Introduction
Paracetamol toxicity is one of the most common
causes of drug-induced hepatotoxicity (1) and acute liver failure (2) due to the worldwide availability of
paracetamol, its affordability and widespread use for the treatment
of pain and fever (3). A number of
studies are underway to clarify the pathophysiology of paracetamol
toxicity and its metabolization and detoxification. Paracetamol is
mainly metabolized in the liver through conjugation with glucuronic
acid and sulfates and is eliminated through the urinary system
(4). A small amount of paracetamol
is metabolized by cytochrome P450 isoenzymes, creating the
extremely toxic metabolite N-acetyl-p-benzoquinone imine (NAPQI).
This metabolite significantly depletes glutathione (GSH) reserves
(5,6) and binds covalently to hepatocyte
membrane proteins. The standard treatment against this mechanism is
N-Acetylcysteine (NAC), which is a precursor influencing GSH
formation and preventing acute liver injury resulting from NAPQI.
However, details of this underlying toxicity mechanism remain to be
elucidated (7).
The renin-angiotensin-aldosterone system (RAAS) is
one of the pathways that serves a role in the pathophysiology of
acute liver injury due paracetamol toxicity. Some studies have
shown that drugs influencing RAAS have potential preventive effects
against paracetamol-induced hepatotoxicity (8,9).
RAAS is a multi-hormonal system that is known to regulate systemic
circulatory homoeostasis, but also contributes to
pathophysiological mechanisms including inflammation and oxidative
stress (10) as well as the
pathogenesis of liver fibrosis (11-13).
Angiotensin II (Ang II), a key mediator in RAAS, releases free
radical precursor enzymes such as nicotinamide adenine dinucleotide
phosphate oxidase and xanthine oxidase into vascular structures and
leads to increased free radicals (14,15).
In the gastrointestinal system, increased Ang II is shown to
promote hepatic inflammation and fibrosis during chronic liver
disease (16). In addition,
reduction in Ang II due to inhibition of angiotensin-converting
enzyme (ACE) prevents oxidative stress and tissue damage (12,17).
For instance, captopril, a well-known ACE inhibitor, is effective
against paracetamol-induced hepatotoxicity (18). Similarly, aliskiren, which directly
inhibits renin by downregulating RAAS, is also shown to promote
antioxidant activity against paracetamol toxicity (19). As such, inhibition of ACE may
potentially prove effective against paracetamol toxicity.
In addition to ACE, the vasoconstrictive response
due to RAAS can also be modulated by neprilysin/neutral
endopeptidase (NEP), an enzyme that degrades natriuretic peptides
(20). Of these peptides, the
concentration of atrial natriuretic peptide (ANP) increases due to
NEP inhibition (21). ANP also
prevents liver damage and mediates hepatoprotective action
(22), as well as promoting
antioxidant activity by enhancing the resistance of hepatocytes
against reactive oxygen species (23). ANP is also known to inhibit the
RAAS pathway (24). Consequently,
an agent able to inhibit both ACE and NEP could provide even
stronger hepatoprotection, which has not been studied or
experimentally demonstrated previously, to the best of the authors'
knowledge.
Omapatrilat, originally proposed as an
antihypertensive agent (25), is a
recently developed drug with effects on the RAAS pathway. Studies
so far have shown that omapatrilat can prevent endothelial
dysfunction (26), provide
cardiovascular and kidney protection and reduce fibrosis (27,28).
In contrast to the aforementioned drugs influencing RAAS,
omapatrilat also inhibits NEP. The increase in natriuretic peptides
due to this NEP inhibition and the decrease in Ang II by ACE
inhibition both have beneficial effects in sepsis, as indicated by
reductions in inflammation and oxidative stress (29-31).
This combined inhibition of ACE and NEP suggests omapatrilat's
hepatoprotective potential against paracetamol-induced toxicity.
The present study aimed to investigate possible effects of
omapatrilat's simultaneous ACE and NEP inhibition on hepatic
pathology and study omapatrilat as a potential therapeutic target
against paracetamol toxicity and acute liver injury for the first
time, to the best of the authors' knowledge.
Materials and methods
Chemicals
Paracetamol, thiopental sodium, N-acetylcysteine and
omapatrilat were purchased from Doğa Ilaç Hammaddeleri Tic. Ltd.
Şti, IE. Ulagay Ilac Sanayii Turk A.S., Hüsnü Arsan İlaçları and
Sigma-Aldrich (Merck KGaA), respectively.
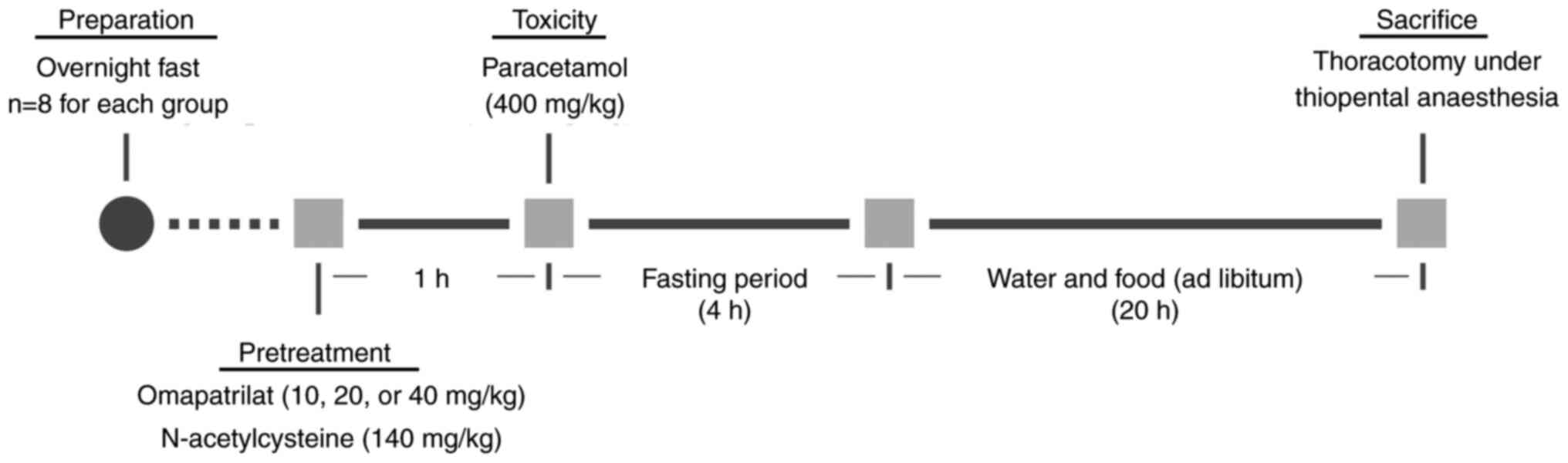
Animals and drug regimen
A total of 56 BALB/c mice aged 14-15 weeks and
weighing 30-35 g were housed in stainless steel cages under
standard conditions (I12-h light/dark cycle; 21±2˚C and 55%
relative humidity) and were given standard pellet feed and water
ad libitum. All animal protocols were approved by
Experimental Animal Ethics Committee of Ataturk University
(approval no. 04.05.2018/108). The mice were fasted overnight,
separated into seven groups (n=8) and administered the following
chemicals according to the schedule depicted in Fig. 1.
Group 1: Control; Group 2 (OMA): Omapatrilat (40
mg/kg); Group 3 (PARA): Paracetamol (400 mg/kg); Group 4 (PARA +
NAC): Paracetamol (400 mg/kg) + NAC (140 mg/kg, 2 doses); Group 5
(PARA + OMA10): Paracetamol (400 mg/kg) + omapatrilat (10 mg/kg),
Group 6 (PARA + OMA20): Paracetamol (400 mg/kg) + omapatrilat (20
mg/kg); Group 7 (PARA + OMA40): Paracetamol (400 mg/kg) +
omapatrilat (40 mg/kg).
