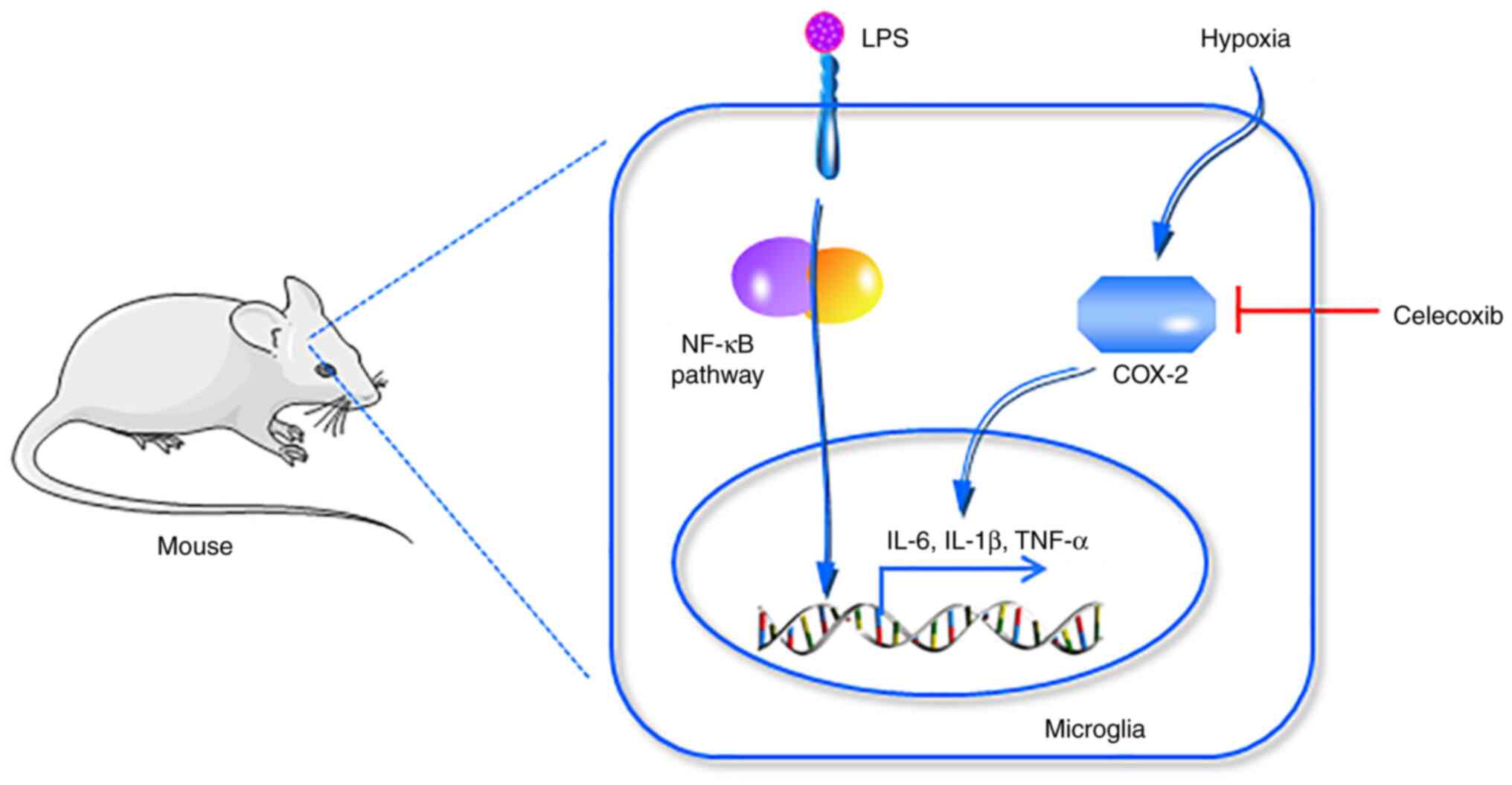
Introduction
Neuroinflammation is involved in the pathophysiology
of several neurological disorders, including stroke, Alzheimer's
disease, Parkinson's disease, amyotrophic lateral sclerosis,
epilepsy, traumatic brain injury and depression (1-3).
During hypoxic-ischemic brain injuries, neuroinflammation can occur
just minutes after damage (4,5). A
number of studies report that hypoxia can aggravate the development
of neuroinflammation. Song et al revealed that pretreatment
with lipopolysaccharide (LPS) followed by hypoxia exposure markedly
increases TNF-α and IL-1β levels compared with treatment of LPS
alone in the plasma and brain cortex of rats (6). Another study previously reported
that, compared with LPS alone, pretreatment with LPS followed by
hypobaric hypoxia increases the development of neuroinflammation in
a mouse model (7). These results
indicate that hypoxia can aggravate LPS-induced neuroinflammation.
However, the underlying mechanisms remain unclear.
Under hypoxic conditions, the activation of the
hypoxia inducible factor (HIF) pathway is considered to be involved
in the inflammatory process (8,9). The
relationship between HIF-1 target genes that are activated by
hypoxia and neuroinflammation has attracted considerable attention
from researchers. Some studies suggest that hypoxia activates
cyclooxygenase-2 (COX-2) expression in a HIF-1α-dependent manner,
as a functional hypoxia response element has been identified in the
COX-2 promoter sequence (10-12).
Moreover, COX-2 is a key mediator of the inflammatory process
(13). In response to growth
factors, cytokines and pro-inflammatory molecules, COX-2 is rapidly
expressed at mRNA levels and has emerged as the isoform (COX-1 is
considered to be constitutively expressed) responsible for
prostanoid production in acute and chronic inflammatory conditions.
The selective inhibition of COX-2 may relieve the symptoms of
inflammatory diseases (14). COX-2
inhibition has been reported to suppress the upregulation of IL-6,
both at the mRNA and protein expression levels in BV2 cells. At the
same time, the upregulation of IL-1β, TNF-α and monocyte
chemoattractant protein-1 is also blocked (15). These results confirmed that
inhibition of COX-2 could suppress neuroinflammation response.
Whether inducible COX-2 plays an important role in the
hypoxia-induced aggravation of neuroinflammation needs to be
determined.
In the present study, the role of COX-2 in
hypoxia-induced aggravation of neuroinflammation was investigated
using both in vitro (microglial BV2 cells) and in
vivo (C57BL/6 mice) models of neuroinflammation induced by LPS
under hypoxic conditions. In addition, the current study
demonstrated that celecoxib, a COX-2 inhibitor, attenuated
neuroinflammation both in vivo and in vitro. The
present results suggested COX-2 is a promising therapeutic target
in future strategies for the treatment of neuroinflammatory
diseases.
