Introduction
According to GLOBOCANE 2020(1), esophageal cancer ranks seventh in
incidence and sixth in mortality among all types of cancer
worldwide. Esophageal squamous cell carcinoma (ESCC), the
predominant histological subtype of esophageal cancer, has high
incidence in China (1). For
resectable advanced ESCC, surgical resection remains the most
common treatment approach, particularly in patients treated with
neoadjuvant chemoradiotherapy (nCRT). Therefore, nCRT is considered
the standard treatment strategy for ESCC (2,3). The
CROSS (2) study also suggested
that patients with ESCC may benefit from preoperative CRT combined
with concurrent radiotherapy. The standard chemotherapy regimen
consists of carboplatin (2 mg/ml/min) and paclitaxel (50
mg/m2/day; days, 1, 8, 15, 22, 29 from inpatient
admission) for five weekly cycles, accompanied by increasing doses
of radiation using 41.4 Gy in 23 fractions, five days/week
(4). The CROSS trial also
illustrated a significant advantage in disease-free survival in
patients with ESCC. nCRT is also beneficial in decreasing tumor
burden and size, which are associated with pathological regression
(5). Additionally, the
NEOCRTEC5010 randomized clinical trial revealed that the survival
rate of patients with locally advanced ESCC treated with nCRT +
surgery was significantly improved compared with those treated with
surgery alone (6).
It has been previously reported that targeted drugs,
such as those targeting programmed cell death-1 (PD-1), improve
clinical response of patients with cancer to nCRT (7,8). The
Keystone-002 trial demonstrated that patients with locally advanced
ESCC exhibit better outcome when treated with nCRT combined with
pembrolizumab (9). Potential
effects of other factors such as the gene expression profile,
microarray or clinical parameters, have been evaluated in terms of
the clinical response of patients with esophageal cancer to nCRT
(10). A study suggested that
evaluation of visual residual tumor cells may be considered as a
significant predictor of tumor regression in patients with ESCC
treated with nCRT (11). In
addition, heterogeneity has been observed in the expression of
microRNAs associated with the pathological response of patients
with ESCC to nCRT, while several promising biomarkers have been
identified (12-15).
However, the currently available prognostic biomarkers for patients
with ESCC undergoing neoadjuvant therapy are limited.
It has been reported that the dysregulation of
epigenetic modifications, such as 5-position of cytosine (5-mC) and
5-hydroxymethylcytosine (5-hmC), serves a crucial role in
tumorigenesis (16). 5-mC and
5-hmC are stable, heritable epigenetic marks governed by
methyltransferases and demethylases (17,18).
5-hmCs have been detected in the gene bodies of promoters and
enhancers [such as pepsinogen A4 (PGA4)] (19). A study showed that tissue-specific
5-hmCs serve a key role in gene expression and function (19). Additionally, a study on circulating
cell-free DNA suggested that tissue levels of 5-hmC reads may be
used as diagnostic biomarkers in patients with esophageal cancer
(20).
Abnormal levels of DNA methylation are involved in
gene silencing during carcinogenesis, thus providing novel insights
into the effects of methylation on cancer progression. Several key
epigenetic biomarkers, such as two DNA
(cytosine-5)-methyltransferase (DNMT) inhibitors (azacitidine and
decitabine) for the treatment of myelodysplastic syndrome (MDS),
have been identified for cancer screening, diagnosis, prognosis and
treatment (21). DNA
hypomethylation and hypermethylation have been identified as the
most common types of methylation abnormality. The members of the
ten-eleven translocation (TET) family convert 5-mC to 5-hmC via DNA
demethylation. It has been previously reported that abnormal levels
of 5-hmC serve a key role in carcinogenesis, including ESCC. Three
TET genes, namely TET1, TET2 and TET3 are responsible for
conversion of 5-mC to 5-hmC. The dysregulation of TETs has been
associated with carcinogenesis (17,21).
Therefore, it was hypothesized that abnormal TET mRNA and protein
expression levels may be associated with altered 5-hmC levels via
5mC oxidization, thus contributing to altered hydroxymethylated and
differentially unmethylated regions (19).
Cancer epigenetics provide novel strategies in the
treatment and prognosis of esophageal cancer (22,23).
Therefore, evaluating expression of genes in hydroxymethylated
regions, such as 5-mC and 5-hmC, is of importance. DNA methylation
in ESCC could increase understanding on the effects of specific
hydroxymethylated regions in response to chemotherapy and
radiotherapy. However, specific hydroxymethylated regions in
patients with ESCC remain unknown, particularly in patients treated
with nCRT.
The present study aimed to analyze genomic
methylation and hydroxymethylation status of patients with ESCC
using sequence-based approaches, including methylated DNA
immunoprecipitation sequencing (MeDIP-seq) and hydroxymethylated
(hMe)DIP-seq to detect methylated or hydroxymethylated DNA regions.
Distribution of methylated and hydroxymethylated DNA regions was
determined in cancer tissue derived from patients with ESCC treated
with nCRT.
Materials and methods
Patient samples and clinical data
A total of 12 patients with ESCC from Changzhou
Tumor Hospital (Changzhou, in China) were enrolled from April 2015
to Dec 2018 in the present study. Patients with lymph node or
distant metastasis were excluded. Patients were treated with
standard nCRT combined with esophagectomy, according to the Dutch
guidelines for treatment of ESCC (v3.0; 2010; update 2014)
(2,3). Tumor tissue samples were obtained
after surgery. Among the 12 patients with ESCC, routinely
followed-up on an outpatient basis, six patients showed a
pathological complete response following nCRT (nCRT-well group),
while six patients exhibited incomplete pathological response after
nCRT (nCRT-poor group). A total of five females and seven males
with ESCC were included, with a mean age of 54.25±6.1 years (range,
48-72 years). Written informed consent was obtained from all
patients and the study was approved (No. 2021055) by the Ethics
Committee of Changzhou Tumor Hospital.
MeDIP and hMeDIP in ESCC tumor
tissue
Genomic DNA extraction from tumor tissue was
performed using kit (Takara MiniBEST Universal Genomic DNA
Extraction kit, cat. no. #9761; Takara Biotechnology Co., Ltd.).
DNA samples were fragmented to 200-800 bp with a Diagenode
Bioruptor. NanoDrop ND-1000 (Nanodrop, Inc., USA) instrument was
used for the measurement of concentration (ABS 260) and protein
contamination (ratio ABS260/ABS280) of total DNA samples. A total
of 1 ug DNA fragments with mixed libraries were generated by
single-stranded DNA molecules following denaturation with 0.1 M
NaOH. The concentration of each library was adjusted to 10 nM
before cluster generation. Sequencing library was determined by
Agilent 2100 Bioanalyzer using the Agilent DNA 1000 chip kit
(Agilent, part #5067-1504), which is followed by amplification
using the HiSeq3000/4000 PE Cluster kit (cat. no. PE-410-1001;
Illumina, Inc.). Paired end sequencing was performed using the
Illumina HiSeq4000 platform (Illumina, Inc.) by running 150 cycles
use HiSeq 3000/4000 SBS Kit (300 cycles) (#FC-410-1003, Illumina)
according to the manufacturer's instructions. The completed
libraries were quantified with the Agilent 2100 Bioanalyzer
(Agilent Technologies, Inc.).