All chemicals were administered orally by gastric
gavage. Omapatrilat was administered at 10, 20 and 40 mg/kg doses,
according to previous experimental studies which showed that
omapatrilat can influence oxidative stress parameters at similar
doses (31-33).
For NAC, two doses were administered at 140 mg/kg each, as per its
recommended therapeutic application against paracetamol poisoning
(7,34). After 1 h following pretreatment
(32,34,35)
by omapatrilat or NAC, paracetamol was administered at 400 mg/kg,
the standard dosage used in acute liver injury models (35-37).
To avoid drug-food interactions, overnight fasting was applied
(38,39). The mice were also fasted for 4 h
after paracetamol administration to avoid possible interactions
that could change drug bioavailability between groups. Finally, 24
h after paracetamol was administered, mice were given a 50 mg/kg
dose of thiopental (intraperitoneal) anesthesia. Blood samples were
collected into heparinized bottles by cardiac puncture, as is
recommended for terminal stage of the study to collect a single,
good quality and large volume of blood from the experimental
animals. A total of ~1-2 ml of blood per mouse was collected by
thoracotomy, which caused mortality. Livers were removed
immediately after sacrifice.
Histopathological imaging
Liver tissue samples obtained from six mice per
group were fixed in 10% formalin solution for 48 h at room
temperature (22-24˚C), dehydrated using alcohols with increasing
concentrations and cleared in xylene. In histopathological
analyses, occasionally tissues can be lost during paraffinization
and/or staining procedures. Multiple slides were made from each
tissue, and histopathological damage was scored and analyzed after
the best slides were selected. Samples were embedded in paraffin
and sectioned to 5 µm slices using a Leica RM2235 microtome and
disposable Leica 819 metal blades. The sections were stained with
hematoxylin (5 min) and eosin (2 min) at room temperature (22-24˚C)
and imaged using a light microscope. Images were then evaluated for
the severity of tissue damage by an independent researcher who was
blinded to the treatment groups, using the following scores:
Apoptotic and necrotic cells in 5 different areas in each organ
were counted and scored as 0 if apoptotic and necrotic cells were
absent (0%), 1 for few (0-33%), 2 for moderate (33-66%) and 3 for
more (66-100%). This score for each animal in each group was
evaluated statistically based on the scores obtained from each
slide. Necrotic cells are a form of cell swelling (oncosis) and
burst due to loss of osmotic pressure. Apoptotic cells loose cell
contacts and changes shape. Chromatin condenses in the nucleus and
moves toward the nuclear envelope. Loss of water results in
significant cell shrinkage and blebbing of the plasma membrane with
little or no morphological changes to the other cellular
organelles. The present study defined cells as apoptotic and
necrotic according to these criteria, in the light of previous
literature (7,38).
Biochemical measurements
In biochemical analyses there were seven samples for
Control, eight samples for control + OMA and six samples for the
rest of all PARA groups. The reason why there were different
numbers of serum samples was loss of samples during collection (as
a result of hemolysis). Serum samples were separated by a 10 min
1,800 x g centrifuge at 4˚C within 1 h of collection and were
stored at -86˚C. For hepatic function assessment, alanine
transaminase (ALT) and aspartate transaminase (AST) activities were
characterized using Wuhan USCN Business Co., Ltd. ELISA kits (cat.
nos. E90207Ra and E91214Ra) according to the manufacturer's
instructions. Approximately 75 mg of ground liver tissue was
homogenized in 1 ml of phosphate-buffered saline (PBS) using a
homogenizer (TissueLyser II; Qiagen GmbH) and then centrifuged at
(4˚C) 1,500 x g for 15 min. Total protein concentrations were
measured using the Lowry method (total protein kit; cat. no.
TP0300-1KT; MilliporeSigma). GSH levels were determined using an
Mouse GSH ELISA kit (cat. no. E13068m; Cusabio Technology LLC)
according to the manufacturer's instructions. Superoxide dismutase
(SOD) activity (40) and
malondialdehyde (MDA) levels (41)
were measured manually from the supernatants, according to modified
methods of the ELISA reader as previously described (42). Finally, levels of ACE (cat. no.
E04492m; Cusabio Technology LLC) and NEP were measured (in ng/ml
and pg/ml) using ELISA kits (cat. no. YLA1760MO; Shanghai YL
Biotech Co., Ltd.) with a BioTek Epoch Microplate
Spectrophotometer.
Statistical analyses
Results from biochemical measurements were analyzed
using one-way analysis of variance (ANOVA) and Tukey's multiple
comparison test using SPSS (version 20; IBM) and expressed as mean
± standard deviations. When the CONTROL group was compared with the
other groups, *P<0.05, **P<0.01 and
***P<0.001 marks were used; when the PARA group was
compared with the other groups, &P<0.05,
&&P<0.01 and
&&&P<0.001 symbols were used and when the
PARA + OMA groups are compared within themselves,
#P<0.05, ##P<0.01 and
###P<0.001 marks were used.
Results from histopathological scoring were analyzed
using Kruskal-Wallis followed by Dunn's test using SPSS (version
20; IBM), expressed as minimum to maximum. When the PARA group was
compared with the other groups, &P<0.05,
&&P<0.01 and
&&&P<0.001 symbols were used.
P<0.05 was considered to indicate a statistically
significant difference.
Results
With omapatrilat administration in the OMA40 group
(group 2), no adverse effects that would have prompted the
discontinuation of this study were observed (such as >15% weight
loss and/or the loss of the ability to walk or properly consume
food/water).