Materials and methods
Cell culture
Mouse microglial BV2 cells (Cell Resource Center of
the Chinese Academy of Medical Sciences) were cultured in DMEM
(Gibco; Thermo Fisher Scientific, Inc.) with 10% FBS (Beijing
Aoqing Biotechnology Co., Ltd.) and 1% penicillin/streptomycin at
37˚C in a humidified atmosphere with 5% CO2 and the
medium was changed every 2 days. Cells were split with 0.125%
trypsin when they reached 80% confluence and the passages 2-10 were
used for carrying out experiments. For LPS treatment, the cells
were treated with 100 ng/ml LPS (cat. no. L2630; Sigma-Aldrich;
Merck KGaA) for the indicated times. To perform the hypoxia
exposure experiments, BV2 cells seeded in 60-mm dishes
(8x105 cells/dish) were put in the cell culture chamber
with 1 or 3% oxygen (O2) at 37˚C for the indicated
times. For the FG-4592 (Selleck Chemicals) treatment, cells were
treated with 10 µM FG-4592 for 6, 12 and 24 h at 37˚C. For the
celecoxib (Selleck Chemicals) treatment, cells were pretreated with
10 and 20 µM celecoxib for 1 h at 37˚C.
Animal treatment
C57BL/6 mice (8-week-old; male; 18±2 g) were
purchased from the Laboratory Animal Center of Vital River
Experimental Animal Company. Mice were housed under a 12-h
light/dark cycle at 22±2˚C with humidity 50±5% and with free access
to standard rodent chow and water. The mice were maintained under
specific pathogen-free conditions. All animal experimental
procedures fully complied with the related laboratory animal
regulations. A total of 16 mice were equally divided into four
groups: i) Normoxia; ii) celecoxib; iii) LPS + hypoxia; and iv)
celecoxib/LPS + hypoxia). The mice were intraperitoneally injected
with celecoxib (20 mg/kg). After 30 min, mice were
intraperitoneally injected with LPS (0.5 mg/kg). Subsequently, the
mice were placed for 6 h in a hypobaric hypoxia chamber mimicking
6,000 m of altitude. After the treatment, the mice were
anesthetized with sodium pentobarbital (50 mg/kg) via
intraperitoneal injection, followed by cardiac perfusion with
prechilled saline solution (0.9%) for 2 min. Thereafter, tissue
samples were collected.
Western blotting
BV2 cells were collected and homogenized using RIPA
lysis buffer (50 mM Tris HCl, 150 mM NaCl, 1% NP-40, 0.5% Sodium
Deoxycholate, 1.0 mM EDTA at a pH of 7.4) supplemented with 50X
Protease Inhibitor Cocktail; Roche Diagnostics) on ice. The protein
concentration was determined using the BCA assay (Applygen
Technologies, Inc.). The total protein (20 µg/per lane) from each
sample was separated using 8% SDS-PAGE. The proteins were
transferred onto polyvinylidene difluoride membranes. After 1 h of
blocking with 5% non-fat milk at room temperature, the membranes
were incubated with primary antibodies against COX-2 (1:1,000; cat.
no. A3560; ABclonal Biotech Co., Ltd.), HIF-1α (1:1,000; cat. no.
36169; Cell Signaling Technology, Inc.) and β-actin (1:5,000; cat.
no. A2228; Sigma-Aldrich; Merck KGaA) overnight at 4˚C. After
washing three times, the membranes were incubated with
anti-mouse/rabbit IgG HRP-conjugated secondary antibodies (1:2,000;
cat. no. 7076 and 7074, respectively; Cell Signaling Technology,
Inc.) for 2 h at room temperature. Protein bands were visualized
using an ECL detection kit (Bio-Rad Laboratories, Inc.).
Quantification of the band intensities was performed using ImageJ
Software (version 1.8.0; National Institutes of Health) and
normalized to the band intensities of β-actin.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA in BV2 cells or hippocampus tissue was
isolated using the TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.), according to the manufacturer's
instructions. Reverse transcription was performed using a HiScript
III All-in-one RT SuperMix kit (Vazyme Biotech Co., Ltd.) according
to the manufacturer's instructions. qPCR was subsequently performed
using the ChamQ SYBR qPCR Master Mix (Vazyme Biotech Co., Ltd.) and
a CFX96 Real-Time PCR Detection System (Bio-Rad). The thermal
cycling conditions include an initial denaturation step at 95˚C for
10 min, followed by 40 cycles at 95˚C for 30 sec, 60˚C for 30 sec
and 72˚C for 30 sec. The mRNA levels were quantified using the
2-ΔΔCq method and normalized to the internal reference
gene β-actin (16). The primer
sequences used for qPCR are presented in Table I.
 | Table IPrimer sequences used for reverse
transcription-quantitative PCR. |
Table I
Primer sequences used for reverse
transcription-quantitative PCR.
| Genes | Forward
(5'-3') | Reverse
(5'-3') |
|---|
| TNF-α |
CCCTCACACTCAGATCATCTTCT |
GCTACGACGTGGGCTACAG |
| IL-1β |
TTCAGGCAGGCAGTATCACTC |
GAAGGTCCACGGGAAAGACAC |
| IL-6 |
AGTCCTTCCTACCCCAATTTCC |
TTGGTCCTTAGCCACTCCTTC |
|
Cyclooxygenase-2 |
AGGTCATTGGTGGAGAGGTG |
CCTGCTTGAGTATGTCGCAC |
| β-actin |
ACTGTCGAGTCGCGTCCA |
GTCATCCATGGCGAACTGGT |
Immunofluorescence staining
After the hypoxia treatment, the mice were collected
from the hypoxia chamber, which was previously brought back to the
local altitude. The mice were immediately anesthetized with sodium
pentobarbital (50 mg/kg) via intraperitoneal injection and perfused
with prechilled saline (0.9%) to remove circulating blood cells.
The brain was fixed in 4% paraformaldehyde overnight. Each brain
was dehydrated with 15 and 30% sucrose solutions, and then frozen
sectioned (-20˚C) at a thickness of 40 µm. Thereafter, sections
were permeabilized with 5% Triton X-100 at room temperature and
blocked with 5% BSA for 30 min at room temperature. The sections
were incubated with a specific primary antibody against ionized
calcium-binding adapter molecule 1 (IBA1; 1:1,500; cat. no.
