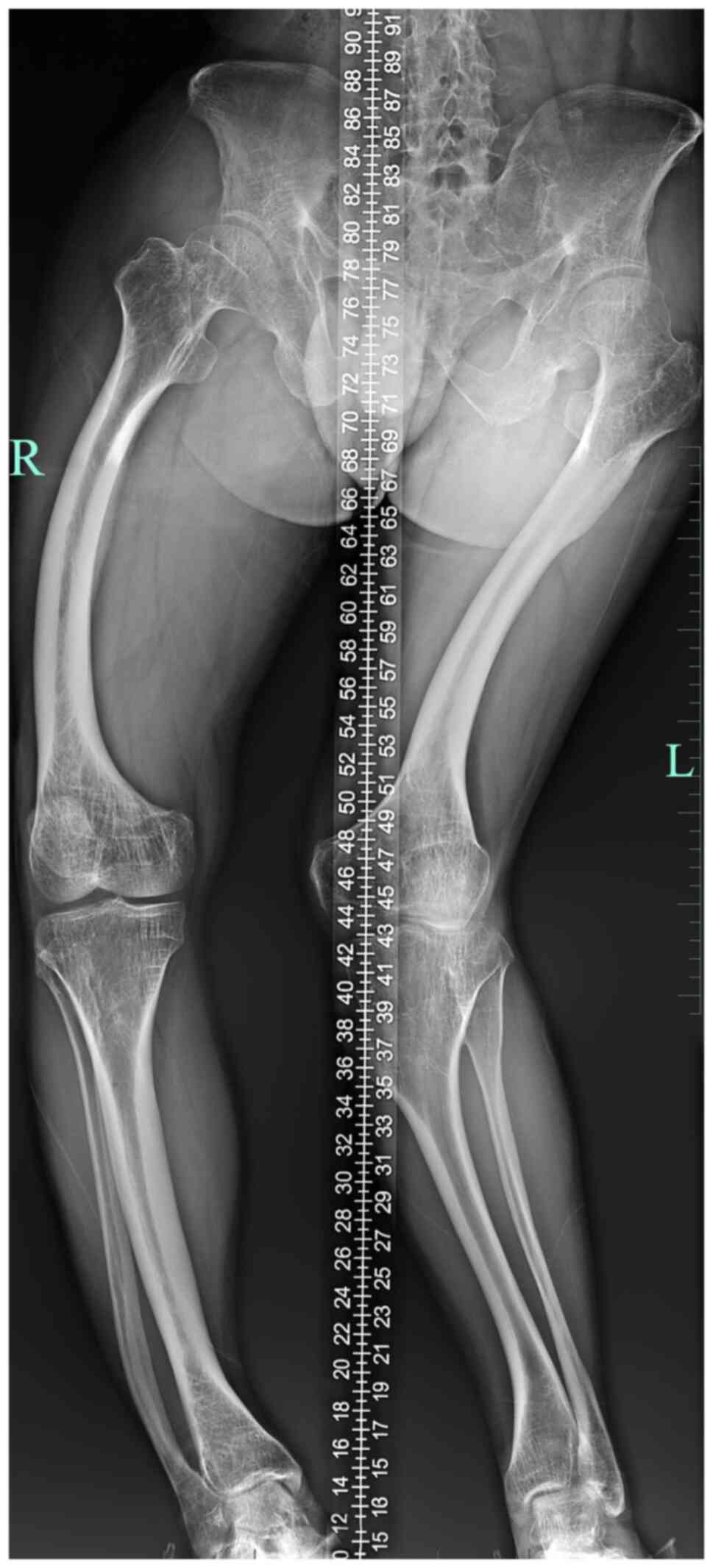
revealed that both of his lower limbs were bent to the right
(Fig. 1). The total lengths of his
right and left lower limbs were 690 and 663 cm, respectively.
Lateral joint space narrowing was observed in the left knee joint.