Histopathological results
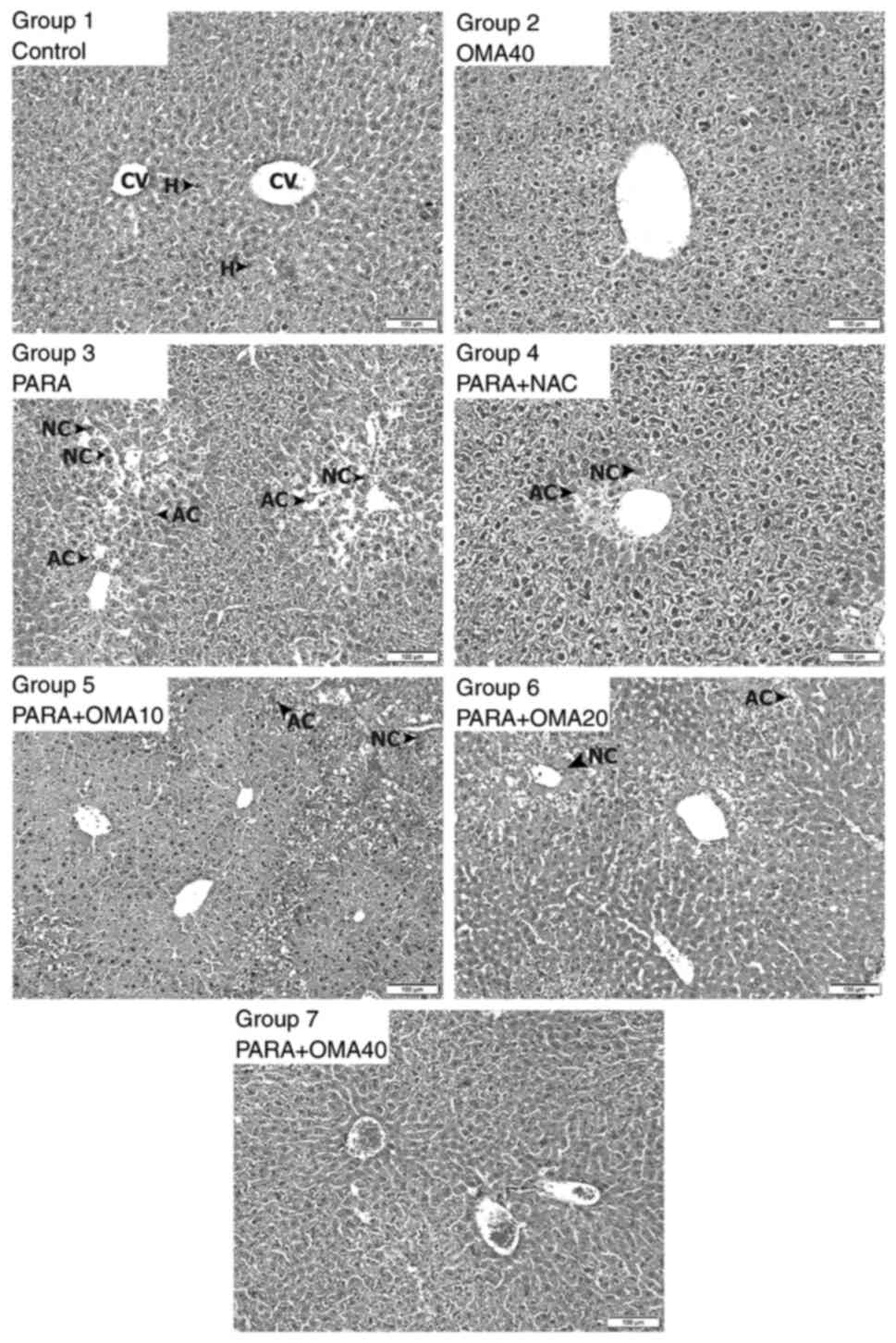
Samples of micrographs of the liver slices are shown
in Fig. 2. There were no visible
differences between the tissues extracted from mice that belonged
to the same experimental group. All hepatic lobules in the Control
group (group 1) presented normal size and morphology with healthy
portal triads and central veins. No histopathological anomalies
were observed for the hepatocytes and sinusoids in this group.
Images from the OMA40 group (group 2) also showed similar portal
triads, central veins, hepatocytes and sinusoids as the Control
group, with no histopathological findings. By contrast, significant
damage was observed in the lobules of the PARA group (group 3). A
number of apoptotic cells with pyknotic nuclei and necrotic cells
with abundant eosinophilic cytoplasm were recorded among the
hepatocytes surrounding the portal vein. With the standard NAC
treatment, a smaller number of apoptotic and necrotic cells were
observed, despite some still remaining near the central vein.
Compared to the PARA group, samples from the PARA + OMA10 group (10
mg/kg omapatrilat; group 5) showed fewer apoptotic and necrotic
cells. This group exhibited sinusoidal dilatation and erythrocyte
infiltration around the portal area. With 20 mg/kg omapatrilat
(PARA + OMA20; group 6), significant differences from the PARA
group were observed. Some eosinophilic cells were observed around
the central vein, similar to the results from the PARA + NAC group
(group 4). Finally, PARA + OMA40 group micrographs (40 mg/kg
omapatrilat; group 7) showed no histopathological anomalies, with
hepatocytes exhibiting almost identical appearance to those in the
Control group. These assessments are summarized with comparative
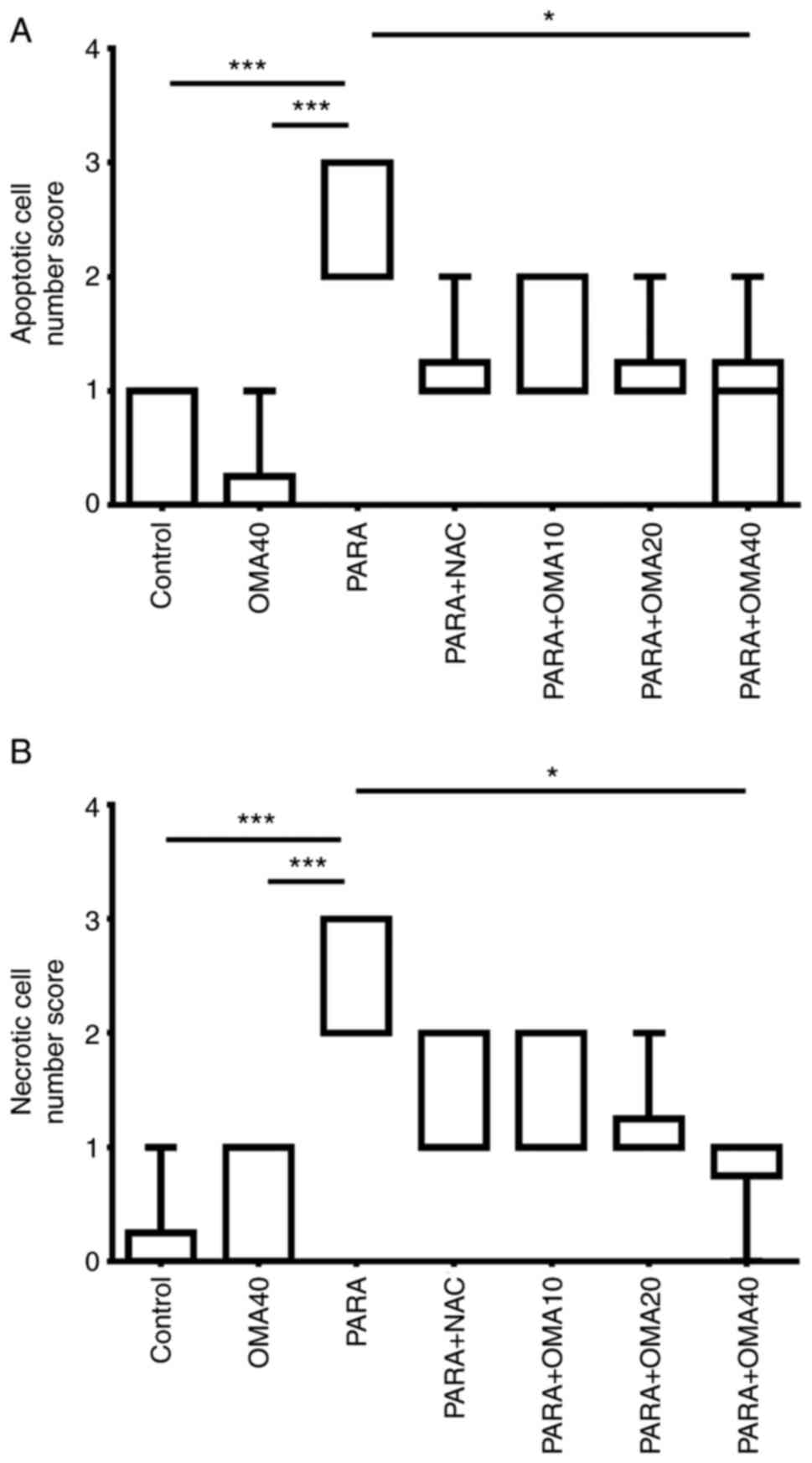
scores in Fig. 3 with relative
indications of damage severity due to apoptotic and necrotic cells.
From the histopathological scoring results it was determined that
PARA group had the highest number of necrotic and apoptotic cells.
A 40 mg/kg dose of OMA decreased these cell scores significantly.
NAC and OMA20 groups has decreased number of apoptotic cells when
compared to PARA group; however these comparisons were borderline
significant (P=0.0501 for PARA + NAC and PARA + OMA20 groups when
compared to PARA according to Dunn's test).