019-19741; Wako Chemicals USA, Inc.) at 4˚C overnight and then
incubated with Alexa Fluor 594-conjugated donkey anti-goat
secondary antibodies (1:1,000; cat. no. A32758; Thermo Fisher
Scientific, Inc.) for 60 min at room temperature. The nuclei were
counterstained with DAPI-containing mounting medium (ZSGB-BIO;
OriGene Technologies, Inc.) for 15 min at room temperature. Images
were captured using a scanning confocal microscope (Nikon
Corporation).
Statistical analysis
Data were analyzed using GraphPad Prism version 7.0
(GraphPad Software, Inc.). Data are presented as the arithmetic
mean ± standard error of the mean and the experiments were
performed three times. Statistically significant differences
between groups were determined using one-way ANOVA followed by
Tukey's test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Hypoxia increases the expression of
cytokines stimulated by LPS in BV2 cells
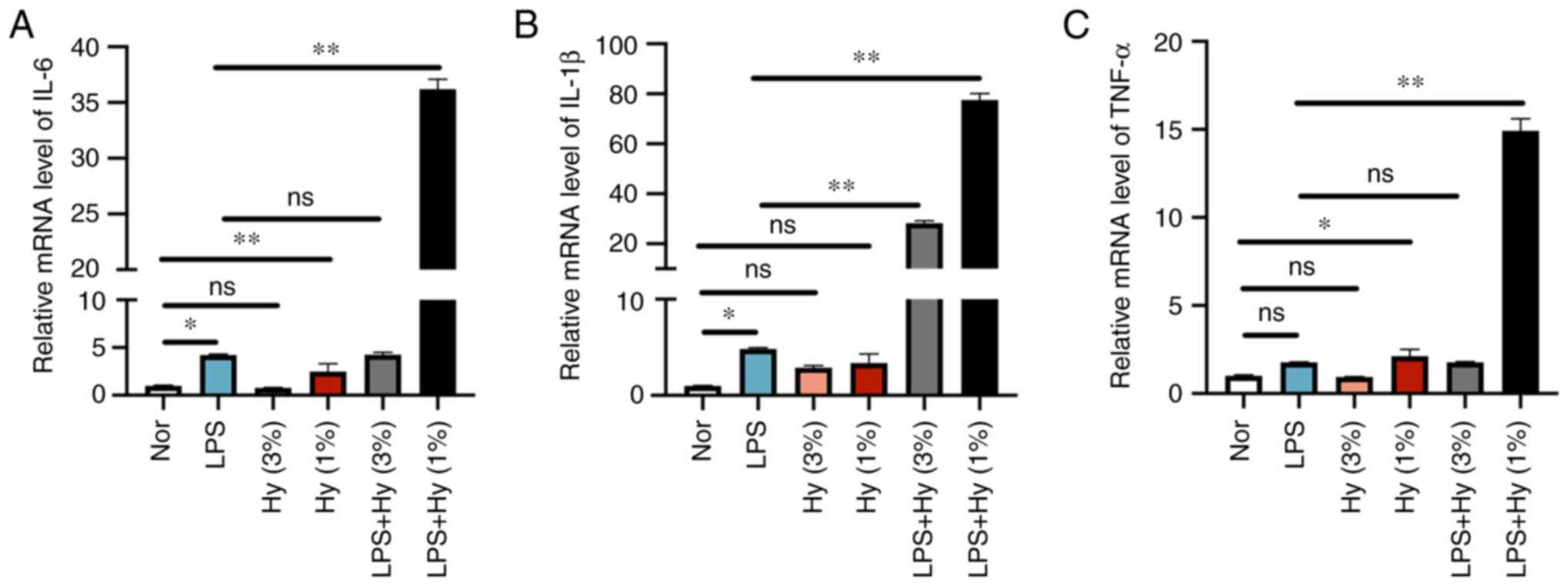
The impact of different hypoxic conditions on
neuroinflammatory response stimulated by LPS in BV2 cells was
determined by measuring the mRNA levels of IL-6, TNF-α and IL-1β
via RT-qPCR analysis. The LPS treatment alone induced a significant
increase in mRNA levels of the three cytokines IL-1β, TNF-α and
IL-6 (Fig. 1A-C). When the cells
were treated with LPS combined with hypoxia with 3% O2,
the mRNA levels of IL-1β were increased compared with those in the
LPS treatment group. However, when the cells were treated with LPS
combined with hypoxia with 1% O2, the mRNA levels of
IL-6, TNF-α and IL-1β were all increased compared with those in the
LPS treatment group (Fig. 1A-C).
Hypoxia with 1% O2 without LSP treatment also induced a
marked increase in the mRNA levels of these three cytokines, which
was related to heavier hypoxia exposure. These results might
indicate that hypoxia aggravated the inflammatory response in BV2
cells. Since hypoxia with 1% O2 aggravated LPS-induced
inflammation response more evidently than hypoxia with 3%
O2, 1% O2 was selected as the representative
hypoxia treatment condition for the subsequent experiments.
