Introduction
Brain ischemia episodes facilitate the onset of
dementia, with 10% of patients developing dementia and cognitive
impairment soon after their first stroke (1). Studies have indicated that ischemia
and the primary type of dementia, Alzheimer's disease (AD), are
statistically correlated (2-4).
For example, a previous clinical study revealed that stroke is an
independent risk factor for AD (4). Numerous animal studies have also
suggested a higher occurrence rate of AD following brain
ischemia/stroke (5,6). Thus, it is important to investigate
the molecular association between brain ischemia and AD to reduce
AD occurrence following a brain ischemia.
Ischemic neurotoxicity leads to extensive neuronal
impairments in certain regions of the forebrain, such as the cortex
and hippocampus. These regions are associated with cognitive
function (7). A previous study
showed that dendritic spine dysfunction is the earliest
neurotoxicity symptom following brain ischemia (8). More importantly, dendritic spine
dysfunction is a biomarker predicting AD occurrence (9,10).
Based on these previous findings, it was hypothesized that
protecting dendritic spine function following brain ischemia might
relieve neuronal death following stroke, which might ultimately
reduce AD occurrence following brain ischemia.
The function and dynamics of dendritic spines are
tightly regulated by cytoskeletal proteins, such as microtubules
and their upstream regulator, Rho-GTPases (11). Members of the protein kinase C
(PKC) family are known for their functions in regulating the
activity of Rho-GTPase. The ε isoform of PKC (PKCε) has been found
to regulate the cytoskeleton in cardiocytes and to protect cells
from mitochondria damage following myocardial ischemia (12). However, the role of PKCε in
neuronal cells following ischemia remains unclear. Oxygen-glucose
deprivation (OGD) is a well-established cell-based method for
simulating brain ischemia in vitro (13) that has been used extensively in
basic and preclinical stroke studies (14-16).
In the present study, OGD was applied to primary hippocampus and
cortical neurons to investigate the role of PKCε in dendritic spine
dysfunction following ischemia. The results of the present study
may suggest novel therapeutic targets for cognitive function
protection following brain ischemia.
Materials and methods
Rat euthanasia
Pregnant Sprague-Dawley rats at E17 were purchased
from the vivarium facility of WuXi AppTec Co., Ltd. Upon arrival,
rats were left in the procedure room overnight and euthanized on
E18. A total of 20 pregnant female rats (age between 8-12 weeks and
body weight around 250 g were used. For euthanasia, rats were put
in a large transparent plastic box for 10 min and inhalant
anaesthesia was performed by placing a 50-ml centrifuge tube
containing 15 ml liquid isoflurane (99.9%; cat. no. H19980141;
Ruitaibio Co.) into the plastic box. The plastic box was then
sealed with the lid to avoid the evaporation of isoflurane. The
final isoflurane percentage was 3% for both induction and
maintenance of anaesthesia. Generally, rats would be anaesthetized
in 10-15 min. Subsequently, the abdomen skin of anaesthetized rats
was sterilized with 70% ethanol. Following sterilization, an
incision was made along the midline of the abdomen using clinical
scissors, from the small intestine up to the heart. Finally, rats
were sacrificed by cutting off a piece of the left heart ventricle
and decapitation with surgical scissors. Death was verified by loss
of heartbeat. Rats were under anaesthesia for the whole euthanasia
process. Embryos were collected into a 100-cm dish for hippocampus
and cortex dissection in a tissue culture room.
The animal welfare and husbandry were conducted at
the vivarium room of WuXi AppTec Co., Ltd. Pregnant rats were
housed in separate cages, with ad libitum access to food and
water supply, and 12/12-h light/dark cycles at 24-26˚C and 30-70%
humidity. Animal health was monitored daily. Physiological or
behavioural symptoms, such as severe pain, overt distress, moribund
and beyond the point where recovery appears reasonable, were used
as humane endpoint criteria resulting in animal euthanasia to
minimize suffering by inhalation of 100% CO2 with 70%
chamber volume /min influx rate. Fetuses were euthanized by
decapitation with surgical scissors.