Biochemical results
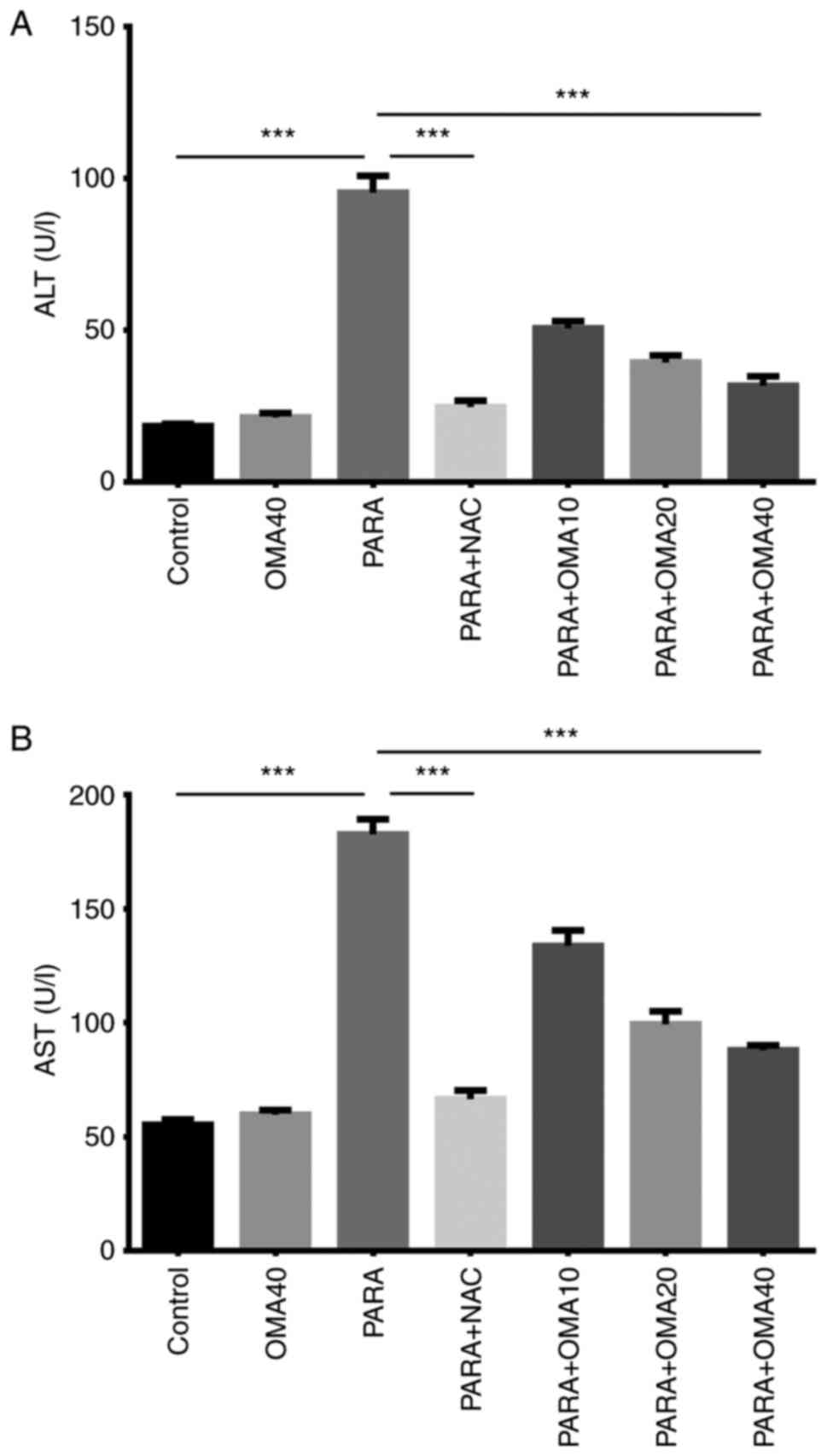
According the liver function tests, significant
increases in the serum activities of ALT and AST were observed in
the PARA group compared with the Control and OMA40 groups, as shown
in Fig. 4. The specific treatment
was found to reduce ALT and AST activities in the PARA + NAC group,
bringing them closer to those observed in the Control group. In
groups pretreated with omapatrilat, it was seen that increasing
omapatrilat dosage gradually reduced the ALT and AST activities,
with the PARA + OMA40 group being the closest to the Control group.
Comparing PARA + NAC and PARA + OMA administered groups, it was
determined that 40 mg/kg dose of omapatrilat showed the closest
results to the NAC-treated group. However, PARA + NAC administered
group still had significantly lower AST and ALT levels than those
in the PARA + OMA40 group.
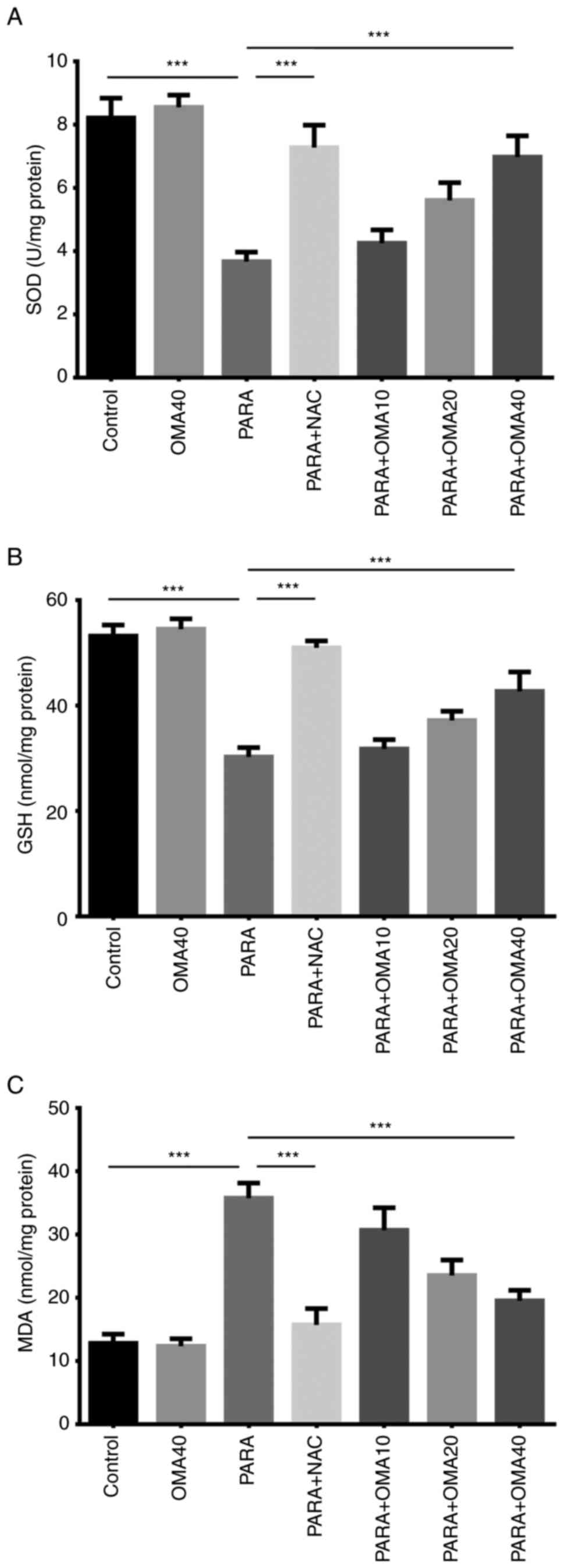
As shown in Fig. 5,
SOD activity and GSH levels in control and OMA40 groups were also
statistically similar. Administration of paracetamol significantly
reduced the SOD activity and the GSH level due to toxicity. This
reduction was reversed with NAC pretreatment in the PARA + NAC
group. Of the groups pretreated with omapatrilat, with increasing
omapatrilat dosage from 10 to 40 mg/kg, it was seen that SOD
activity and GSH level increased towards that measured in the
control group, with the PARA + OMA40 group exhibiting the closest
SOD and GSH levels to the control and OMA40 groups. Measurements of
the oxidative stress biomarker MDA yielded similar results. Due to
toxicity, introduction of paracetamol significantly increased the
MDA level that was lower in the Control and OMA40 groups. Specific
pretreatment in the PARA + NAC group also yielded results similar
to the Control group. As with SOD and GSH, of the three groups
pretreated with omapatrilat, PARA + OMA40 showed the closest
results to the Control group, with increasing omapatrilat dosage
gradually reversing the increase in the MDA level. Comparing the
oxidative stress parameters and antioxidant status of PARA + NAC
and PARA + OMA administered groups, it was determined that 40 mg/kg
dose of omapatrilat was closest to the NAC-treated group. There was
no significant difference between SOD and MDA levels in the PARA +
NAC and PARA + OMA40 groups. Moreover, in comparison to the PARA
group, the PARA + NAC group had increased GSH levels matching that
of healthy mice. The next highest GSH levels were measured in the
PARA + OMA40 group, below the control and PARA + NAC groups.