Bioinformatics
To identify mRNAs significantly associated with
hMeDIP-enriched regions (peaks), aligned reads were assessed using
MACS2 software (cut-off, q-value ≤1x10-5; Ver 2.2.7,
https://pypi.org/project/MACS2/). mRNAs
associated with-hMeDIP enriched regions were annotated to the
nearest genes using the University of California Santa Cruz RefSeq
database (https://www.ncbi.nlm.nih.gov/refseq/). The
statistically significant mRNA-associated differentially
hydroxymethylated regions (DhMRs) within the promoter sequence
between two samples were identified using diffReps software
(version 1.55.6) (cut-off, log2FC ≥1;
P≤1x10-4) (https://code.google.com/p/diffreps/under). Gene
Ontology (GO) (geneontology.org; cut-off, P≤0.05), was also used in
the present study. Kyoto Encyclopedia of Genes and Genomes (KEGG;
genome.jp/kegg/pathway.html) significantly enriched pathways were
determined using EASE-score or Fisher's or hypergeometric-P-value
(cut-off, P≤0.05). GO enrichment analysis included at least one
differentially methylated region (DMR). In addition, python
(version 2.70) (https://www.python.org/), R language (version 3.4.1)
(https://www.r-project.org/), FastQC
(version 0.11.5) (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/),
Cutadapt (version 1.14; github.com/marcelm/cutadapt/), Hisat2 (version 2.1.0;
daehwankimlab.github.io/hisat2/), MACS2 (version
2.1.1) and diffReps (version 1.55.6) software were used for
bioinformatics.
DiseaseMeth 3.0
DiseaseMeth version 3.0 (diseasemeth.edbc.org/) contains DNA methylation
information updated from 1 October 2015 to 31 January 2021. Data
sources include public databases and literature. A total of 4,708
samples in 247 high-throughput datasets were collected from the
Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) and The Cancer
Genome Atlases databases (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga).
Literature data were searched manually in PubMed resulting in 2,210
references (pubmed.ncbi.nlm.nih.gov/).
Immunohistochemical (IHC)
staining
Surgical specimens were immersed in 10% formalin
solution for 25˚C, 24 h, dehydrated using a serial alcohol
gradient, and embedded in paraffin. Longitudinal cuts were made
along the specimens, and then were cut for 5-micrometer-thick
sections. Sections were blocked with 1% BSA) (cat. nos. 37525,
Thermo Scientific™, USA) for 25˚C, 15 min. The sections
were recovered by a high pH solution for 20 min at 95˚C, then
incubating 5 min in a 3% hydrogen peroxide solution, dewaxed in
xylene, rehydrated using decreasing concentrations of ethanol, and
washed in PBS. Tissue was incubated with primary antibodies against
TET1, 2 and 3 (cat. nos. ab272900, ab99432 and ab153724,
respectively; all Abcam), at 1:500 dilution for 60 min, 37˚C, then
with HRP Anti-Rabbit IgG antibody (ab6759, Abcam, USA) or HRP
Anti-Goat IgG antibody (ab6858, Abcam) secondary antibodies, at
1:1,000 dilution for 20 min, 37˚C. A chromogenic reaction is
performed by eBioscience™ DAB Advanced Chromogenic Kit
(cat. no. 8801-4965-72, Invitrogen™, USA). And
counterstained by 1% hematoxylin Stain Solution (cat. nos. Q38803,
Thermo Scientific™), for 25˚C, 8 min. Nuclear TET1-, 2-
and 3-positive expression was defined as weak or strong when ≤30%
or >30% of cells were stained, respectively. IHC staining score
was independently calculated by two pathologists, according to the
IHC scoring system of HER2(24)
due to lack of standards for TET protein expression. Images were
scanned by the Leica DM RXA2 light microscope (Software: Leica LAS
3.8) at a magnification of x200.
Determination of TET mRNA expression
levels
Total RNA was extracted from tissue using TRIzol
reagent (cat. no. 15596026; Thermo Fisher Scientific, Inc.),
according to the manufacturer's instructions. Subsequently, total
RNA was reverse transcribed into cDNA (37˚C, 15 min and 85˚C, 5
sec) and reverse transcription-quantitative (q)PCR kit (cat. no.
RR036Q; Takara Biomedical Technology Co., Ltd.) was performed on
the Mx3000P qPCR System (Stratagene; Agilent Technologies, Inc.)
using the corresponding SYBR Green qPCR kit, according to the
manufacturer's instructions (cat. no. 9767; Takara Biomedical
Technology Co., Ltd.). qPCR for TETs mRNA expression was performed
under the following conditions: 5 min at 95˚C, 40 cycles of 28 sec
at 95˚C, 30 sec at 60˚C, and 1 min at 72˚C. The mRNA expression
levels of TETs were normalized to those of β-actin. Cq values for
triplicate reactions were averaged and relative TETs expression was
determined with the comparative
2-ΔΔCq method (25), using average Cq values for TETs and
β-actin. The primers used are listed in Table SI.
Statistical analysis
All statistical analysis was performed out using
SPSS 17.0 software (SPSS, Inc.). All P-values were two-sided. The
differences in mean TET mRNA expression levels (mean ± SEM of three
independent repeats) between two groups were compared using
unpaired t test. The overall survival was estimated using the
Kaplan-Meier method and results were compared by log-rank
(Mantel-Cox) test. P<0.05 was considered to indicate a
statistically significant difference.
Results
TET expression is higher in patients
in nCRT-well than nCRT-poor group
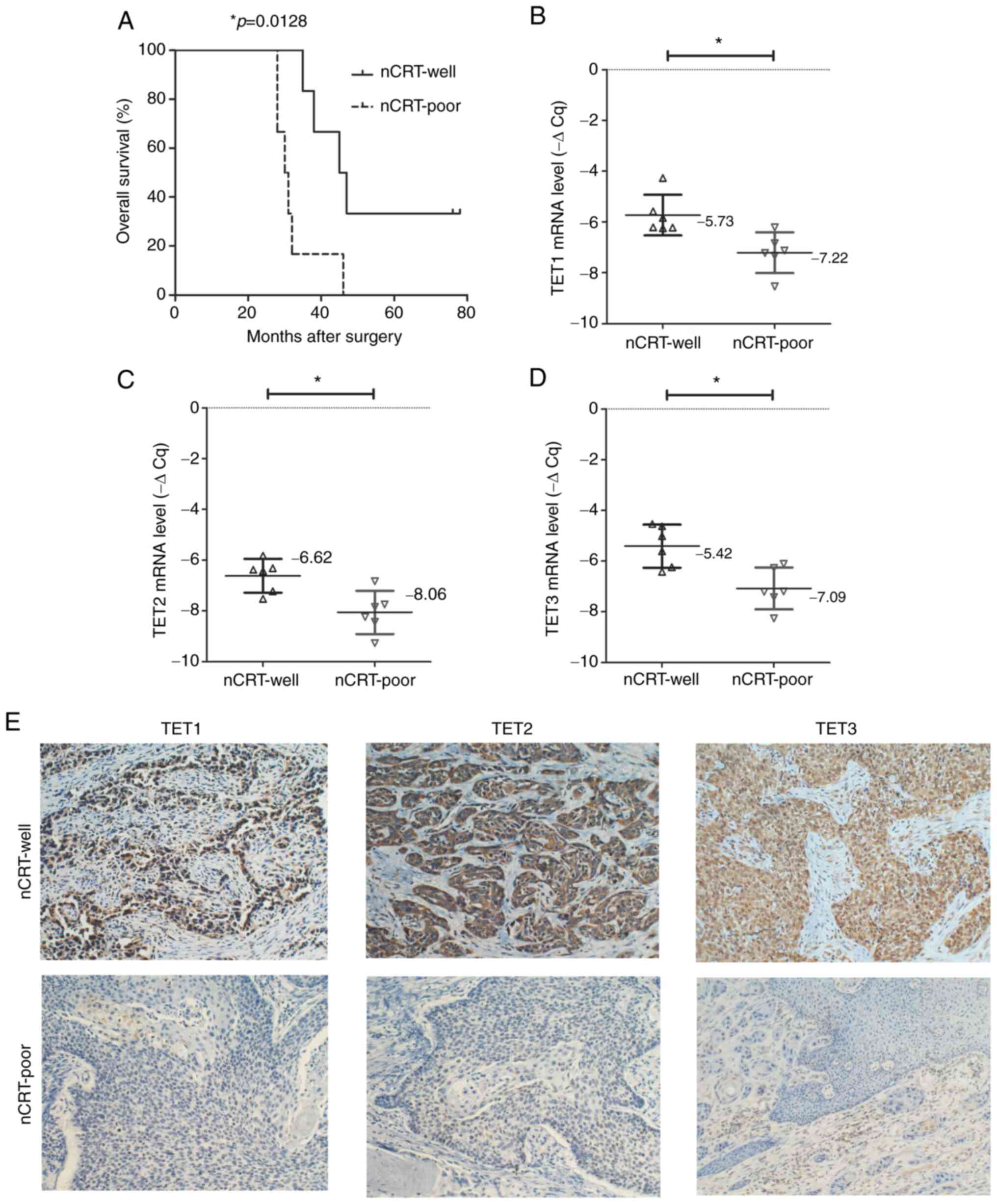
The overall survival of patients in the nCRT-poor
group was shorter compared with that in the nCRT-well group
(median, 30.5 vs. 46 months; P=0.0128; HR=0.1494; 95CI%
0.0334-0.6678; Fig. 1A). This was
not associated with clinicopathological parameters, including
regional lymph node and distant metastasis and recurrence rate, but
the survival rate were statistically significant (Fig. 1A).