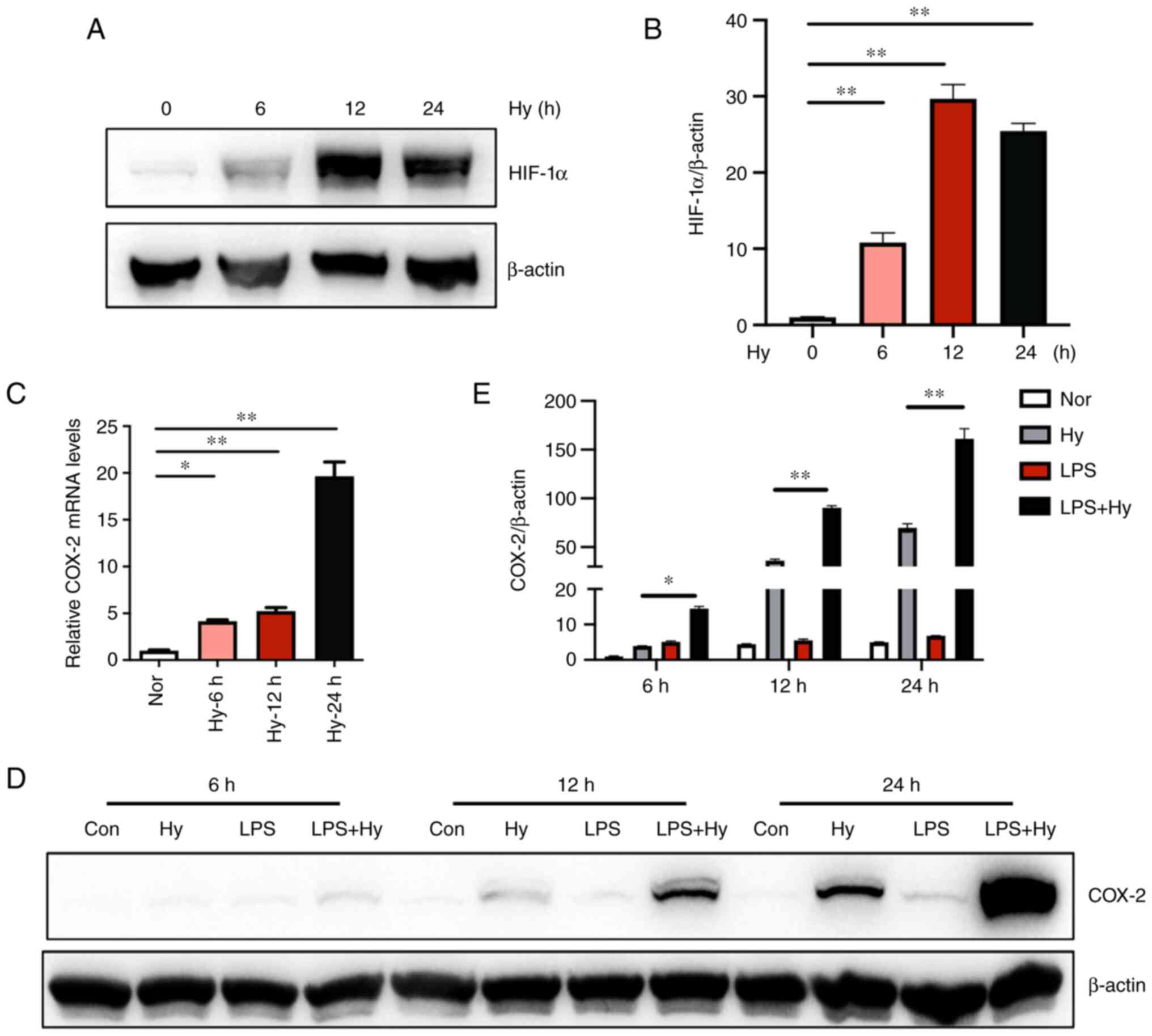
Hypoxia exposure increases the
expression of COX-2 at both the mRNA and protein levels in BV2
cells
The activation of HIF-1 signaling pathway was first
detected because it is a well-known marker responding to hypoxia
exposure in microglia. Western blotting demonstrated that the
protein levels of HIF-1α increased significantly following hypoxia
exposure (Fig. 2A and B). To determine the effect of hypoxia on
COX-2 expression, the mRNA levels of COX-2 were measured via
RT-qPCR. Exposure to hypoxia for 6, 12 and 24 h significantly
induced the upregulation of COX-2 by 4.1, 5.2 and 19.7-fold,
respectively, compared with that in the normoxia control group
(Fig. 2C). Moreover, western
blotting demonstrated that exposure to hypoxia (1% O2)
for 6, 12 and 24 h increased the protein levels of COX-2, while
there were no significant differences in the groups that were
treated with LPS alone for 6, 12 and 24 h. Notably, the combination
of hypoxia and LPS significantly increased the expression of COX-2
compared with that in the hypoxia group (Fig. 2D and E). These results might indicate that
hypoxia and LPS treatment had a synergic effect on the expression
of COX-2 protein.
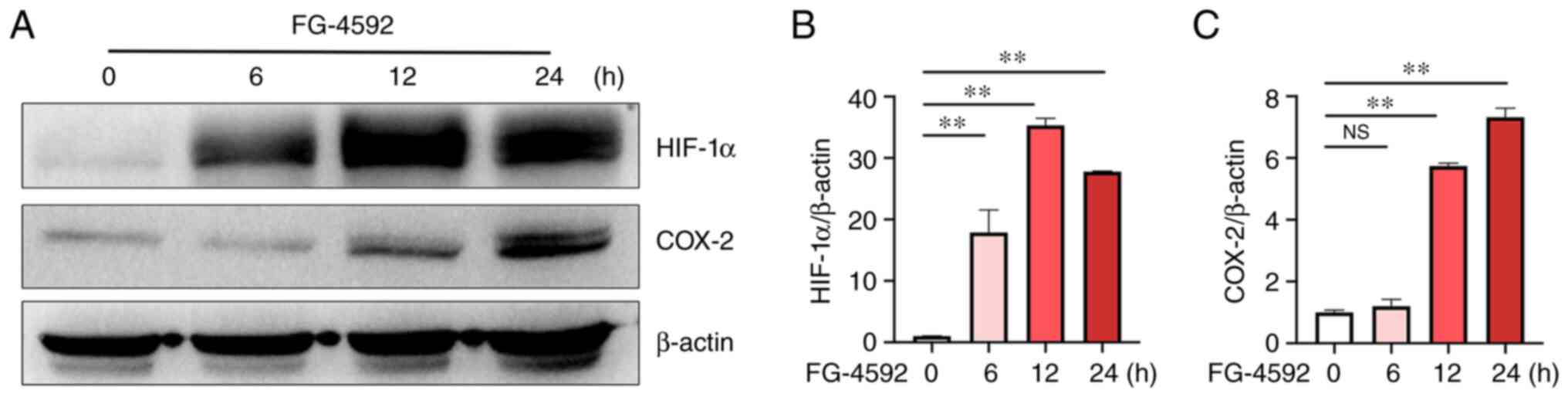
HIF-1 activator FG-4592 induces the
expression of COX-2
To further confirm the inducible effect of hypoxia
on the expression of COX-2 in microglia, FG-4592, a prolyl
hydroxylase (PHD) inhibitor, was applied. FG-4592 can stabilize
HIF-1 and promote the expression of its target genes. Western
blotting (Fig. 3A and B) demonstrated that FG-4592 significantly
increased the protein levels of HIF-1α at 6, 12 and 24 h compared
with that at 0 h (Fig. 3B).
Moreover, the protein levels of COX-2 were also significantly
increased at 12 and 24 h compared with that at 0 h (Fig. 3C), indicating that hypoxia induced
the COX-2 expression via the HIF-1 pathway in BV2 cells.