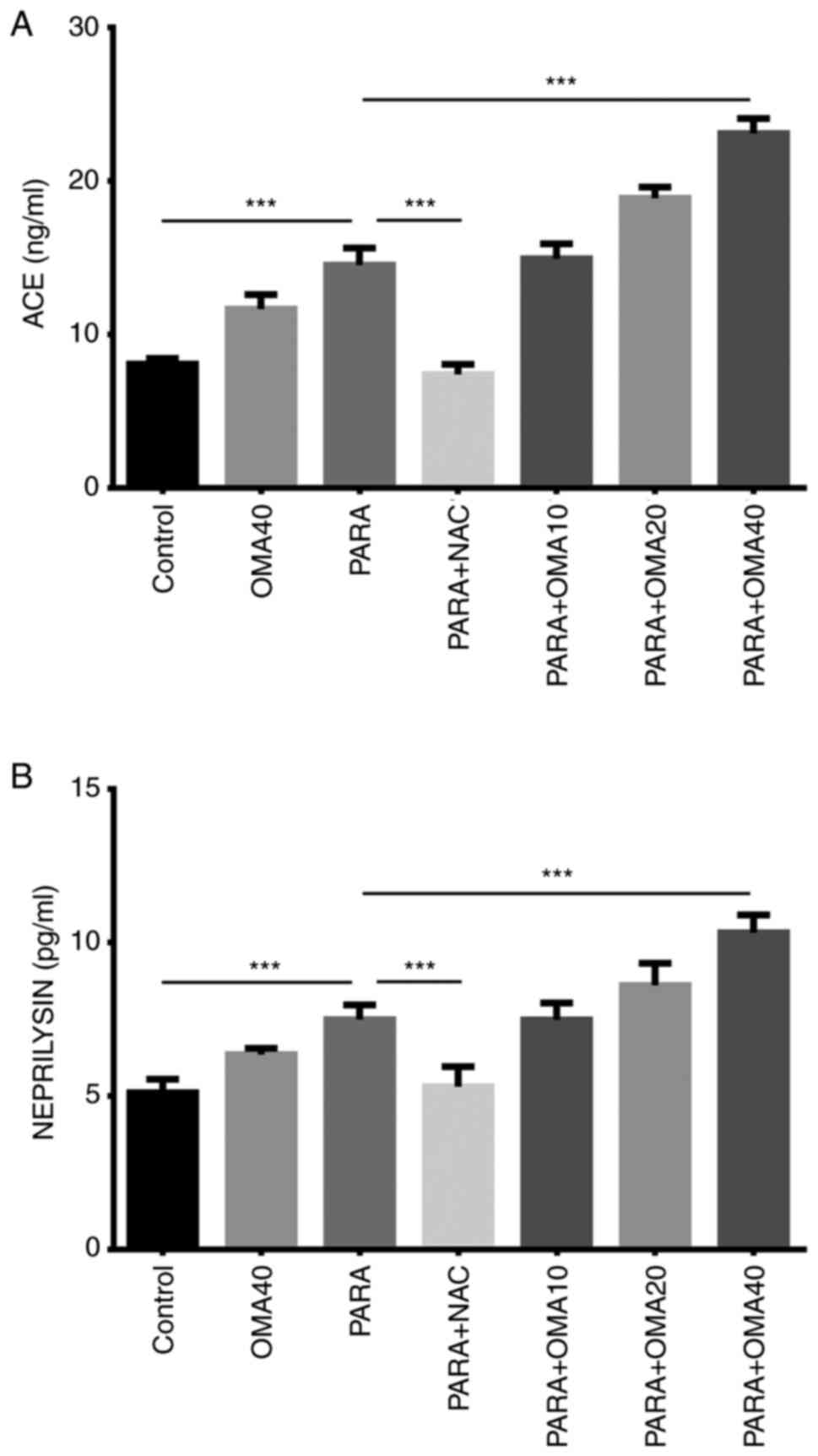
ACE and NEP levels shown in Fig. 6 were statistically higher in the
PARA group than the Control and were both reduced by the standard
treatment in the PARA + NAC group. In comparison to the control
group, statistical increases in both ACE and NEP activities were
observed with increasing omapatrilat dosage. All doses of
omapatrilat resulted in significantly higher levels of ACE and NEP
when compared with the PARA + NAC group.
Discussion
The present study investigated the protective
effects of omapatrilat against paracetamol-induced hepatotoxicity.
It was found that omapatrilat suppressed the acute liver damage
caused by paracetamol, as evidenced by the histopathological
results and the measurements of biochemical indicators. The
histopathological results showed that in the case of specific
treatment by NAC, no damaged cells were found, whereas the samples
from the paracetamol toxicity group contained numerous apoptotic
and necrotic cells. In groups pretreated with omapatrilat,
histopathological results approached those of the Control group
with increasing omapatrilat dosage. At 40 mg/kg of omapatrilat
dosage, the tissue samples closely resembled the Control group and
no histopathological findings were recorded. Also in the
histopathological scoring results, the highest necrotic and
apoptotic cell scores were determined in PARA groups. When PARA +
NAC and PARA + OMA40 groups were evaluated, it demonstrated
significant decrease in apoptotic and necrotic cell ratios. These
findings were supported with biochemical analyses in which AST and
ALT, serum biomarkers of liver function, were measured.
ALT and AST are enzymes synthesized in hepatocytes
and are used in the evaluation of hepatocellular damage and are
sensitive markers for the diagnosis of liver diseases. Previous
studies have demonstrated significant changes in ALT and AST
activities due to oxidative stress and liver damage resulting from
the intake of toxic doses of paracetamol. For instance, paracetamol
poisoning was shown to increase ALT and AST activities in rabbits
(43). Another study on the
effects of ACE inhibitor enalapril on paracetamol-induced liver
damage has found significantly reduced ALT and AST activities in
mice treated with enalapril (44).
The present study investigated the effects of another ACE
inhibitor, omapatrilat, on ALT and AST activities. The results also
indicated that paracetamol administration results in much higher
ALT and AST activities due to liver damage. The reduction of these
enzymes with omapatrilat pretreatment towards those measured in the
Control group is the primary evidence of omapatrilat's
hepatoprotective activity.
Oxidative stress occurs because of increased free
radical production, weakening antioxidant defense and a shift in
the balance between oxidants and antioxidants in favor of oxidants.