In addition, mRNA expression levels of TET1
were significantly higher in the nCRT-well group (mean-ΔCq=-5.73)
compared with the nCRT-poor group (mean-ΔCq=-7.22; P=0.0072;
Fig. 1B). Additionally, mRNA
expression levels of TET2 were significantly different
between the nCRT-well (mean-ΔCq=-6.62) and nCRT-poor groups
(mean-ΔCq=-8.06; P=0.0064; Fig.
1C). Consistently, TET3 was significantly upregulated in
the nCRT-well group (mean-ΔCq=5.42) compared with the nCRT-poor
group (mean-ΔCq=-7.09; P=0.0048; Fig.
1D). Furthermore, IHC staining revealed that expression of
TET1, TET2 and TET3 was stronger in tumor tissue derived from
patients in the nCRT-well group compared with that in the nCRT-poor
group (Fig. 1E).
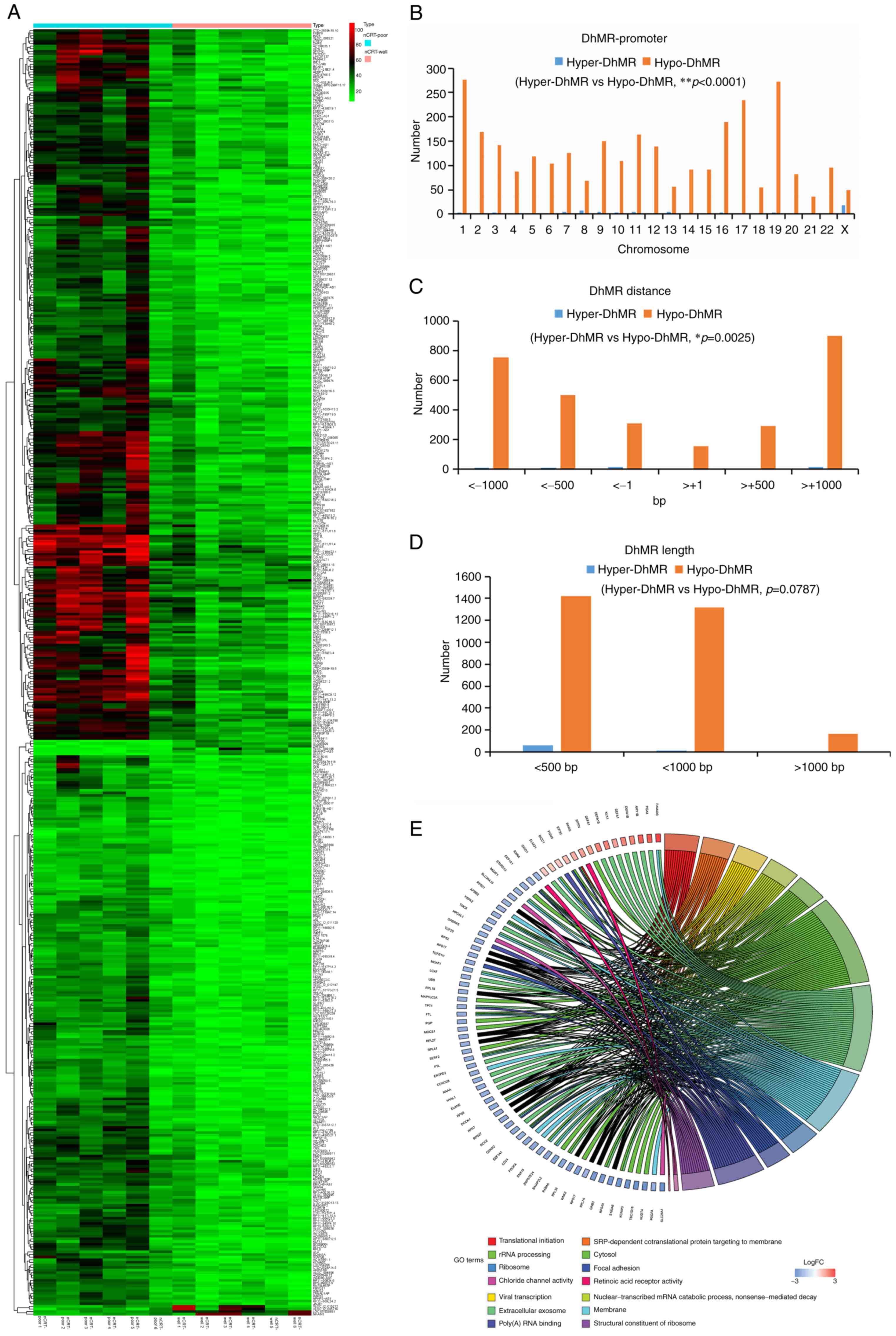
mRNA-associated DhMRs in promoter
sequences
A total of 2,925 mRNA-associated hypo-DhMRs and 292
mRNA-associated hyper-DhMRs were identified in the promoter
sequences between the nCRT-well and nCRT-poor groups (Fig. 2A). The hypo-(0~18 genes) and
hyper-DhMRs (36-277 genes) were detected in the promoter regions on
different chromosomes (chromosome 1-22, X; Fig. 2B). The analysis also revealed that
the distance of DhMRs from the transcription start site varied from
1,000 bp downstream to ~1,000 bp to upstream (Fig. 2C). Additionally, the length of
DhMRs was varied from <500 bp to >1,000 bp (Fig. 2D). DhMRs were significantly
enriched in different GO terms in the nCRT-well group compared with
the nCRT-poor group, such as ribosome, cytosol, rRNA processing,
translational initiation, focal adhesion, membrane' (Fig. 2E).