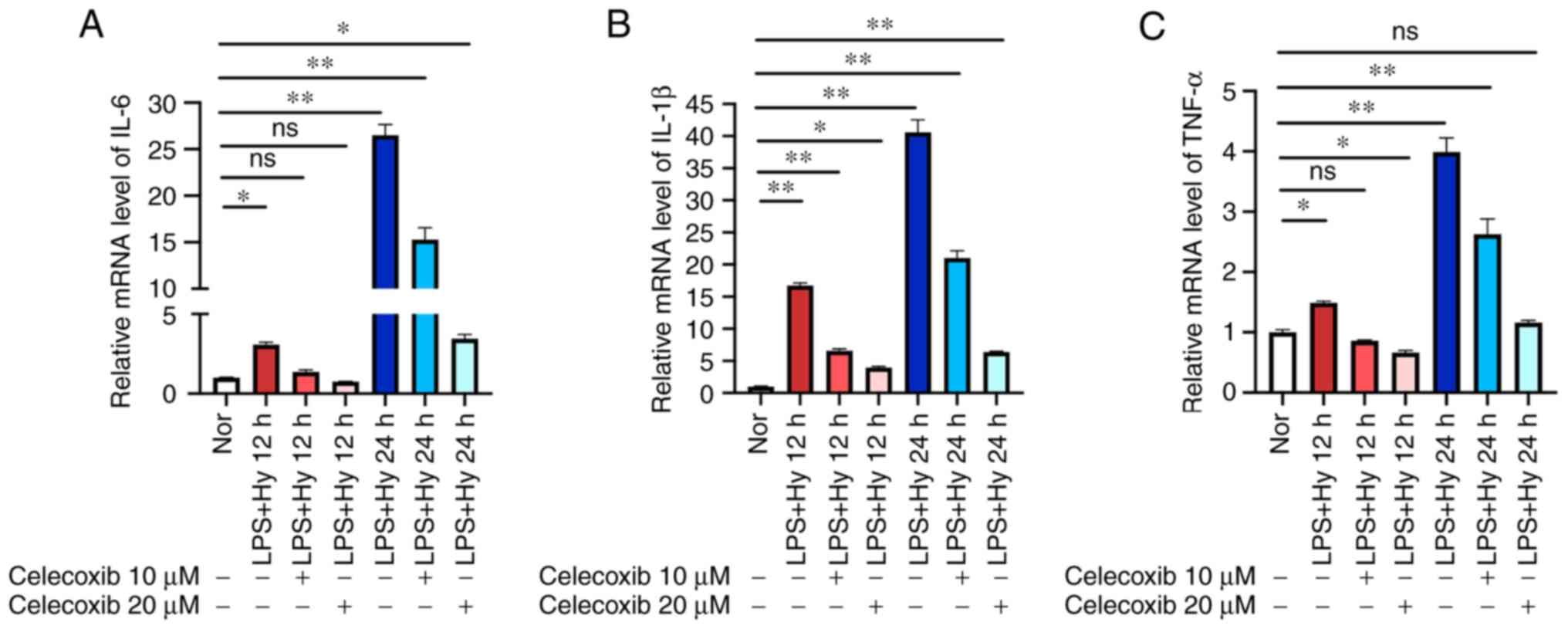
Celecoxib inhibits the inflammatory
response in BV2 cells
To determine whether COX-2 was involved in
neuroinflammation under hypoxic conditions, the effect on
inflammation of the COX-2 selective inhibitor celecoxib was
investigated in BV2 cells. RT-qPCR demonstrated that exposure to
hypoxia (1% O2) combined with LPS (100 ng/ml) for 12 and
24 h significantly increased the expression of proinflammatory
cytokines, such as IL-6, TNF-α and IL-1β. By contrast, pretreatment
with celecoxib dose-dependently inhibited the increased mRNA levels
of these cytokines caused by the combination of LPS treatment and
hypoxia (Fig. 4A-C). These results
might indicate that COX-2 was involved in the neuroinflammatory
response under hypoxic conditions in BV2 cells.
Celecoxib suppresses microglial
activation and decreases the mRNA levels of proinflammatory
cytokines in mice
The effect of celecoxib on inflammation in mice was
investigated. To evaluate neuroinflammation, immunofluorescence
staining for IBA1 was performed to measure microglial activation.
The proportion of IBA1-positive microglia was significantly
increased by the combination of LPS treatment and hypoxia compared
with the normoxic group, but this effect was significantly
inhibited by celecoxib treatment (Fig.
5A and B). In addition, the
mRNA levels of the three proinflammatory cytokines in the
hippocampal region of the brain were measured. The RT-qPCR results
indicated that celecoxib treatment significantly attenuated the
significantly increased mRNA levels of TNF-α caused by the
combination of LPS treatment and hypoxia (Fig. 5C-E). The mRNA levels of IL-6 and
IL-1β were also decreased, but without statistical significance.
These results indicated that blocking COX-2 inhibited the
neuroinflammatory response under hypoxic conditions in a mouse
model.