Oxidative stress and its biomarker MDA are also associated with
paracetamol-induced liver damage (7). Paracetamol toxicity promotes reactive
oxygen species production and initiates lipid peroxidation which
causes tissue damage by affecting membrane structure and cell
contents (45). As MDA is one of
the end products of lipid peroxidation, elevated MDA levels in the
paracetamol toxicity group of the present study indicated oxidative
stress-induced liver damage. As such, the reduction in the MDA
levels for the omapatrilat groups demonstrated reduced oxidative
stress and a strong hepatoprotective response by omapatrilat.
Glutathione (GSH), a crucial antioxidant, protects
cells against oxidative damage by reacting with free radicals and
peroxides (46). It is known that
the amount of GSH significantly drops with paracetamol overdose and
that N-acetylcysteine is the standard treatment for toxicity due to
this glutathione depletion (47).
This drop in GSH levels was also confirmed in the PARA group in the
present study. Its measurements revealed that, with increasing
omapatrilat dosage from 10 to 40 mg/kg, GSH content significantly
increased. According to these results, it can be concluded that the
increased amount of GSH prevented liver damage due to oxidative
stress and renders the toxic metabolite NAPQI harmless.
SOD measurements also confirmed these findings and
proved the effectivity of omapatrilat against paracetamol toxicity.
As SOD is an antioxidant enzyme, similar to the GSH measurements,
SOD activity was also found to significantly decrease with
paracetamol toxicity. As with GSH, increasing omapatrilat dosage
also increased the SOD activity, with the PARA + OMA40 group
approaching the Control group the closest. It has previously been
shown that antioxidant activity in thiol-carrying antioxidants is
directly influenced by the specific number of thiol groups and the
oxidation state of sulfur atoms in the antioxidant molecule
(48). Therefore, in addition to
the antioxidant increase observed with the MDA, GSH and SOD
results, the existence of a thiol group in omapatrilat may also
contribute to the reduction of oxidative stress by promoting
antioxidant activity.
Previous studies have found drugs effecting RAAS
such as enalapril and aliskiren successful in increasing
antioxidant levels and preventing liver damage due to oxidative
stress (19,44). Of these drugs, omapatrilat, a more
recently developed RAAS-acting agent, is the first one that is
known to inhibit both ACE and NEP enzymes simultaneously. As such,
the present study investigated the possible effects of ACE/NEP
pathway on acute liver injury due to paracetamol toxicity and noted
significant increases in the activities of both enzymes in the PARA
group. This increase in ACE and NEP activities can be interpreted
as a defense mechanism accompanying liver damage. Similar increases
in ACE activity are also reported in case of acute pancreatitis in
which the activation of local RAAS components in peripheral tissues
are interpreted as differential mechanisms for regulation of
physiological and pathophysiological functions (49). The potential pathophysiological
role of increased ACE activity in acute coronary syndrome has also
been previously shown (50). These
earlier studies support our results that ACE activity can be
influenced by acute phenomena. Moreover, the continued
dose-dependent increase of ACE and NEP enzymes in omapatrilat
groups may indicate deterioration of the negative feedback
mechanism controlled by Ang II. These findings reveal the role of
ACE and NEP enzymes and the preventive effects of omapatrilat in
paracetamol toxicity.
There were limitations to the present study: i)
Administration of omapatrilat before paracetamol demonstrated its
protective effect, rather than its curative effect, on
paracetamol-induced hepatotoxicity; ii) Lack of investigation on
the potential combined effects between omapatrilat and NAC before
and after paracetamol toxicity; and iii) the lack of molecular
assays (immunofluorescence or western blotting) for the assessment
of both ROS signaling and ACE-NEP pathway.
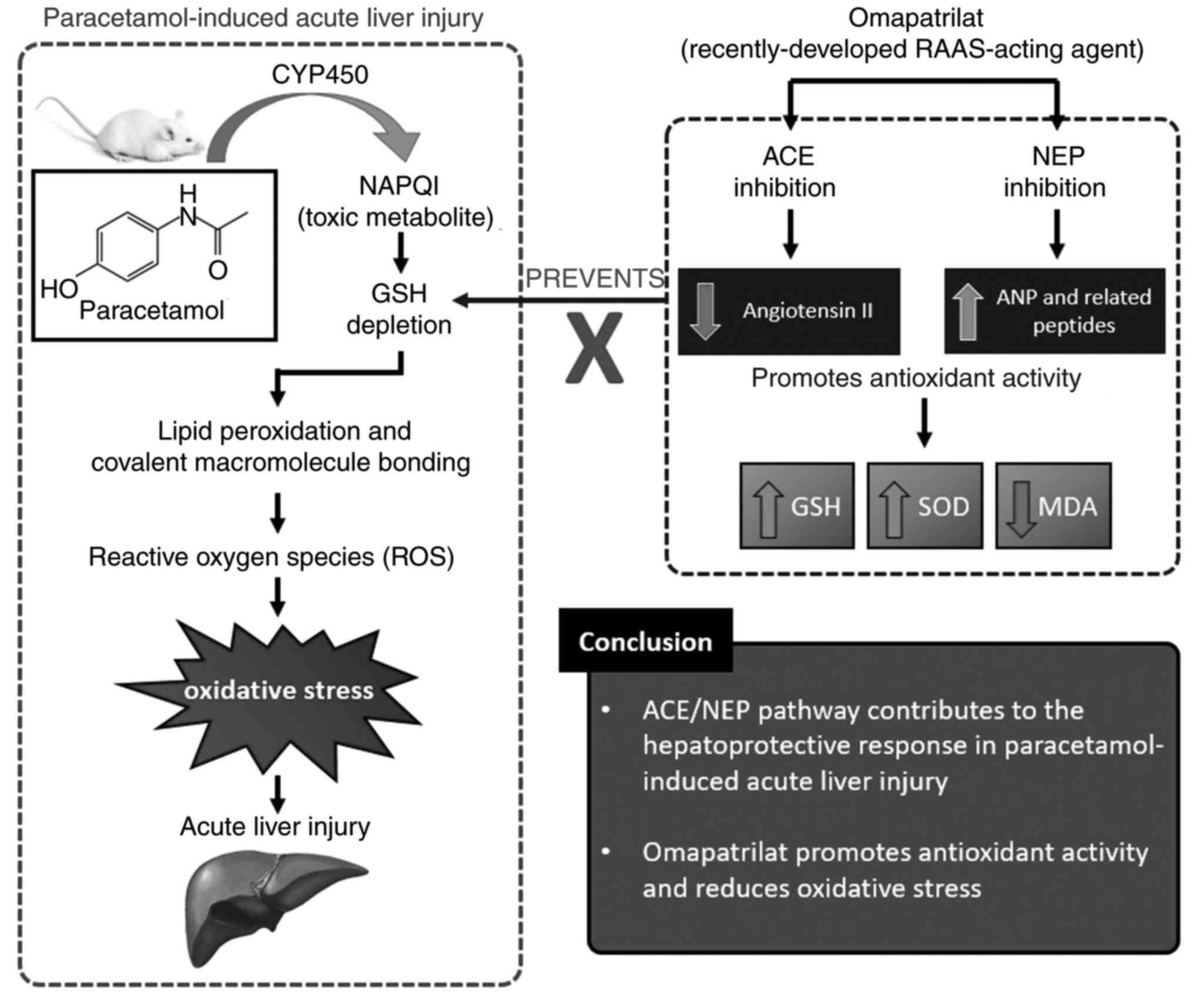
In summary, the results indicated that omapatrilat
can be an effective hepatoprotective drug for paracetamol-induced
hepatotoxicity. It showed that toxicity-induced mice pretreated
with 40 mg/kg omapatrilat exhibited histopathological and
biochemical results similar to the control group. It also showed
that highest dose of omapatrilat (40 mg/kg) resulted in similar
protective effects as NAC. Omapatrilat, acting through the ACE/NEP
pathway, demonstrated beneficial effects by correcting the
antioxidant parameters and the GSH depletion resulting from
paracetamol toxicity. Additionally, it was found that omapatrilat
increased the antioxidant SOD activity and reduced the MDA oxidant
levels by suppressing the oxidative stress associated with acute
liver injury. Moreover, increase of ACE and NEP enzymes in
omapatrilat groups may indicate deterioration of the negative
feedback mechanism controlled by Ang II (Fig. 7). These measurements indicated the
potential hepatoprotective effects of the ACE/NEP pathway on
physiopathology of paracetamol toxicity. The aim of the present
study our was not directly to suggest omapatrilat as a first aid
agent after paracetamol toxicity, but to investigate possible
contribution of ACE/NEP pathway during paracetamol toxicity. The
present study cannot conclude that omapatrilat as an antidote but
it shed light for future studies on omapatrilat and also other new
agents targeting the ACE/NEP pathway.