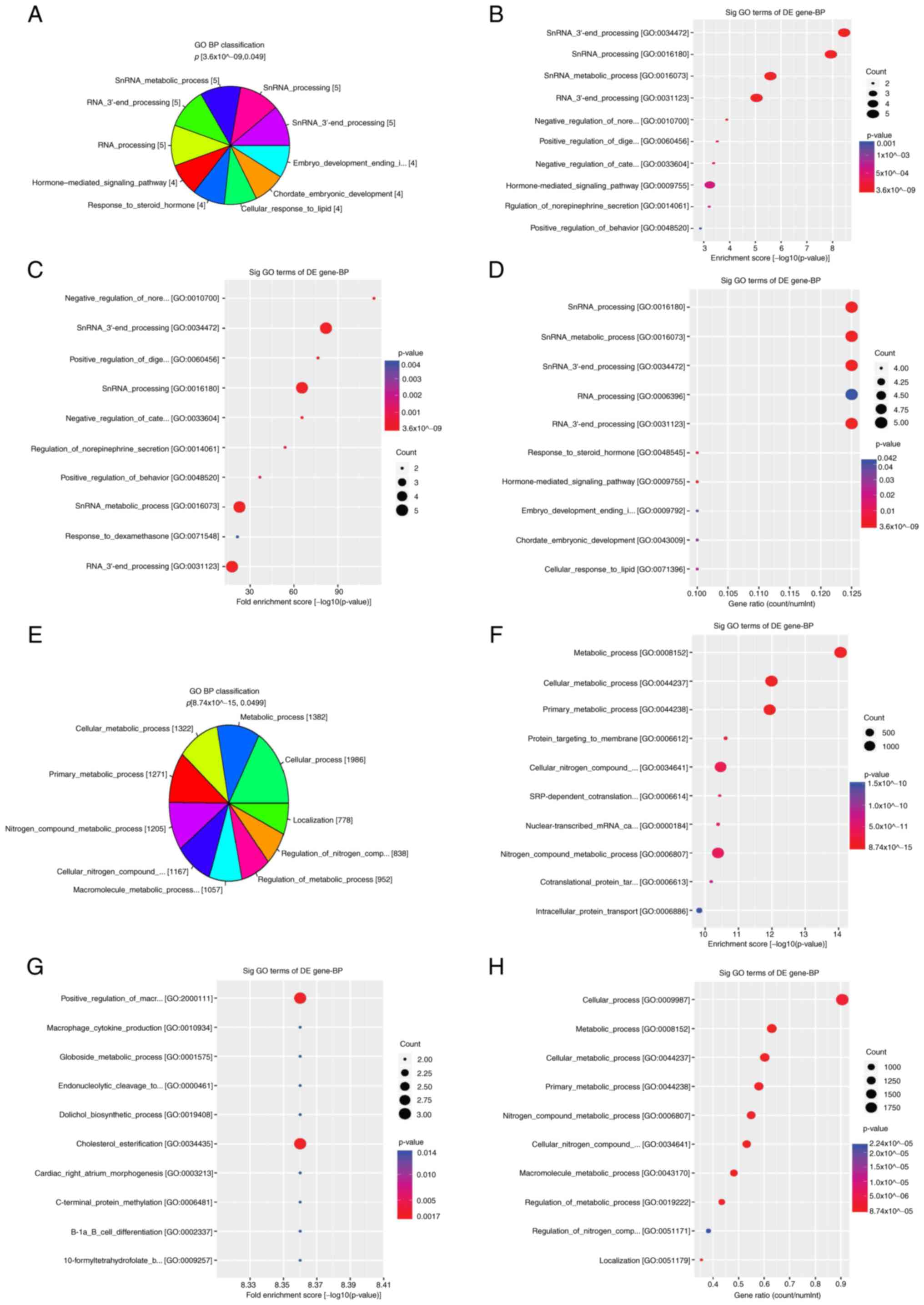
Biological processes in GO
Functional pathway analysis revealed that the
differentially hydroxymethylated CpGs were enriched in biological
pathways. Hyper-DhMRs were significantly enriched in biological
processes, including ‘small nuclear RNA (snRNA) 3'-end processing’,
‘snRNA processing’, ‘snRNA metabolic process’, ‘RNA 3'-end
processing’ [cancer/testis antigen family 45 (CT45), CT45A2,
CT45A5, CT45A6, CT45A8 and CT45A9], ‘hormone-mediated signaling
pathway’, ‘response to steroid hormone’, ‘cellular response to
lipid’, ‘cellular response to hormone stimulus’
[corticotropin-releasing hormone (CRH), defensin alpha 1 (DEFA1),
growth hormone secretagogue receptor (GHSR) and retinoic acid
receptor, gamma (RARG)], ‘embryo development ending in birth’ and
‘chordate embryonic development’ [matrix metallopeptidase 16
(MMP16), RARG, SKI like proto-oncogene (SKIL) and zinc finger
protein 568 (ZNF568); Fig. 3A-D].
Furthermore, hypo-DhMRs were primarily enriched in the following
biological processes: ‘Nitrogen compound metabolic process’,
‘macromolecule metabolic process’, ‘cellular metabolic process’,
‘cellular nitrogen compound metabolic process’, ‘metabolic
process’, ‘localization’, ‘cellular process’, ‘regulation of
metabolic process’ and ‘regulation of nitrogen compound metabolic
process’ such as alpha 1,4-galactosyltransferase [A4GALT], aladin
WD repeat nucleoporin [AAAS], adipogenesis associated Mth938 domain
containing [AAMDC], etc (Fig.
3E-H).
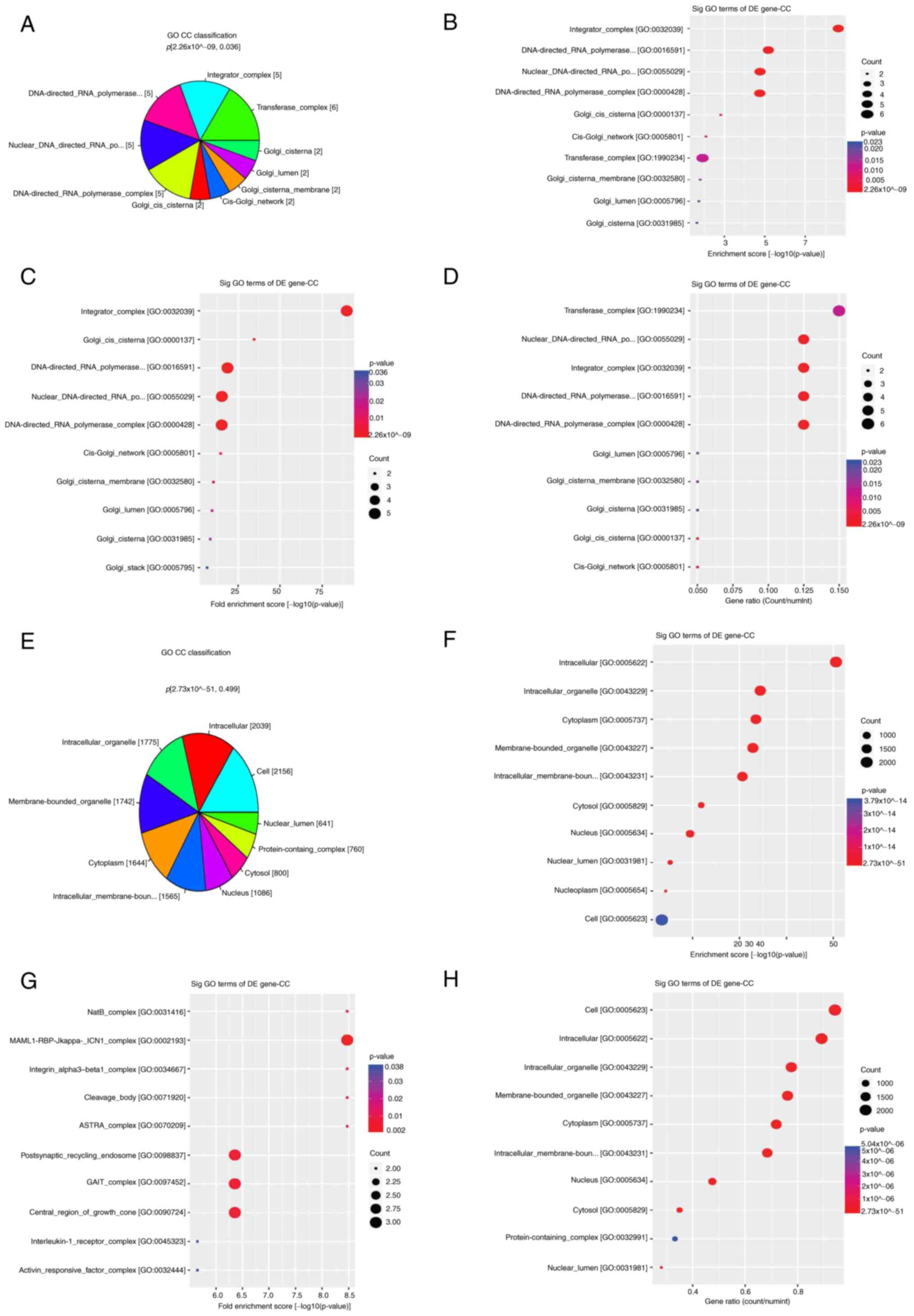
Cellular component classification in
GO
Hyper-DhMRs were enriched in cellular components,
including ‘transferase complex’ (CT45A2, CT45A5, CT45A6, CT45A8,
CT45A9 and gigaxonin), ‘integrator complex’, ‘DNA-directed RNA
polymerase II’, ‘nuclear DNA-directed RNA polymerase complex’ and
‘DNA-directed RNA polymerase complex’ (CT45A2, CT45A5, CT45A6,
CT45A8 and CT45A9), ‘Golgi cis cisterna’, ‘cis-Golgi network’,
‘Golgi cisterna membrane’, ‘Golgi cisterna’, ‘Golgi stack’ [golgin
A8 family member GOLGA8)A and GOLGA8B] and ‘Golgi lumen’ (DEFA1 and
MMP16; Fig. 4A-D). In addition,
hypo-DhMRs were primarily enriched in ‘cytoplasm’, ‘intracellular
organelle’, ‘membrane-bounded organelle’, ‘intracellular
membrane-bounded organelle’, ‘intracellular’, ‘nucleus’, ‘cell’,
‘cytosol’, ‘protein-containing complex’ and ‘nuclear lumen’
cellular components (Fig.