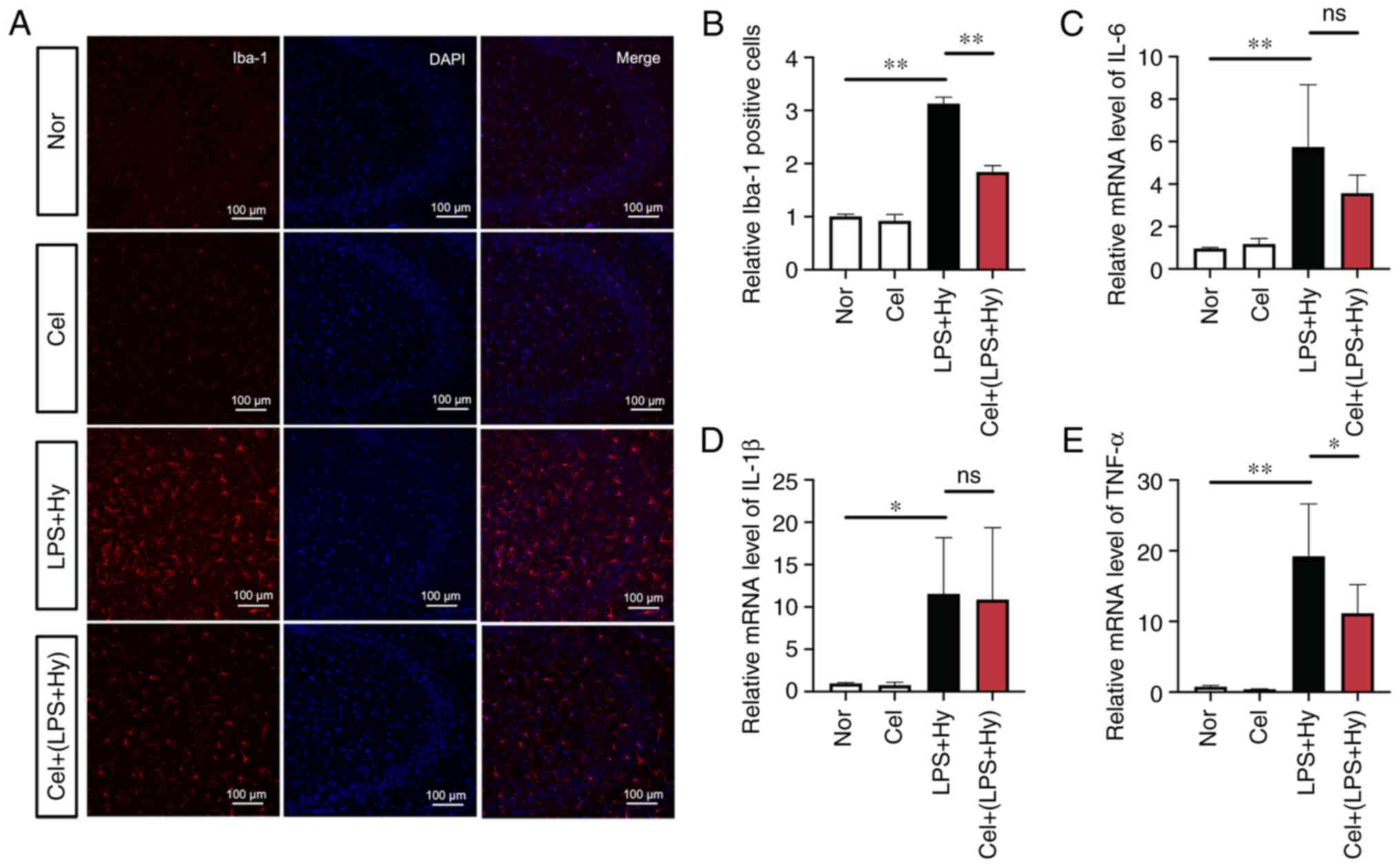 | Figure 5Celecoxib inhibits microglial
activation and decreases the mRNA levels of proinflammatory
cytokines in the mouse model. (A) Mice were pretreated with
celecoxib (20 mg/kg) and then treated with lipopolysaccharide
injection (0.5 mg/kg) and hypoxia exposure (mimicking 6,000 m of
high altitude) for 24 h. The brain slices were subjected to
immunofluorescence staining. Representative images of ionized
calcium-binding adapter molecule 1 staining in the hippocampus. The
nucleus was stained with DAPI. Scale bar, 100 µm. (B) Statistical
analysis of microglial activation, as presented in A. The mRNA
levels of (C) IL-6, (D) IL-1β and (E) TNF-α were measured via
reverse transcription-quantitative PCR assay. The results are
expressed as the mean ± SEM (n=4). *P<0.05 and
**P<0.01. SEM, standard error of the mean; IBA1,
ionized calcium-binding adapter molecule 1; LPS,
lipopolysaccharide; Nor, normoxia; Hy, hypoxia; Cel, celecoxib; ns,
not significant. |
Discussion
Microglia, resident mononuclear macrophage-like
cells in the CNS, play an important role in cerebral inflammation
(17). Microglia activation can be
triggered in various injury processes, such as hypoxia, ischemia
and immune responses (18).
Hypoxia-induced neuroinflammation is the key pathological mechanism
involved in acute mountain sickness (due to high altitude) or other
various neural diseases, such as Alzheimer's disease, multiple
sclerosis and traumatic brain injury. In addition, the peripheral
immune system contributes to this process (19). Hypoxia has a direct role in
microglial activation. It has been reported that hypoxia triggers
the M1 phenotype in BV2 cells through the activation of STAT1
signaling (20). Zhang et
al also revealed that acute hypoxia induces an imbalanced M1/M2
activation of microglia through the NF-κB signaling pathway
(21). Furthermore, even
preexposure to hypoxia for 3-6 days can lead to persistent and
aberrant inflammatory responses (22). The present study revealed that
compared with 3% O2, 1% O2 increased the
expression of proinflammatory cytokines, including IL-6, TNF-α and
IL-β. In addition, hypoxia markedly increased the neuroinflammatory
response to LPS stimulation. Hypoxia and inflammation are two major
pathogenic mechanisms of brain injury (23,24).
Prompted by this phenomenon, the current study attempted to explore
the underlying mechanisms of inflammation response under hypoxia
conditions.
Numerous observational and experimental studies have
indicated that there are several mechanisms involved in
neuroinflammation under hypoxia (25,26).
A recent study demonstrated that autophagy participates in
neuroinflammation induced by hypoxia (26). In addition, it has been reported
that membrane receptors are involved in neuroinflammation (27). Moreover, crosstalk between the
NF-κB and HIF-1 signaling pathways may play important roles in this
process (28). MicroRNAs also are
involved in hypoxia-induced neuroinflammation (29). For example, induction of miR-3473b,
which likely targets suppressor of cytokine signaling 3,
contributes to stroke pathogenesis by enhancing poststroke
neuroinflammation injury (30).
Therefore, based on these different pathophysiological models and
cell types and several treatment parameters, increasing numbers of
signaling mediators have been revealed.
COX-2 and prostaglandin E2 are well-known in
vitro and in vivo inflammatory inducers. Previous
studies have indicated that the expression of COX-2 is induced by
the hypoxic microenvironment in various systems, such as tumors and
epithelial cells (11,31). COX-2 is a direct HIF-1 target gene
and acts as a mediator of both inflammation and angiogenesis
(32,33). However, the contribution of COX-2
to pro-inflammatory responses under hypoxic conditions remains
unclear. The present study revealed that LPS produced no
significant effect on COX-2 expression, but hypoxia induced COX-2
expression. This was further confirmed by treating cells with
FG-4592, a well-known PHD inhibitor, which resulted in the
activation of the HIF-1 signaling pathway. In addition, the
increase in COX-2 expression observed upon treatment with both
hypoxia and LPS was concomitant with the cytokine burst, indicating
that COX-2 probably mediated hypoxia-induced aggravation of
LPS-induced neuroinflammation. Consistently, celecoxib effectively
inhibited the expression of cytokines induced by the combination
treatment of hypoxia and LPS and a similar result was observed in
the mouse neuroinflammation model. These results demonstrated that
COX-2 served as an important mediator of hypoxia-induced
aggravation of LPS-induced neuroinflammation.