Osteopenia was evident. A plain CT scan (Optima CT-540; GE
Healthcare) revealed osteopenia in all components of the pelvic
bone and scoliosis. A lumbar spine X-ray revealed that the spinal
alignment was curved to the right. The T-scores for bone mineral
density (BMD) (Lunar Prodigy; GE Healthcare) were -2.0 at the left
femoral neck and -2.2 at the left hip, which indicates
osteopenia.
Whole-exome Sanger sequencing was performed in the
peripheral venous blood of this patient and his parents owing to a
suspected genetic contribution. Peripheral venous blood from this
patient and his parents was taken. DNA was isolated from peripheral
blood with CWE9600 Automated Nucleic Acid Extraction System using
CWE2100 Blood DNA Kit V2 (cat. no. CW2553; CoWin Biosciences). The
primer 1-F (5'-TGTGGCCAGTAGGGGACTT-3'), 1-R
(5'-CCAGACGCTGGTCACTCTG-3'), and the primer 2-F
(5'-ATCTGCAGGATCTCCACACC-3'), 2-R (5'-CATGACCCAGACCCTCAAGT-3') were
designed for CYP27B1 gene using Primer Premier 5.0 (Premier
Biosoft International) before PCR (Phanta Max Super-Fidelity DNA
Polymerase; Vazyme Biotech Co., Ltd) was performed to amplify the
fragments covering the mutated sites in a LifeECO Thermal Cycler
TC-96/G/H(b)C (Hangzhou Bioer Co., Ltd.). PCR reaction conditions
for these primers were as follows: 95˚C for 5 min (once only),
followed by 25 cycles consisting of 95˚C for 30 sec, 60˚C for 30
sec and 72˚C for 40 sec, followed by 20 cycles at 95˚C for 30 sec,
50˚C for 30 sec and 72˚C for 40 sec, then a final 10 min extension
step was performed at 72˚C, and maintained at 4˚C. The PCR products
were further purified with 2% agarose gel electrophoresis, stained
with Gelstain Red (cat. no. S2009L; Shanghai Bioscience Technology
Co., Ltd.), and then sequenced by a ABI 3730XL DNA Sequencer
(Applied Biosystems; Thermo Fisher Scientific, Inc.). Sanger
sequencing results were analyzed by Chromas Lite v2.01