4E-H).
Molecular function classification in
GO
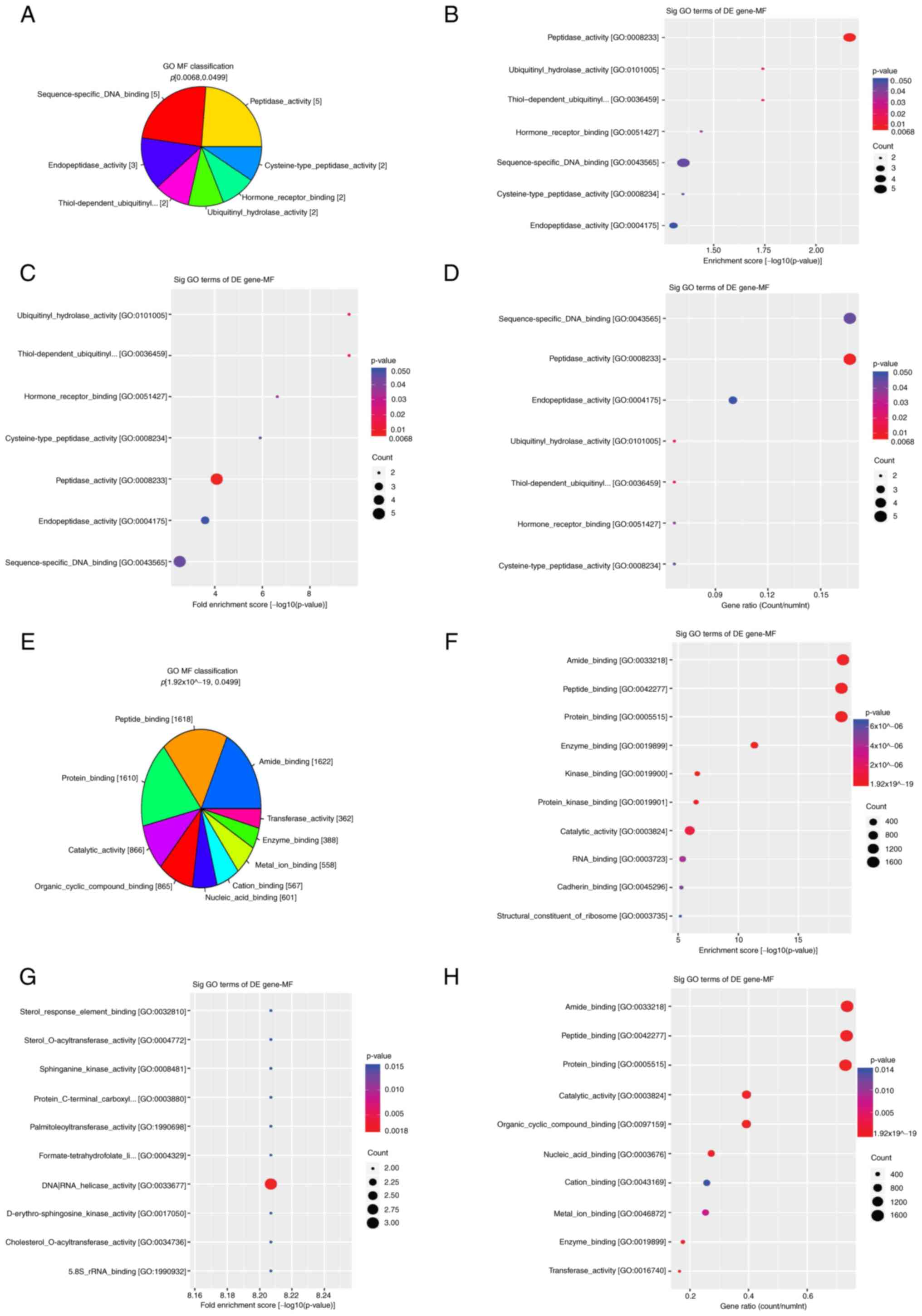
GO enrichment analysis revealed that hyper-DhMRs
were enriched in molecular functions, such as ‘peptidase activity’
(KLK1, MMP16, PGA3, USP17L11 and USP17L18), ‘ubiquitinyl hydrolase
activity’, ‘thiol-dependent ubiquitinyl hydrolase activity’ and
‘cysteine-type peptidase activity’ (USP17L11 and USP17L18),
‘endopeptidase activity’ (KLK1, MMP16 and PGA3), ‘hormone receptor
binding’ (CRH and RARG) and ‘sequence-specific DNA binding’
(FOXD4L3, FOXD4L4, RARG, SKIL and ZNF568; Fig. 5A-D). Hypo-DhMRs were enriched in
the following molecular functions: ‘Peptide binding’, ‘protein
binding’, ‘nucleic acid binding’, ‘organic cyclic compound
binding’, ‘catalytic activity’, ‘cation binding’, ‘amide binding’,
‘metal ion binding’, ‘enzyme binding’ and ‘transferase activity’,
such as acetoacetyl-CoA synthetase [AACS], A4GALT, ATP binding
cassette subfamily D member 1 [ABCD1], etc (Fig. 5E-H).