Acknowledgements
The abstract was presented at the 6th International
Gevher Nesibe Health Sciences Conference Nov 13-15 2020 in Ankara
with title ‘The effects of omapatrilat on paracetamol-induced acute
liver failure in mice’ and published in ‘Proceedings Book of
International Gevher Nesibe Health Sciences Conference-VI’,
p244-245: 2020.
Funding
Funding: The present study was supported by Ataturk University
Scientific Research Projects Coordination (grant no.
TDK-2018-6840).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZBAM, ZH and EC conceived the study and designed the
experiments. ZBAM, ZH and RAU conducted the animal experiments
under the supervision of EC. ET performed the histopathological
imaging and analysis. ZBAM analyzed the data and wrote the
manuscript with revisions from all authors. ZH and EC supervised
the study. ZBAM, ET and RAU confirm the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All animal protocols were approved by Experimental
Animal Ethics Committee of Ataturk University (approval no.
04.05.2018/108).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Penna A and Buchanan N: Paracetamol
poisoning in children and hepatotoxicity. Br J Clin Pharmacol.
32:143–149. 1991.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kurtovic J and Riordan S:
Paracetamol-induced hepatotoxicity at recommended dosage. J Intern
Med. 253:240–243. 2003.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Morthorst BR, Erlangsen A, Nordentoft M,
Hawton K, Hoegberg LCG and Dalhoff KP: Availability of paracetamol
sold over the counter in Europe: A descriptive cross-sectional
international survey of pack size restriction. Basic Clin Pharmacol
Toxicol. 122:643–649. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Steventon GB, Mitchell SC and Waring RH:
Human metabolism of paracetamol (acetaminophen) at different dose
levels. Drug Metabol Drug Interact. 13:111–117. 1996.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Jollow DJ, Mitchell JR, Potter WZ, Davis
DC, Gillette JR and Brodie BB: Acetaminophen-induced hepatic
necrosis. II. Role of covalent binding in vivo. J Pharmacol Exp
Ther. 187:195–202. 1973.PubMed/NCBI
|
|
6
|
Nilsson JLÅ, Blomgren A, Nilsson UJ,
Högestätt ED and Grundemar L:
N,N'-Bis(2-mercaptoethyl)isophthalamide binds electrophilic
paracetamol metabolites and prevents paracetamol-induced liver
toxicity. Basic Clin Pharmacol Toxicol. 123:589–593.
2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yayla M, Halici Z, Unal B, Bayir Y,
Akpinar E and Gocer F: Protective effect of Et-1 receptor
antagonist bosentan on paracetamol induced acute liver toxicity in
rats. Eur J Pharmacol. 726:87–95. 2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
El-Demerdash E, Salam OM, El-Batran SA,
Abdallah HM and Shaffie NM: Inhibition of the renin-angiotensin
system attenuates the development of liver fibrosis and oxidative
stress in rats. Clin Exp Pharmacol Physiol. 35:159–167.
2008.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Stokkeland K, Lageborn CT, Ekbom A, Höijer
J, Bottai M, Stål P and Söderberg-Löfdal K: Statins and
angiotensin-converting enzyme inhibitors are associated with
reduced mortality and morbidity in chronic liver disease. Basic
Clin Pharmacol Toxicol. 122:104–110. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Aroor AR, Demarco VG, Jia G, Sun Z,
Nistala R, Meininger GA and Sowers JR: The role of tissue
renin-angiotensin-aldosterone system in the development of
endothelial dysfunction and arterial stiffness. Front Endocrinol
(Lausanne). 4(161)2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Bataller R, Gäbele E, Schoonhoven R,
Morris T, Lehnert M, Yang L, Brenner DA and Rippe RA: Prolonged
infusion of angiotensin II into normal rats induces stellate cell
activation and proinflammatory events in liver. Am J Physiol.
285:G642–G651. 2003.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Bataller R, Gäbele E, Parsons CJ, Morris
T, Yang L, Schoonhoven R, Brenner DA and Rippe RA: Systemic
infusion of angiotensin II exacerbates liver fibrosis in bile
duct-ligated rats. Hepatology. 41:1046–1055. 2005.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Friedman SL: Mechanisms of hepatic
fibrogenesis. Gastroenterology. 134:1655–1669. 2008.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Mollnau H, Wendt M, Szöcs K, Lassègue B,
Schulz E, Oelze M, Li H, Bodenschatz M, August M, Kleschyov AL, et
al: Effects of angiotensin II infusion on the expression and
function of NAD(P)H oxidase and components of nitric oxide/cGMP
signaling. Circ Res. 90:E58–E65. 2002.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Landmesser U, Spiekermann S, Preuss C,
Sorrentino S, Fischer D, Manes C, Mueller M and Drexler H:
Angiotensin II induces endothelial xanthine oxidase activation:
Role for endothelial dysfunction in patients with coronary disease.
Arterioscler Thromb Vasc Biol. 27:943–948. 2007.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Bataller R, Ginès P, Nicolás JM, Görbig
MN, Garcia-Ramallo E, Gasull X, Bosch J, Arroyo V and Rodés J:
Angiotensin II induces contraction and proliferation of human
hepatic stellate cells. Gastroenterology. 118:1149–1156.
2000.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Hitomi H, Kiyomoto H and Nishiyama A:
Angiotensin II and oxidative stress. Curr Opin Cardiol. 22:311–315.
2007.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yeung JH: Effect of sulphydryl drugs on
paracetamol-induced hepatotoxicity in mice. Drug Metabol Drug
Interact. 6:295–301. 1988.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Karcioglu SS, Palabiyik SS, Bayir Y,
Karakus E, Mercantepe T, Halici Z and Albayrak A: The role of RAAS
inhibition by aliskiren on paracetamol-induced hepatotoxicity model
in rats. J Cell Biochem. 117:638–646. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
McDowell G, Coutie W, Shaw C, Buchanan KD,
Struthers AD and Nicholls DP: The effect of the neutral
endopeptidase inhibitor drug, candoxatril, on circulating levels of
two of the most potent vasoactive peptides. Br J Clin Pharmacol.
43:329–332. 1997.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Mangiafico S, Costello-Boerrigter LC,
Andersen IA, Cataliotti A and Burnett JC Jr: Neutral endopeptidase
inhibition and the natriuretic peptide system: An evolving strategy
in cardiovascular therapeutics. Eur Heart J. 34:886–893c.