(Technelysium Pty Ltd.).
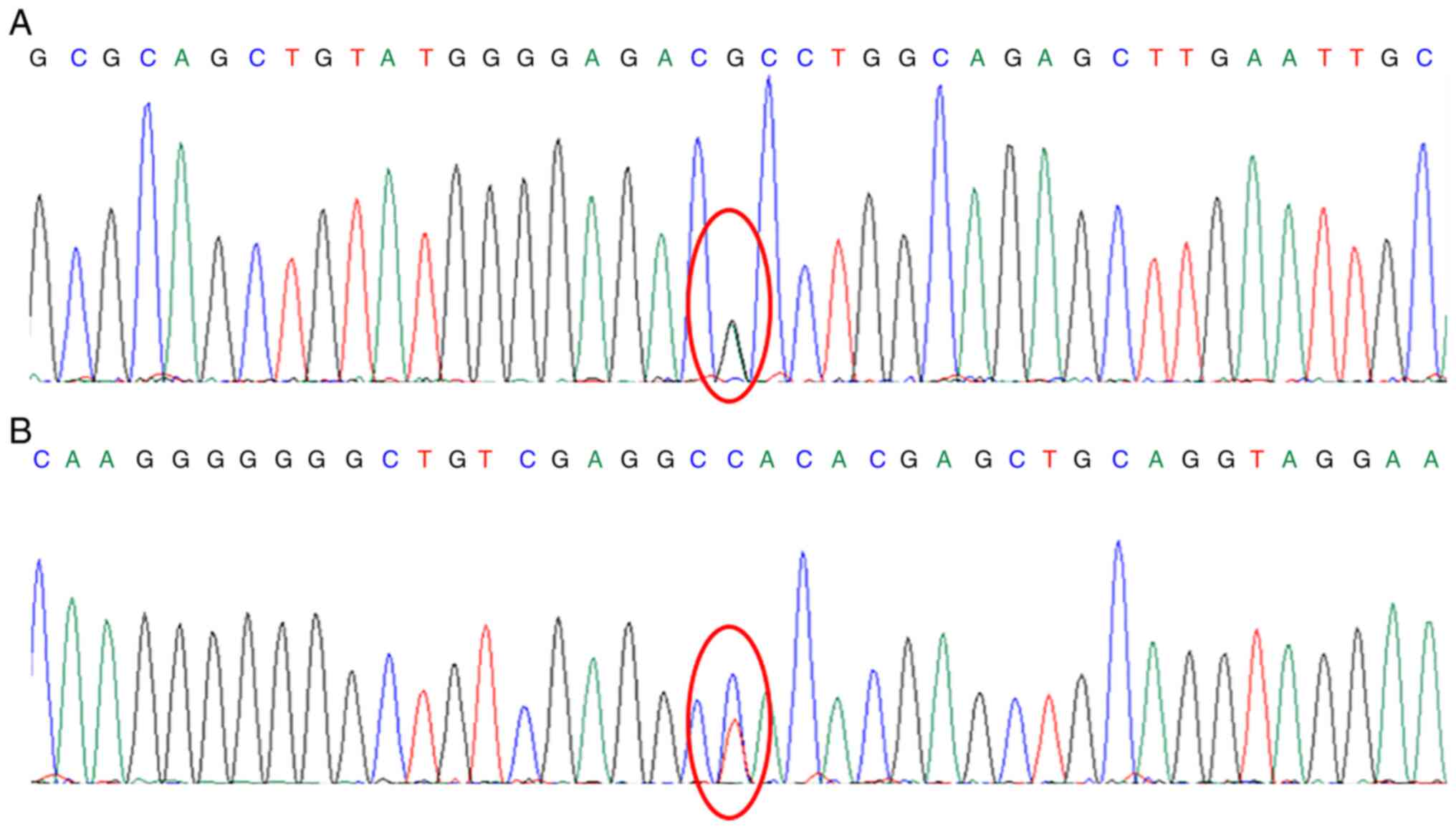
A genetic analysis of this patient revealed two
heterozygous mutations in CYP27B1, namely exon 8
c.1376G>A (p.R459H) and exon 1 c.182T>C (p.L61P) (Fig. 2). The c.1376G>A (p.R459H)
mutation indicates that nucleotide 1,376 in the coding region was
mutated from guanine to adenine, resulting in amino acid 459 being
changed from arginine to histidine. The c.182T>C (p.L61P)
mutation indicates that nucleotide 182 in the coding region was
mutated from thymine to cytosine, resulting in amino acid 61 being
changed from leucine to cytosine. To the best of our knowledge, to
date there are no previous studies available on exon 1 c.182T>C
causing VDDR-1A. The heterozygous mutation in exon 8 [c.1376G>A
(p.R459H)] was inherited from the patient's father (Fig. S1), who has no mutation in exon 1
at nucleotide 182. By contrast, the mutation in exon 1 [c.182T>C
(p.L61P)] was inherited from the mother, who has no mutation in
exon 8 at nucleotide 1376 (Fig.
S2). The parents only carry the mutations and do not have
VDDR-1A. The patient carried both mutations. Based on the genetic
sequencing results, the patient was diagnosed with VDDR-1A. The
standard clinical treatment, 0.5 µg 1,25(OH)2D twice
daily and 0.6 g calcium carbonate once daily (6), were administered to improve the
patient's serum marker levels and BMD. Following 1 month of
treatment, follow-up examinations revealed a PTH concentration of
38.4 pg/ml and a serum calcium level of 2.36 mmol/l. After 1 year
of treatment, the serum PTH and serum calcium concentration
remained at normal levels, and BMI were -1.6 at the left femoral
neck and -1.8 at the left hip, which suggested that it was improved
compared with the previous year. However, since the diagnosis was
established in adulthood, the patient had already suffered
irreversible deformities.