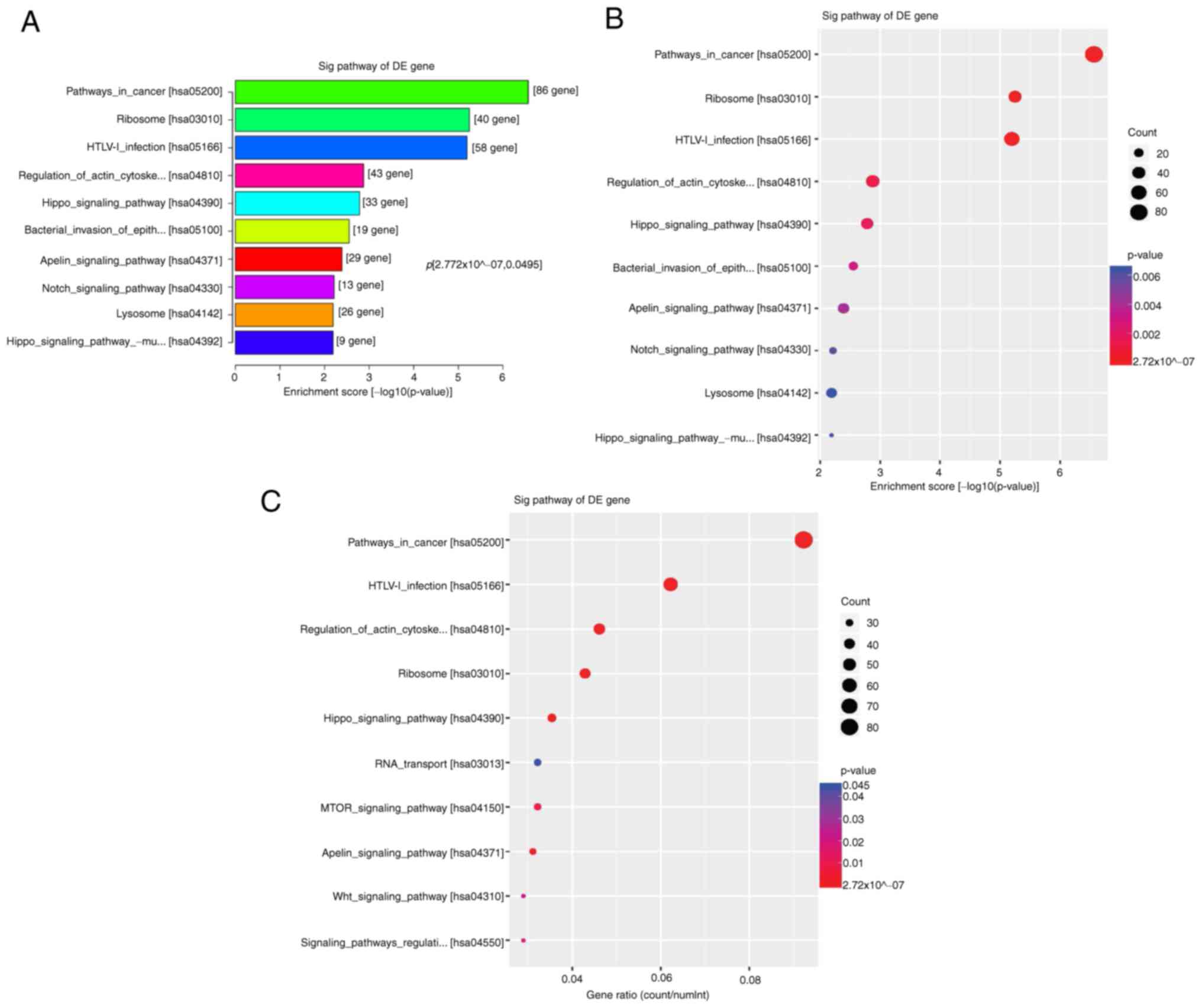
KEGG pathway analysis
KEGG enrichment analysis showed that hyper-DhMRs
were not significantly enriched in any cancer-associated pathway.
However, hypo-DhMRs were enriched in cancer-associated pathways,
including ‘ribosome’, ‘HTLV-1 infection’, ‘hippo signaling
pathways’, ‘regulation of actin cytoskeleton’, ‘bacterial invasion
of epithelial cells’, ‘apelin signaling pathway’ and ‘Notch
signaling pathway’, such as mitochondrial ribosomal protein L23
[MRPL23], adenylate cyclase 1 [ADCY1], actin beta [ACTB], abl
interactor 2 [ABI2], a disintegrin and metallopeptidase domain 17
[ADAM17], etc (Fig. 6A-C).
Discussion
Esophageal cancer is a key cause of
cancer-associated mortality worldwide and is characterized by
inter- and intra-tumoral genomic heterogeneity. Since the
diagnostic strategies are limited, the majority of patients with
ESCC first present with lymph node or distal metastasis, thus
leading to poor outcomes. Therefore, understanding tumor
heterogeneity may be beneficial for the clinical management of
patients with esophageal cancer (2,26,27).
How to assess tumor response after nCRT has raised
discussion. A study suggested that the mRNA expression levels of
PD-ligand 1 and CD8B may be used as prognostic markers in patients
with ESCC treated with nCRT (28).
However, assessment of response to nCRT via rebiopsy, endoscopy and
endoscopic ultrasound cannot accurately predict clinical outcomes
(29). Wang et al (11) suggested that the percentage of
visual residual tumor cells may be used to evaluate the response of
patients with ESCC to nCRT in clinical practice.
It has been reported that genomic alterations, such
as somatic mutations and copy number alteration, are involved in
the molecular regulation of esophageal cancer. Furthermore,
epigenetic changes, particularly specific DNA methylation
alterations such as 5-mC and 5-hmC, have been established as
targets for therapeutic intervention in several types of cancer,
such as MDS (21,30,31).
The results of the present study showed that levels of abnormal DNA
methylation were significantly different, especially
hypo-hydroxymethylation, which is 2925 hypo-DhMRs and 292
hyper-DhMRs in promoter between nCRT-well and nCRT-poor patients.
Additionally, mRNA and protein expression levels of TET1, 2 and 3
were notably increased in tumor tissue derived from patients in the
nCRT-well group compared with those in the nCRT-poor group. These
findings suggested that enhanced expression levels of TETs may be
involved in abnormal levels of 5-hmC. There are some genome-wide
hydroxymethylation analyses (20,22,23),
but, to the best of our knowledge, there is no study for nCRT-well
and nCRT-poor patients with ESCC. The present results contribute to
understanding DhMRs and may facilitate development of novel
invasive tools for clinical response to nCRT.
Following Illumina HiSeq 4000 sequencing, a total of
2,925 hypo-DhMRs and 292 hyper-DhMRs were identified between the
nCRT-well and nCRT-poor groups. The hyper-DhMRs were enriched in
biological processes such as ‘snRNA processing’, ‘hormone-mediated
signaling pathway’ and ‘cellular response’. Consistently,
hypo-DhMRs were also enriched in metabolic processes. Hyper-DhMRs
were enriched in the cellular component ‘transferase complex’,
associated with CT45 genes. On the other hand, hypo-DhMRs were
primarily enriched in the term ‘intracellular organelle’.
Additionally, hyper-DhMRs were enriched in the molecular functions
‘peptidase activity’, ‘USP17L’ and ‘DNA binding’, and hypo-DhMRs in
‘peptide binding’, ‘amide binding’, ‘protein binding’ and
‘catalytic activity’. KEGG pathway enrichment analysis revealed
that hypo-hydroxymethylated CpG-associated genes were primarily
enriched in ‘apelin signaling pathway’, ‘hippo pathways’ and ‘Notch
pathways’. The aforementioned findings suggested that the GO and
KEGG databases may provide more information regarding the molecular
mechanisms of DhMRs in ESCC (32).
Profiling of cell-free DNA 5-mCs may provide
facilitate development of epigenetic genomic markers for the early
diagnosis and surveillance of cancer (33). DhMRs contribute to the development
of novel invasive tools for evaluating clinical response to nCRT.
Here, several differentially expressed genes, such as DES, TF,
TFEB, CD53, MAD1L1, GPX7, HIVEP3, TRIM71, CDKN1C, ARHGAP25 and
CT45, were identified between the nCRT-well and nCRT-poor groups.
The methylation modifications in the aforementioned genes were
assessed using DiseaseMeth version 3.0(34). The results showed that both GPX7
and TRIM71 genes were hypermethylated. Peng et al (35) demonstrated that GPX7 is frequently
downregulated in esophageal adenocarcinoma and that GPX7 promoter
is hypermethylated in more than half of esophageal adenocarcinoma
samples. A significant inverse correlation between DNA methylation
and mRNA expression levels of GPX7 has been observed (36). To the best of our knowledge, there
are no studies on methylation status of TRIM71 in patients with
ESCC. However, Qu et al (37) revealed a specific group of
risk-associated DMRs located near TRIM71 gene in patients with
acute myeloid leukemia. To the best of our knowledge, there are no
studies on methylation status and mRNA expression levels of TRIM71
in patients with ESCC.
Although the present study found differential
expression of the aforementioned genes, more samples are required
for confirmation; this is a limitation of the present study. These
genes may play an important role during tumorigenesis, such as
cancer stem-like pathway signaling. It is necessary to confirm
expression of these genes and DNA hydroxymethylation in nCRT-well
and nCRT-poor patient groups. Taken together, the aforementioned
studies support the present results regarding the key role of
abnormal gene promoter methylation in mRNA gene expression and its
association with response of patients with ESCC to nCRT.