2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Gerwig T, Meissner H, Bilzer M, Kiemer AK,
Arnholdt H, Vollmar AM and Gerbes AL: Atrial natriuretic peptide
preconditioning protects against hepatic preservation injury by
attenuating necrotic and apoptotic cell death. J Hepatol.
39:341–348. 2003.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Kiemer AK, Gerbes AL, Bilzer M and Vollmar
AM: The atrial natriuretic peptide and cGMP: Novel activators of
the heat shock response in rat livers. Hepatology. 35:88–94.
2002.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Sekino M, Makita T, Ureshino H, Sungsam C
and Sumikawa K: Synthetic atrial natriuretic peptide improves
systemic and splanchnic circulation and has a lung-protective
effect during endotoxemia in pigs. Anesth Analg. 110:141–147.
2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Kostis JB, Packer M, Black HR, Schmieder
R, Henry D and Levy E: Omapatrilat and enalapril in patients with
hypertension: The omapatrilat cardiovascular treatment vs enalapril
(OCTAVE) trial. Am J Hypertens. 17:103–111. 2004.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Quaschning T, d'Uscio LV, Shaw S and
Lüscher TF: Vasopeptidase inhibition exhibits endothelial
protection in salt-induced hypertension. Hypertension.
37:1108–1113. 2001.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Dong Y, Zhou H, Shaffer E, Atamas N, Liao
WC and Wei C: The cardiovascular actions of omapatrilat in
spontaneously hypertensive rats. Curr Hypertens Rep. 3 (Suppl
2):S1–S5. 2001.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Taal MW, Nenov VD, Wong W, Satyal SR,
Sakharova O, Choi JH, Troy JL and Brenner BM: Vasopeptidase
inhibition affords greater renoprotection than
angiotensin-converting enzyme inhibition alone. J Am Soc Nephrol.
12:2051–2059. 2001.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Song Z, Cui Y, Ding MZ, Jin HX and Gao Y:
Protective effects of recombinant human brain natriuretic peptide
against LPS-Induced acute lung injury in dogs. Int Immunopharmacol.
17:508–512. 2013.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Kostakoglu U, Topcu A, Atak M, Tumkaya L,
Mercantepe T and Uydu HA: The protective effects of
angiotensin-converting enzyme inhibitor against cecal ligation and
puncture-induced sepsis via oxidative stress and inflammation. Life
Sci. 241(117051)2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ugan RA, Un H, Gurbuz MA, Kaya G,
Kahramanlar A, Aksakalli-Magden ZB, Halici Z and Cadirci E:
Possible contribution of the neprilysin/ACE pathway to sepsis in
mice. Life Sci. 258(118177)2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Hayek T, Hamoud S, Keidar S, Pavlotzky E,
Coleman R, Aviram M and Kaplan M: Omapatrilat decreased macrophage
oxidative status and atherosclerosis progression in atherosclerotic
apolipoprotein E-deficient mice. J Cardiovasc Pharmacol.
43:140–147. 2004.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Lapointe N, Nguyen QT, Desjardins JF,
Marcotte F, Pourdjabbar A, Moe G, Calderone A and Rouleau JL:
Effects of pre-, peri-, and postmyocardial infarction treatment
with omapatrilat in rats: Survival, arrhythmias, ventricular
function, and remodeling. Am J Physiol Heart Circ Physiol.
285:H398–H405. 2003.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Canayakin D, Bayir Y, Baygutalp NK,
Karaoglan ES, Atmaca HT, Ozgeris FBK, Keles MS and Halici Z:
Paracetamol-induced nephrotoxicity and oxidative stress in rats:
The protective role of Nigella sativa. Pharm Biol. 54:2082–2091.
2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Randle LE, Sathish JG, Kitteringham NR,
Macdonald I, Williams DP and Park BK: alpha(1)-Adrenoceptor
antagonists prevent paracetamol-induced hepatotoxicity in mice. Br
J Pharmacol. 153:820–830. 2008.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Patterson AD, Shah YM, Matsubara T, Krausz
KW and Gonzalez FJ: Peroxisome proliferator-activated receptor
alpha induction of uncoupling protein 2 protects against
acetaminophen-induced liver toxicity. Hepatology. 56:281–290.
2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Scialis RJ, Ghanem CI and Manautou JE: The
modulation of transcriptional expression and inhibition of
multidrug resistance associated protein 4 (MRP4) by analgesics and
their primary metabolites. Curr Res Toxicol. 1:34–41.
2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Palabiyik SS, Karakus E, Akpinar E, Halici
Z, Bayir Y, Yayla M and Kose D: The role of urotensin receptors in
the paracetamol-induced hepatotoxicity model in mice: Ameliorative
potential of urotensin II antagonist. Basic Clin Pharmacol Toxicol.
118:150–159. 2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Ferah I, Halici Z, Bayir Y, Demirci E,
Unal B and Cadirci E: The role of infliximab on paracetamol-induced
hepatotoxicity in rats. Immunopharmacol Immunotoxicol. 35:373–381.
2013.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Sun Y, Oberley LW and Li Y: A simple
method for clinical assay of superoxide dismutase. Clin Chem.
34:497–500. 1988.PubMed/NCBI
|
|
41
|
Ohkawa H, Ohishi N and Yagi K: Assay for
lipid peroxides in animal tissues by thiobarbituric acid reaction.
Anal Biochem. 95:351–358. 1979.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Ugan RA, Cadirci E, Halici Z, Toktay E and
Cinar I: The role of urotensin-II and its receptors in
sepsis-induced lung injury under diabetic conditions. Eur J
Pharmacol. 818:457–469. 2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Cigremis Y, Turel H, Adiguzel K, Akgoz M,
Kart A, Karaman M and Ozen H: The effects of acute acetaminophen
toxicity on hepatic mRNA expression of SOD, CAT, GSH-Px, and levels
of peroxynitrite, nitric oxide, reduced glutathione, and
malondialdehyde in rabbit. Mol Cell Biochem. 323:31–38.
2009.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Betto MRB, Lazarotto LF, Watanabe TT,
Driemeier D, Leite CE and Campos MM: Effects of treatment with
enalapril on hepatotoxicity induced by acetaminophen in mice.
Naunyn Schmiedebergs Arch Pharmacol. 385:933–943. 2012.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Bessems JG and Vermeulen NP: Paracetamol
(acetaminophen)-induced toxicity: Molecular and biochemical
mechanisms, analogues and protective approaches. Crit Rev Toxicol.
31:55–138. 2001.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Karatas E, Bayraktutan Z and Cadirci E:
Investigation of the effects of amlodipine on paracetamol-induced
acute kidney toxicity in rats. Clin Exp Health Sci. 12:155–161.
2022.
|
|
47
|
Kalsi SS, Dargan PI, Waring WS and Wood
DM: A review of the evidence concerning hepatic glutathione
depletion and susceptibility to hepatotoxicity after paracetamol
overdose. Open Access Emerg Med. 3:87–96. 2011.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Castañeda-Arriaga R, Pérez-González A
and Galano A: Chemical protectors against the toxic effects of
paracetamol (acetaminophen) and its meta analogue: Preventing
protein arylation. ACS Omega. 3:18582–18591. 2018.
|
|
49
|
Ip SP, Kwan PC, Williams CH, Pang S,
Hooper NM and Leung PS: Changes of angiotensin-converting enzyme
activity in the pancreas of chronic hypoxia and acute pancreatitis.
Int J Biochem Cell Biol. 35:944–954. 2003.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Hoshida S, Kato J, Nishino M, Egami Y,
Takeda T, Kawabata M, Tanouchi J, Yamada Y and Kamada T: Increased
angiotensin-converting enzyme activity in coronary artery specimens
from patients with acute coronary syndrome. Circulation.
103:630–633. 2001.PubMed/NCBI View Article : Google Scholar
|