Discussion
The patient described in the present case report had
exhibited symptoms of rickets since his childhood and had visited
the orthopedic department several times. However, the treatment and
diagnosis have not changed over time. The patient was diagnosed
with rickets induced by calcium deficiency. Following irregular
treatments with oral calcium carbonate and vitamin D3, the patient
developed severe deformities in his lumbar spine and lower limb, in
addition to osteoporosis inconsistent with his age. VDDR-1A was
confirmed through physical examination, laboratory tests, imaging
and genetic testing. The patient was then supplemented with calcium
and 1,25(OH)2D based on the cause.
Sanger sequencing in the present case revealed a
mutation in the CYP27B1 gene. CYP27B1 is located on
chromosome 12q13.3, spans 4,859 bases and is 5 kb in length
(9). It encodes 1α-hydroxylase
(10), which regulates calcium
metabolism by synthesizing 1,25(OH)2D in the kidneys.
Homozygous or compound heterozygous alterations of this gene have
been previously associated with VDDR-1A (11). The mutations in the present patient
are located in the CYP27B1 gene and the clinical
manifestations are consistent with the symptoms of VDDR-1A.
Therefore, it can be speculated that these mutations are
meaningful. Due to the rarity of this disease and the fact that the
mutation sites in each patient differs, it is highly difficult to
study this relationship. The patient in the present report carried
two heterozygous mutations in this gene, namely exon 8
[c.1376G>A(p.R459H)] and exon 1 [c.182T>C (p.L61P)]. The
mutation site inherited from the patient's father was consistent
with the amino acid position of the mutation site reported in the
literature (12). This pathogenic
variant previously reported is c.1375C>T (p.R459C), which
results in the substitution of cysteine into arginine (12). However, this nucleic acid variant
site was not consistent with that of the patient in the present
report. Another previous study reported mutations at position R459
in a Chinese patient, suggesting that this genetic region may be a
mutation hotspot within the Chinese population (13). By contrast, the exon 1 [c.182T>C
(p.L61P)] mutation originating from the patient's mother has not
been previously recorded in the Human Gene Mutation Database (HGMD;
https://www.hgmd.cf.ac.uk/ac/index.php). Therefore,
this suggests that novel mutations were discovered in the present
report, which expanded the knowledge on existing mutation
sites.
In total, ~100 CYP27B1 gene mutations have
been reported in the ClinVar (www.ncbi.nlm.nih.gov/clinvar/), the most common of
which are missense mutations. However, they also occasionally
include nonsense, deletion and splicing mutations. Different
mutations result in varying levels of disease severity. The
patient's genetic sequencing results were consistent missense
mutations. Partial mutations in the CYP27B1 gene, such as
Q65H, R107H and P112L, can cause loss of enzyme activity, resulting
in severe phenotypes (14). Other
mutations, such as G57V, G73W and R459C, are associated with
moderate to severe phenotypes (14). VDDR-1A was first reported
internationally in 1961(15), in
which children carrying mutations exhibited symptoms from 6 months
to 2 years of age. This disease is rarely reported in the Chinese
population. A previous study in 2020(16) cited 11 reports of cases involving
Chinese patients, in which all had heterozygous compound mutations.