De Klerk et al (38) identified several candidate
epigenetic biomarkers, such as NDRG4, TFPI2, RUNX3, MGMT, CHFR,
CDKN2A, MLH1 and RASSF1 genes, in 75 patients with adenocarcinoma
and 16 patients with ESCC associated with response to nCRT. Iwabu
et al (39) also
demonstrated using genome-wide DNA methylation analysis that FGF5
methylation is significantly associated with response to definitive
CRT in 117 patients with ESCC. The aforementioned findings
indicated that identifying potential methylation biomarkers
associated with response to nCRT may be beneficial for development
of individualized therapy.
Coscia et al (40) suggested that cancer/testis antigen
45 (CT45) may be considered as an independent prognostic factor in
patients with ovarian cancer. The aforementioned study showed that
CT45 is associated with resistance to platinum-based chemotherapy
via regulating protein phosphatase 4 activity. Therefore, increased
CT45 protein levels enhance DNA damage and platinum sensitivity via
activating cytotoxic T cells and killing tumor cells. Zhang et
al (41) demonstrated that the
expression of CT45 is regulated via promoter hypomethylation in
epithelial ovarian cancer. Herein, CT45 was identified as a
mRNA-associated DhMR also associated with response to nCRT.
DNA hydroxymethylation is one of the most common
processes of epigenetic regulation, which affects expression of
both oncogenes and tumor suppressor factors (42). The results of the present study
showed that global 5-hmC content was notably dysregulated, thus
affecting the response of patients with ESCC to nCRT. This
indicated that the levels of 5-hmC undergo highly dynamic changes
during CRT.
However, the current study has limitations. Due to
the high cost of hMeDIP-seq, the sample size was small and all
patients were of the same background (no lymph node and distant
metastasis), thus resulting some variance in DhMRs. However,
hydroxymethylation profiling was performed in tissue derived from
patients with ESCC treated with nCRT. Key hydroxymethylated genes
were identified in different biological and metabolic pathways.
Further studies with larger sample size should be performed in
future to uncover the molecular mechanisms involved in the
aforementioned processes.
The results of the present study suggested that
hyper- and hypo-DhMRs affect molecular pathways, such as the hippo
and Notch signaling pathways, thus providing basic information on
epigenetic modifications associated with the clinical response to
nCRT. The hyper- and hypo-DhMRs identified in the present study may
serve as potential biomarkers for nCRT in patients with esophageal
cancer.
Supplementary Material
Primer sequences used for reverse
transcription-quantitative PCR.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Suzhou Science
and Technology Program (grant no. SLT202005), Suzhou Municipal
Commission of Health and Family Planning (grant no. LCZX202031) and
Suzhou New District Science and Technology Program (grant no.
2019Z009).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the Bioproject repository for the
National Center for Biotechnology Information [ncbi.nlm.nih.gov/bioproject/?term=(Bioproject No.)],
Bioproject nos. PRJNA885773, PRJNA885596, PRJNA885597, PRJNA885605,
PRJNA885611, PRJNA885751, PRJNA885771, PRJNA885877, PRJNA885896,
PRJNA885912, PRJNA886044 and PRJNA886049.
Authors' contributions
CZ and MW conceived and designed the study. ML, XZ
and JZ collected data and analyzed data. CZ analyzed and
interpreted data. All authors wrote the manuscript. All authors
have read and approved the final manuscript. CZ and MW confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
The study was conducted in accordance with the
amended Declaration of Helsinki and was approved by the Ethic
Committee of the Second Affiliated Hospital of Nanchang University,
Changzhou Cancer Hospital and Suzhou Science and Technology Town
Hospital (approval no. 2021055). All patients gave written informed
consent for participation in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Shapiro J, van Lanschot JJB, Hulshof MCCM,
van Hagen P, van Berge Henegouwen MI, Wijnhoven BPL, van Laarhoven
HWM, Nieuwenhuijzen GAP, Hospers GAP, Bonenkamp JJ, et al:
Neoadjuvant chemoradiotherapy plus surgery versus surgery alone for
oesophageal or junctional cancer (CROSS): Long-term results of a
randomised controlled trial. Lancet Oncol. 16:1090–1098.
2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Li J, Zhao Q, Ge X, Song Y, Tian Y, Wang
S, Liu M and Qiao X: Neoadjuvant chemoradiotherapy improves
survival in locally advanced adenocarcinoma of esophagogastric
junction compared with neoadjuvant chemotherapy: A propensity score
matching analysis. BMC Surg. 21(137)2021.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Yang Y, Zhu L, Cheng Y, Liu Z, Cai X, Shao
J, Zhang M, Liu J, Sun Y, Li Y, et al: Three-arm phase II trial
comparing camrelizumab plus chemotherapy versus camrelizumab plus
chemoradiation versus chemoradiation as preoperative treatment for
locally advanced esophageal squamous cell carcinoma (NICE-2 study).
BMC Cancer. 22(506)2022.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Liu J, Yang Y, Liu Z, Fu X, Cai X, Li H,
Zhu L, Shen Y, Zhang H, Sun Y, et al: Multicenter, single-arm,
phase II trial of camrelizumab and chemotherapy as neoadjuvant
treatment for locally advanced esophageal squamous cell carcinoma.
J Immunother Cancer. 10(e004291)2022.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Yang H, Liu H, Chen Y, Zhu C, Fang W, Yu
Z, Mao W, Xiang J, Han Y, Chen Z, et al: Long-term efficacy of
neoadjuvant chemoradiotherapy plus surgery for the treatment of
locally advanced esophageal squamous cell carcinoma: The
NEOCRTEC5010 randomized clinical trial. JAMA Surg. 156:721–729.
2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yang P, Zhou X, Yang X, Wang Y, Sun T,
Feng S and Ma X: Neoadjuvant camrelizumab plus chemotherapy in
treating locally advanced esophageal squamous cell carcinoma
patients: A pilot study. World J Surg Oncol. 19(333)2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Liu J, Li J, Lin W, Shao D, Depypere L,
Zhang Z, Li Z, Cui F, Du Z, Zeng Y, et al: Neoadjuvant camrelizumab
plus chemotherapy for resectable, locally advanced esophageal
squamous cell carcinoma (NIC-ESCC2019): A multicenter, phase 2
study. Int J Cancer. 151:128–137. 2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Shang X, Zhang W, Zhao G, Liang F, Zhang
C, Yue J, Duan X, Ma Z, Chen C, Pang Q, et al: Pembrolizumab
combined with neoadjuvant chemotherapy versus neoadjuvant
chemoradiotherapy followed by surgery for locally advanced
oesophageal squamous cell carcinoma: Protocol for a multicentre,
prospective, randomized-controlled, phase III clinical study
(Keystone-002). Front Oncol. 12(831345)2022.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Huang RW, Chao YK, Wen YW, Chang HK, Tseng
CK, Chan SC and Liu YH: Predictors of pathological complete
response to neoadjuvant chemoradiotherapy for esophageal squamous
cell carcinoma. World J Surg Oncol. 12(170)2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wang X, Wang H, Wang H, Huang J, Wang X,
Jiang Z, Tan L, Jiang D and Hou Y: Prognostic value of visual
residual tumour cells (VRTC) for patients with esophageal squamous
cell carcinomas after neoadjuvant therapy followed by surgery. BMC
Cancer. 21(111)2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Lin D, Chen X and Tan L: The predictive
value of microRNAs for pathological response after neoadjuvant
treatment in esophageal squamous cell carcinoma: A systematic
review. Ann Transl Med. 9(420)2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Niwa Y, Yamada S, Sonohara F, Kurimoto K,
Hayashi M, Tashiro M, Iwata N, Kanda M, Tanaka C, Kobayashi D, et
al: Identification of a serum-based miRNA signature for response of
esophageal squamous cell carcinoma to neoadjuvant chemotherapy. J
Transl Med. 17(1)2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wen J, Luo K, Liu H, Liu S, Lin G, Hu Y,
Zhang X, Wang G, Chen Y, Chen Z, et al: MiRNA expression analysis
of pretreatment biopsies predicts the pathological response of
esophageal squamous cell carcinomas to neoadjuvant
chemoradiotherapy. Ann Surg. 263:942–948. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Slotta-Huspenina J, Drecoll E, Feith M,
Habermehl D, Combs S, Weichert W, Bettstetter M, Becker K and
Langer R: MicroRNA expression profiling for the prediction of
resistance to neoadjuvant radiochemotherapy in squamous cell
carcinoma of the esophagus. J Transl Med. 16(109)2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Shekhawat J, Gauba K, Gupta S, Choudhury
B, Purohit P, Sharma P and Banerjee M: Ten-eleven translocase: Key
regulator of the methylation landscape in cancer. J Cancer Res Clin
Oncol. 147:1869–1879. 2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Bachman M, Uribe-Lewis S, Yang X, Williams
M, Murrell A and Balasubramanian S: 5-Hydroxymethylcytosine is a
predominantly stable DNA modification. Nat Chem. 6:1049–1055.