Primary neuronal culture
The present study was collaborative research between
the Huzhou Third Municipal Hospital, The Affiliated Hospital of
Huzhou University (Huzhou, Zhejiang, China) and WuXi AppTec Co.,
Ltd. Procedures involving animals were approved by the
Institutional Animal Care and Use Committee of the WuXi AppTec Co.,
Ltd. company at the Qidong (Jiangsu, China) site (IACUC approval
no. GP01-QD089-2022v1.0). Cell culture was performed as previously
described (17). In brief, before
dissection, 35-mm cell culture dishes were pre-treated with 1 ml
Poly-L-Lysine (0.5 mg/ml; cat. no. P4707; MilliporeSigma) overnight
at 37˚C and washed with sterile water three times. Pregnant
Sprague-Dawley rats at embryonic day 18 (E18) were sacrificed and
embryos were decapitated with surgical scissors and dissected for
hippocampus and cortex neuron culture. Low-density hippocampus
(1x104 cells for a total of three coverslips/35 mm dish)
or high-density cortical cultures (1x107 cells/35 mm
dish) were grown in neurobasal medium (cat no. 21103049;
Invitrogen; Thermo Fisher Scientific, Inc.) supplemented with 1X
B-27 (cat no. 17504044; Invitrogen; Thermo Fisher Scientific, Inc.)
and 2 mM GlutaMAX™ (cat no. 35050061; Invitrogen; Thermo Fisher
Scientific, Inc.). At 6 days in vitro (DIV), primary neurons
were transfected with enhanced green fluorescent protein (eGFP)
plasmids (pcDNA3-eGFP, cat no. 13031 Addgene) for visualization
using calcium phosphate precipitation, as previously described
(17). In brief, 2 µg eGFP plasmid
was transfected overnight at 37˚C using Calcium Phosphate
Transfection Kit (cat no. K278001, Thermo Fisher). Fresh medium was
added and primary neurons were incubated in 37˚C until use on DIV
17.
OGD model establishment
The OGD model was established as previously
described (18). To simulate
ischemic stroke in vitro, primary neurons were washed gently
with PBS (pH 7.4; cat. no. 10010023; Invitrogen; Thermo Fisher
Scientific, Inc.) twice. Neurons were cultured in DMEM with no
glucose (cat. no. 11966025; Invitrogen; Thermo Fisher Scientific,
Inc.) or supplements and incubated in a humidified oxygen control
CO2 incubator (type-i160; Thermo Fisher Scientific,
inc.) with 1% O2, 5% CO2 and 94%
N2 at 37˚C for 2 h.
Confocal imaging and
quantification
Confocal images were obtained using a Zeiss LSM 800
confocal microscope [Carl Zeiss IMT (Shanghai) Co., Ltd.] with an
x63 oil objective (1.0 numerical aperture) with a sequential
acquisition setting. Following OGD treatment, primary hippocampus
neurons were washed with PBS and fixed with 4% paraformaldehyde at
room temperature for 20 min (cat. no. P0099; Beyotime Institute of
Biotechnology). Fixed cells were visualized with 488 nm excitation
for enhanced green-fluorescent protein. Up to 10 positive neurons
were observed per dish. Dendritic spines resembled mushroom-shaped
protrusions. Spine length and width were measured using ImageJ
(version 1.50i; National Institutes of Health). Spine length was
defined as the distance from the tip of the spine head to the point
where the spine starts to grow at the dendrite. Spine width was
defined as the maximal width of the spine head perpendicular to the
long axis of the spine neck.
For PKCε inhibiting assay, PKCε-specific inhibiting
peptide, EAVSLKPT, was used here (cat no. 539522; MilliporeSigma).
In brief, hippocampus neurons were transfected with 2 µg of eGFP
plasmid at DIV6 and treated with 1 mM inhibitor at DIV16. After 24
h treatment at 37˚C, cells were moved to an OGD chamber for 2 h.
After OGD treatment, cells were fixed and observed under confocal
microscope in the way as stated above.
While performing quantifications, all green positive
neurons in each dish that have intact cell membrane were imaged and
used for quantification. All dendritic spines identified on each
imaged neuron were quantified for number, length and width. There
were at least 30 positive neurons from 3-5 repeated experiments
used for quantification and 1,200-1,500 dendritic spines were
quantified in total in each group (50-70 spines per neuron). The
quantifications were performed by two individuals (LL and GS) who
were blind to the experimental conditions and the results were
averaged.