This is consistent with the patient in the present study. This
suggests that Chinese patients with VDDR-1A are predominantly
heterozygous for this mutation. A previous report in 2020(14) described two brothers from India
with VDDR-1A diagnosed at 30 and 15 months, respectively. They
presented with complaints of slow growth, inability to stand,
hypotonia and rickets, where genetic testing revealed a homozygous
c.1294C>T (p.R432C) mutation. This mutation is associated with
severe enzyme inactivation, which in turn results in a severe
phenotype. A previous study (11)
reported that 38 of the 63 CYP27B1 mutations in the database were
missense. This same study also reported that a 13-month-old Saudi
Arabian girl homozygous for the nonsense c.1510C>T (p.Q504X)
mutation developed a premature stop codon, leading to a severe
phenotype. Kaygusuz et al (4) analyzed 183 patients with VDDR-1,
reporting a median age at diagnosis of 2.55 (min, 1.0; max, 12)
years. They also found that the c.195+2T > G and p.K192E
mutations resulted in the most and least severe phenotypes,
respectively. Li et al (16) analyzed two Chinese children with
VDDR-1A with the same homozygous mutations in exon 8
(c.1319_1325dupCCCACCC, p.Phe443Profs * 24) who were diagnosed at
33 and 22 months of age, where both patients presented with severe
growth retardation. In particular, one patient experienced a
fracture, which is a severe clinical phenotype. Therefore, there is
highly likely to be a clear genotype-phenotype association in
patients with VDDR-IA. However, another study on nine patients
observed differences in the severity of the clinical presentations
among patients with the same mutation (17). Therefore, larger sample sizes are
required to verify this genotype-phenotype association.
Unlike the patient reported in the present report,
the cases in the aforementioned literature revealed that the
majority of patients were diagnosed during childhood. Results from
previous studies showed that early diagnosis and appropriate
treatment will allow significant improvements in the skeletal
symptoms and serum indicators (4,18).
Owing to a lack of awareness of rare diseases among physicians in
China and a lack of widespread access to genetic testing, the
patient described in the present case report was diagnosed with
VDDR-1A late in adulthood. As a result, the patient experienced
severe physical ailments that negatively affected his quality of
life and caused him significant psychological damage. This case
report may improve awareness of this disease among clinicians and
increase their vigilance. Although the present report is limited to
only one case and therefore the inferred results are not universal,
it remains to be recommended that the early genetic screening of
children with calcium deficiencies and the early diagnosis of rare
diseases are performed to reduce the burden of these disorders on
these individuals, families and society.
Supplementary Material
Genetic testing result of the
patient's father. He has the mutation in exon8 (c.1376G>A) but
does not have the mutation in exon1 (c.182T>C).
Genetic testing result of the
patient's mother. She has the mutation in exon1 (c.182T>C) but
does not have the mutation inexon8 (c.1376G>A).
Acknowledgements
Not applicable.
Funding
Funding: The present report was supported by the Medical Health
Science and Technology Project of Zhejiang Provincial Health
Commission (grant nos. 2020KY686 and 2019KY487).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CY, JHe, JX, XiaoFZ, XianFZ and JHu contributed to
the acquisition, analysis and interpretation of the data. All
authors read and approved the final manuscript. CY, JHe, JX,
XiaoFZ, XianFZ and JHu confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
The present study was conducted in accordance with
the World Medical Association Declaration of Helsinki and was
approved by the Institutional Ethics Board of the ‘Hangzhou First
People's Hospital, Zhejiang University School of Medicine’. The
patient and the parents of the patient provided written informed
consent for participation in the present report.
Patient consent for publication
The patient and the parents of the patient provided
written consent for publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pang X, Yang Z, Wang J, Duan Y, Zhao L, Yu
D and Lai J: Relationship between serum 25OH-vitamin D2 level and
vitamin D status of children aged 3-5 years in China. Nutrients.
13(4135)2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Goyal A, Anastasopoulou C, Ngu M, et al:
Hypocalcemia. In: StatPearls [Internet]. StatPearls Publishing,
Treasure Island, FL, 2022.
|
|
3
|
Giannakopoulos A, Efthymiadou A and
Chrysis D: A case of vitamin-D-dependent rickets type 1A with
normal 1,25-dihydroxyvitamin D caused by two novel mutations of the
CYP27B1 gene. Horm Res Paediatr. 87:58–63. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kaygusuz SB, Alavanda C, Kirkgoz T, Eltan
M, Abali ZY, Helvacioglu D, Guran T, Ata P, Bereket A and Turan S:
Does genotype-phenotype correlation exist in vitamin D-dependent
rickets type IA: Report of 13 new cases and review of the
literature. Calcif Tissue Int. 108:576–586. 2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Malloy PJ and Feldman D: Genetic disorders
and defects in vitamin D action. Rheum Dis Clin North Am.