2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Pfeifer GP, Xiong W, Hahn MA and Jin SG:
The role of 5-hydroxymethylcytosine in human cancer. Cell Tissue
Res. 356:631–641. 2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
He B, Zhang C, Zhang X, Fan Y, Zeng H, Liu
J, Meng H, Bai D, Peng J, Zhang Q, et al: Tissue-specific
5-hydroxymethylcytosine landscape of the human genome. Nat Commun.
12(4249)2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Tian X, Sun B, Chen C, Gao C, Zhang J, Lu
X, Wang L, Li X, Xing Y, Liu R, et al: Circulating tumor DNA
5-hydroxymethylcytosine as a novel diagnostic biomarker for
esophageal cancer. Cell Res. 28:597–600. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Jin N, George TL, Otterson GA,
Verschraegen C, Wen H, Carbone D, Herman J, Bertino EM and He K:
Advances in epigenetic therapeutics with focus on solid tumors.
Clin Epigenetics. 13(83)2021.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Lin L, Cheng X and Yin D: Aberrant DNA
methylation in esophageal squamous cell carcinoma: Biological and
clinical implications. Front Oncol. 10(549850)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Grady WM, Yu M and Markowitz SD:
Epigenetic alterations in the gastrointestinal tract: Current and
emerging use for biomarkers of cancer. Gastroenterology.
160:690–709. 2021.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Buza N and Hui P: Characteristics of HER2
gene amplification by fluorescence in situ hybridization in
endometrial serous carcinoma. Arch Pathol Lab Med.
146(0)2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Dagogo-Jack I and Shaw AT: Tumour
heterogeneity and resistance to cancer therapies. Nat Rev Clin
Oncol. 15:81–94. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Arnold M, Soerjomataram I, Ferlay J and
Forman D: Global incidence of oesophageal cancer by histological
subtype in 2012. Gut. 64:381–387. 2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Mori T, Kumagai K, Nasu K, Yoshizawa T,
Kuwano K, Hamada Y, Kanazawa H and Suzuki R: Clonal expansion of
tumor-infiltrating T cells and analysis of the tumor
microenvironment within esophageal squamous cell carcinoma relapsed
after definitive chemoradiation therapy. Int J Mol Sci.
22(1098)2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Schneider PM, Metzger R, Schaefer H,
Baumgarten F, Vallbohmer D, Brabender J, Wolfgarten E,
Bollschweiler E, Baldus SE, Dienes HP and Hoelscher AH: Response
evaluation by endoscopy, rebiopsy, and endoscopic ultrasound does
not accurately predict histopathologic regression after neoadjuvant
chemoradiation for esophageal cancer. Ann Surg. 248:902–908.
2008.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Hoshimoto S, Takeuchi H, Ono S, Sim MS,
Huynh JL, Huang SK, Marzese DM, Kitagawa Y and Hoon DS: Genome-wide
hypomethylation and specific tumor-related gene hypermethylation
are associated with esophageal squamous cell carcinoma outcome. J
Thorac Oncol. 10:509–517. 2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Cancer Genome Atlas Research Network;
Analysis Working Group: Asan University; BC Cancer Agency; Brigham
and Women's Hospital; Broad Institute; Brown University; Case
Western Reserve University; Dana-Farber Cancer Institute; Duke
University et al. Integrated genomic characterization of
oesophageal carcinoma. Nature. 541:169–175. 2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Chen X, Cai S, Li B, Zhang X, Li W, Linag
H and Cao X: Identification of key genes and pathways for
esophageal squamous cell carcinoma by bioinformatics analysis. Exp
Ther Med. 16:1121–1130. 2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhou M, Hou P, Yan C, Chen L, Li K, Wang
Y, Zhao J, Su J and Sun J: Cell-free DNA 5-hydroxymethylcytosine
profiles of long non-coding RNA genes enable early detection and
progression monitoring of human cancers. Clin Epigenetics.
13(197)2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Xing J, Zhai R, Wang C, Liu H, Zeng J,
Zhou D, Zhang M, Wang L, Wu Q, Gu Y and Zhang Y: DiseaseMeth
version 3.0: A major expansion and update of the human disease
methylation database. Nucleic Acids Res. 50 (D1):D1208–D1215.
2022.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Peng D, Belkhiri A, Hu T, Chaturvedi R,
Asim M, Wilson KT, Zaika A and El-Rifai W: Glutathione peroxidase 7
protects against oxidative DNA damage in oesophageal cells. Gut.
61:1250–1260. 2012.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Peng DF, Razvi M, Chen H, Washington K,
Roessner A, Schneider-Stock R and El-Rifai W: DNA hypermethylation
regulates the expression of members of the Mu-class glutathione
S-transferases and glutathione peroxidases in Barrett's
adenocarcinoma. Gut. 58:5–15. 2009.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Qu X, Davison J, Du L, Storer B, Stirewalt
DL, Heimfeld S, Estey E, Appelbaum FR and Fang M: Identification of
differentially methylated markers among cytogenetic risk groups of
acute myeloid leukemia. Epigenetics. 10:526–535. 2015.PubMed/NCBI View Article : Google Scholar
|
|
38
|
de Klerk LK, Goedegebuure RSA, van Grieken
NCT, van Sandick JW, Cats A, Stiekema J, van der Kaaij RT, Farina
Sarasqueta A, van Engeland M, Jacobs MAJM, et al: Molecular
profiles of response to neoadjuvant chemoradiotherapy in
oesophageal cancers to develop personalized treatment strategies.
Mol Oncol. 15:901–914. 2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Iwabu J, Yamashita S, Takeshima H, Kishino
T, Takahashi T, Oda I, Koyanagi K, Igaki H, Tachimori Y, Daiko H,
et al: FGF5 methylation is a sensitivity marker of esophageal
squamous cell carcinoma to definitive chemoradiotherapy. Sci Rep.
9(13347)2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Coscia F, Lengyel E, Duraiswamy J,
Ashcroft B, Bassani-Sternberg M, Wierer M, Johnson A, Wroblewski K,
Montag A, Yamada SD, et al: Multi-level proteomics identifies CT45
as a chemosensitivity mediator and immunotherapy target in ovarian
cancer. Cell. 175:159–170.e16. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhang W, Barger CJ, Link PA,
Mhawech-Fauceglia P, Miller A, Akers SN, Odunsi K and Karpf AR: DNA
hypomethylation-mediated activation of cancer/testis antigen 45
(CT45) genes is associated with disease progression and reduced
survival in epithelial ovarian cancer. Epigenetics. 10:736–748.
2015.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Martinez-Useros J, Martin-Galan M,
Florez-Cespedes M and Garcia-Foncillas J: Epigenetics of most
aggressive solid tumors: Pathways, targets and treatments. Cancers
(Basel). 13(3209)2021.PubMed/NCBI View Article : Google Scholar
|