COX-1 is constitutively expressed in a wide range of
tissues, while COX-2 is an inducible enzyme that produces
prostaglandins in inflammatory settings (34). Chauhan et al (35) revealed that COX-1 and COX-2
isoforms contribute to downstream proinflammatory responses in a
high-altitude hypoxia exposure rat model (35). Based on a hypoxic postnatal rat
model, Li et al (36) also
demonstrated that prostaglandin E2 regulates inflammatory mediators
in activated microglia via the prostaglandin E2 receptor-cAMP
signaling pathway under hypoxic conditions. COX-2 is an
intracellular, inducible protein that positively regulates cytokine
signaling in numerous cell types (37). The present report focused on
microglial cells, which are the main components of the innate
immune system in the CNS (38). In
BV2 cells, pretreatment with celecoxib decreased the expression of
cytokines. Additionally, the administration of celecoxib also
inhibited microglial activation in a mouse model injected with LPS
treatment. The neuroprotective roles of celecoxib in other neural
diseases, such as neurodegeneration caused by exposure to high
altitude hypoxia (35),
Alzheimer's disease (39),
neonatal brain injury (40),
autoimmune encephalomyelitis (41)
and ischemic injury (42), should
be considered. The present results indicated the potential
significance of targeting COX-2 in the treatment of
neuroinflammatory diseases, even under hypoxic conditions.
In summary, the current study revealed that COX-2, a
downstream mediator of HIF-1, contributed to neuroinflammation
response to hypoxic conditions. Blocking COX-2 function with
celecoxib effectively inhibited neuroinflammation in vivo
and in vitro (Fig. 6). The
present results demonstrated that COX-2 is an important mediator of
hypoxia-induced aggravation of LPS-induced neuroinflammation.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by the National Natural Science
Foundation of China (grant no. 81930054).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MZ conceived and designed the experiments. YY, YG,
XC, JG and ZS performed experiments. YY and YG analyzed and
interpreted the data. MZ wrote and revised the manuscript. YY, YG
and MZ confirm the authenticity of all the raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
All procedures were approved by the Institutional
Animal Care and Use Committee of the Beijing Institute of Basic
Medical Sciences (approval no. IACUC-DWZX-2021-648).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Straub RH and Schradin C: Chronic
inflammatory systemic diseases: An evolutionary trade-off between
acutely beneficial but chronically harmful programs. Evol Med
Public Health. 2016:37–51. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Han VX, Patel S, Jones HF and Dale RC:
Maternal immune activation and neuroinflammation in human
neurodevelopmental disorders. Nat Rev Neurol. 17:564–579.
2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Mishra A, Bandopadhyay R, Singh PK, Mishra
PS, Sharma N and Khurana N: Neuroinflammation in neurological
disorders: Pharmacotherapeutic targets from bench to bedside. Metab
Brain Dis. 36:1591–1626. 2021.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Lee Y, Lee S, Park JW, Hwang JS, Kim SM,
Lyoo IK, Lee CJ and Han IO: Hypoxia-induced neuroinflammation and
learning-memory impairments in adult zebrafish are suppressed by
glucosamine. Mol Neurobiol. 55:8738–8753. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Algra SO, Groeneveld KM, Schadenberg AW,
Haas F, Evens FC, Meerding J, Koenderman L, Jansen NJ and Prakken
BJ: Cerebral ischemia initiates an immediate innate immune response
in neonates during cardiac surgery. J Neuroinflammation.
10(24)2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Song TT, Bi YH, Gao YQ, Huang R, Hao K, Xu
G, Tang JW, Ma ZQ, Kong FP, Coote JH, et al: Systemic
pro-inflammatory response facilitates the development of cerebral
edema during short hypoxia. J Neuroinflammation.
13(63)2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zhou Y, Huang X, Zhao T, Qiao M, Zhao X,
Zhao M, Xu L, Zhao Y, Wu L, Wu K, et al: Hypoxia augments
LPS-induced inflammation and triggers high altitude cerebral edema
in mice. Brain Behav Immun. 64:266–275. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Palazon A, Goldrath AW, Nizet V and
Johnson RS: HIF transcription factors, inflammation, and immunity.
Immunity. 41:518–528. 2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Walmsley SR, Chilvers ER, Thompson AA,
Vaughan K, Marriott HM, Parker LC, Shaw G, Parmar S, Schneider M,
Sabroe I, et al: Prolyl hydroxylase 3 (PHD3) is essential for
hypoxic regulation of neutrophilic inflammation in humans and mice.
J Clin Invest. 121:1053–1063. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Bruning U, Fitzpatrick SF, Frank T,
Birtwistle M, Taylor CT and Cheong A: NFκB and HIF display
synergistic behaviour during hypoxic inflammation. Cell Mol Life
Sci. 69:1319–1329. 2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Cook-Johnson RJ, Demasi M, Cleland LG,
Gamble JR, Saint DA and James MJ: Endothelial cell COX-2 expression
and activity in hypoxia. Biochim Biophys Acta. 1761:1443–1449.
2006.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Csiki I, Yanagisawa K, Haruki N, Nadaf S,
Morrow JD, Johnson DH and Carbone DP: Thioredoxin-1 modulates
transcription of cyclooxygenase-2 via hypoxia-inducible
factor-1alpha in non-small cell lung cancer. Cancer Res.
66:143–150. 2006.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Cui J and Jia J: Natural COX-2 inhibitors
as promising anti-inflammatory agents: An update. Curr Med Chem.
28:3622–3646. 2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Patrignani P and Patrono C: Cyclooxygenase
inhibitors: From pharmacology to clinical read-outs. Biochim
Biophys Acta. 1851:422–432. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhu J, Li S, Zhang Y, Ding G, Zhu C, Huang
S, Zhang A, Jia Z and Li M: COX-2 contributes to LPS-induced Stat3
activation and IL-6 production in microglial cells. Am J Transl
Res. 10:966–974. 2018.PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Li Q and Barres BA: Microglia and
macrophages in brain homeostasis and disease. Nat Rev Immunol.