38:93–106. 2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Chanchlani R, Nemer P, Sinha R, Nemer L,
Krishnappa V, Sochett E, Safadi F and Raina R: An overview of
rickets in children. Kidney Int Rep. 5:980–990. 2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kaddam IM, Al-Shaikh AM, Abaalkhail BA,
Asseri KS, Al-Saleh YM, Al-Qarni AA, Al-Shuaibi AM, Tamimi WJ and
Mukhtar AM: Prevalence of vitamin D deficiency and its associated
factors in three regions of Saudi Arabia. Saudi Med J. 38:381–390.
2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kim CJ, Kaplan LE, Perwad F, Huang N,
Sharma A, Choi Y, Miller WL and Portale AA: Vitamin D
1alpha-hydroxylase gene mutations in patients with
1alpha-hydroxylase deficiency. J Clin Endocrinol Metab.
92:3177–3182. 2007.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Kitanaka S, Takeyama K, Murayama A and
Kato S: The molecular basis of vitamin D-dependent rickets type I.
Endocr J. 48:427–432. 2001.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Jansen GA, Wanders RJ, Watkins PA and
Mihalik SJ: Phytanoyl-coenzyme A hydroxylase deficiency-the enzyme
defect in Refsum's disease. N Engl J Med. 337:133–134.
1997.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Babiker AM, Al Gadi I, Al-Jurayyan NA, Al
Nemri AMH, Al Haboob AAN, Al Boukai AA, Zahrani AA and Habib HA: A
novel pathogenic mutation of the CYP27B1 gene in a patient with
vitamin D-dependent rickets type 1: A case report. BMC Res Notes.
7(783)2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Cui N, Xia W, Su H, Pang L, Jiang Y, Sun
Y, Nie M, Xing X, Li M, Wang O, et al: Novel mutations of CYP27B1
gene lead to reduced activity of 1alpha-hydroxylase in Chinese
patients. Bone. 51:563–569. 2012.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hu WW, Ke YH, He JW, Fu WZ, Wang C, Zhang
H, Yue H, Gu JM and Zhang ZL: A novel compound mutation of CYP27B1
in a Chinese family with vitamin D-dependent rickets type 1A. J
Pediatr Endocrinol Metab. 27:335–341. 2014.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Dhull RS, Jain R, Deepthi B, Cheong HL,
Saha A, Mehndiratta M and Basu S: Vitamin D-dependent rickets
(VDDR) type 1: Case series of two siblings with a CYP27B1 mutation
and review of the literature. J Bras Nefrol. 42:494–497.
2020.PubMed/NCBI View Article : Google Scholar : (In English,
Portuguese).
|
|
15
|
Prader A, Illig R and Heierli E: An
unusual form of primary vitamin D-resistant rickets with
hypocalcemia and autosomal-dominant hereditary transmission:
Hereditary pseudo-deficiency rickets. Helv Paediatr Acta.
16:452–468. 1961.PubMed/NCBI(In German).
|
|
16
|
Li Y, Yuan X, Chen R, Lin X, Shangguan H,
Yang X and Zhang Y: Clinical and genetic analysis of two Chinese
families with vitamin D-dependent rickets type IA and follow-up.
Orphanet J Rare Dis. 15(273)2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ozden A and Doneray H: The genetics and
clinical manifestations of patients with vitamin D dependent
rickets type 1A. J Pediatr Endocrinol Metab. 34:781–789.
2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Demir K, Kattan WE, Zou M, Durmaz E,
BinEssa H, Nalbantoğlu O, Al-Rijjal RA, Meyer B, Özkan B and Shi Y:
. Novel CYP27B1 gene mutations in patients with vitamin D-dependent
rickets type 1A. PLoS One. 10(e0131376)2015.PubMed/NCBI View Article : Google Scholar
|