18:225–242. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
DiSabato DJ, Quan N and Godbout JP:
Neuroinflammation: The devil is in the details. J Neurochem. 139
(Suppl 2):S136–S153. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Guo L and Zhu L: Multiple roles of
peripheral immune system in modulating ischemia/hypoxia-induced
neuroinflammation. Front Mol Biosci. 8(752465)2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Butturini E, Boriero D, Carcereri de Prati
A and Mariotto S: STAT1 drives M1 microglia activation and
neuroinflammation under hypoxia. Arch Biochem Biophys. 669:22–30.
2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhang F, Zhong R, Li S, Fu Z, Cheng C, Cai
H and Le W: Acute hypoxia induced an imbalanced M1/M2 activation of
microglia through NF-κB signaling in Alzheimer's disease mice and
wild-type littermates. Front Aging Neurosci. 9(282)2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kiernan EA, Ewald AC, Ouellette JN, Wang
T, Agbeh A, Knutson AO, Roopra AS and Watters JJ: Prior hypoxia
exposure enhances murine microglial inflammatory gene expression in
vitro without concomitant H3K4me3 enrichment. Front Cell Neurosci.
14(535549)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Li B, Concepcion K, Meng X and Zhang L:
Brain-immune interactions in perinatal hypoxic-ischemic brain
injury. Prog Neurobiol. 159:50–68. 2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Merelli A, Repetto M, Lazarowski A and
Auzmendi J: Hypoxia, oxidative stress, and inflammation: Three
faces of neurodegenerative diseases. J Alzheimers Dis. 82
(s1):S109–S126. 2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Kaur C, Rathnasamy G and Ling EA: Roles of
activated microglia in hypoxia induced neuroinflammation in the
developing brain and the retina. J Neuroimmune Pharmacol. 8:66–78.
2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Sha S, Tan J, Miao Y and Zhang Q: The role
of autophagy in hypoxia-induced neuroinflammation. DNA Cell Biol.
40:733–739. 2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Chen PZ, He WJ, Zhu ZR GJ, Xu G, Chen DW
and Gao YQ: Adenosine A2A receptor involves in
neuroinflammation-mediated cognitive decline through activating
microglia under acute hypobaric hypoxia. Behav Brain Res.
347:99–107. 2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Peng X, Li C, Yu W, Liu S, Cong Y, Fan G
and Qi S: Propofol attenuates hypoxia-induced inflammation in BV2
microglia by inhibiting oxidative stress and NF-κB/Hif-1α
signaling. Biomed Res Int. 2020(8978704)2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Chen YM, He XZ, Wang SM and Xia Y:
δ-Opioid receptors, microRNAs, and neuroinflammation in cerebral
ischemia/hypoxia. Front Immunol. 11(421)2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wang X, Chen S, Ni J, Cheng J, Jia J and
Zhen X: miRNA-3473b contributes to neuroinflammation following
cerebral ischemia. Cell Death Dis. 9(11)2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Hashemi Goradel N, Najafi M, Salehi E,
Farhood B and Mortezaee K: Cyclooxygenase-2 in cancer: A review. J
Cell Physiol. 234:5683–5699. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Kaidi A, Qualtrough D, Williams AC and
Paraskeva C: Direct transcriptional up-regulation of
cyclooxygenase-2 by hypoxia-inducible factor (HIF)-1 promotes
colorectal tumor cell survival and enhances HIF-1 transcriptional
activity during hypoxia. Cancer Res. 66:6683–6691. 2006.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Lee JJ, Natsuizaka M, Ohashi S, Wong GS,
Takaoka M, Michaylira CZ, Budo D, Tobias JW, Kanai M, Shirakawa Y,
et al: Hypoxia activates the cyclooxygenase-2-prostaglandin E
synthase axis. Carcinogenesis. 31:427–434. 2010.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Kirkby NS, Lundberg MH, Harrington LS,
Leadbeater PD, Milne GL, Potter CM, Al-Yamani M, Adeyemi O, Warner
TD and Mitchell JA: Cyclooxygenase-1, not cyclooxygenase-2, is
responsible for physiological production of prostacyclin in the
cardiovascular system. Proc Natl Acad Sci USA. 109:17597–17602.
2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Chauhan G, Roy K, Kumar G, Kumari P, Alam
S, Kishore K, Panjwani U and Ray K: Distinct influence of COX-1 and
COX-2 on neuroinflammatory response and associated cognitive
deficits during high altitude hypoxia. Neuropharmacology.
146:138–148. 2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Li P, Lu J, Kaur C, Sivakumar V, Tan KL
and Ling EA: Expression of cyclooxygenase-1/-2, microsomal
prostaglandin-E synthase-1 and E-prostanoid receptor 2 and
regulation of inflammatory mediators by PGE(2) in the amoeboid
microglia in hypoxic postnatal rats and murine BV-2 cells.
Neuroscience. 164:948–962. 2009.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Turini ME and DuBois RN: Cyclooxygenase-2:
A therapeutic target. Annu Rev Med. 53:35–57. 2002.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Woodburn SC, Bollinger JL and Wohleb ES:
The semantics of microglia activation: Neuroinflammation,
homeostasis, and stress. J Neuroinflammation.
18(258)2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Mhillaj E, Morgese MG, Tucci P, Furiano A,
Luongo L, Bove M, Maione S, Cuomo V, Schiavone S and Trabace L:
Celecoxib prevents cognitive impairment and neuroinflammation in
soluble amyloid β-treated rats. Neuroscience. 372:58–73.
2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Fan LW, Kaizaki A, Tien LT, Pang Y, Tanaka
S, Numazawa S, Bhatt AJ and Cai Z: Celecoxib attenuates systemic
lipopolysaccharide-induced brain inflammation and white matter
injury in the neonatal rats. Neuroscience. 240:27–38.
2013.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Di Penta A, Chiba A, Alloza I, Wyssenbach
A, Yamamura T, Villoslada P, Miyake S and Vandenbroeck K: A
trifluoromethyl analogue of celecoxib exerts beneficial effects in
neuroinflammation. PLoS One. 8(e83119)2013.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Gou J, Liang S, Cheng W, Wu S, Ye Z, Ma Y,
Yin Y and Wang H: Neuroprotective effect of combined use of
nicotine and celecoxib by inhibiting neuroinflammation in ischemic
rats. Brain Res Bull. 175:234–243. 2021.PubMed/NCBI View Article : Google Scholar
|