Cell viability assay
The viability of primary neurons was detected via
MTT assay (cat no. ST1537; Beyotime Institute of Biotechnology),
according to the manufacturer's instructions. Briefly, following
the OGD treatments, primary cortical neurons were washed with PBS
and incubated with MTT reagent (50 µl/well in a 6-well-plate) at
37˚C for 4 h. 100 µl DMSO was added to dissolve the formazan.
Absorbance was measured at 450 nm using a plate reader (BioTek
Synergy HT Multi-Mode Microplate reader, BioTek China).
Rho-GTPase activity assay
The rho-GTPase activity was measured using different
G-LISA activation assay kits (cat. nos. BK124 for active RhoA,
BK128 for active Rac1 and BK127 for CDC42; Cytoskeleton, Inc.)
following the manufacturer's instructions. Briefly, high-density
cortical neurons with OGD treatment were lysed using the lysis
buffer supplied with the kits. A total of 25 µg total protein/group
was loaded into each well and then incubated with primary and
secondary antibodies that supplied with the G-LISA kits (cat. nos.
BK124 for active RhoA, BK128 for active Rac1 and BK127 for CDC42;
Cytoskeleton, Inc.). According to manufacturer's instruction, the
primary antibody can incubate at room temperature for 1 h and then
the secondary antibody can incubate at room temperature for 1 h.
The reaction plate was incubated with a detection solution at room
temperature for 30 min according to manufacturer's instruction and
the level of activation was determined by measuring the absorbance
at 490 nm (BioTek Synergy HT Multi-Mode Microplate reader, BioTek
China).
Electrophysiology recording
Electrophysiology whole-cell recordings were
performed in voltage-clamp mode using a MultiClamp 700B amplifier
(Molecular Devices, LCC) at a sampling frequency of 50 kHz and
recorded signals were digitized using a Digidata 1440 digitizer
(Molecular Devices, LCC). Patch pipettes were pulled from
borosilicate glass and had a resistance of 3-5 MΩ when filled with
standard intracellular solution (95.0 K-gluconate, 50.0 KCl, 10.0
HEPES, 4.00 Mg-ATP, 0.3 NaGTP and 10.0 mM phosphocreatine; pH 7.2,
300 mOsm). Miniature excitatory postsynaptic current (mEPSC) was
measured in rat hippocampal neurons following OGD at room
temperature in artificial cerebrospinal fluid (126.0 NaCl, 2.5 KCl,
10.0 glucose, 1.25 NaH2PO4, 2.0
MgCl2, 2.0 CaCl2 and 26.0 mM
NaHCO3) with 0.5 µM tetrodotoxin (Sigma-Aldrich; Merck
KGaA).
In vitro PKCε kinase activity
PKCε kinase activity was measured using PKCε Kinase
Enzyme System (cat. no. V4037; Promega Corporation), according to
the manufacturer's instructions. Briefly, high-density cortical
neurons with OGD treatment were lysed with RIPA buffer (cat. no.
P0013B; Beyotime Institute of Biotechnology) and 100 µg total
protein/group was then used for incubation with 5 µM ATP and 0.1
µg/µl substrate for 60 min at room temperature. A total of 25 ng
purified PKCε was used as the positive control. Following
incubation, ADP-Glo™ was added and incubated at room
temperature for 40 min according to manufacturer's instructions.
Luminescence signals were detected using a microplate reader
(BioTek Synergy HT Multi-Mode microplate reader, BioTek China).
Western blotting
Following OGD treatment, primary cortical neurons
were washed twice with PBS and cells were lysed using a protein
lysis buffer (cat. no. P0013B; Beyotime Institute of Biotechnology)
in a cold room at 4˚C for 10 min. Following centrifugation (10,000
x g for 10 min at 4˚C), the supernatant was transferred to a new
tube and protein concentration was quantified using a Pierce BCA
protein assay (cat. no. 23225; Thermo Fisher Scientific, Inc.). An
equal amount of protein from each group was denatured at 95˚C for 5
min and 50 µg protein/lane was separated by SDS-PAGE on a 10% gel.
Following the transfer onto a 0.45-µm PVDF membrane (cat no. 88518;
Thermo Fisher Scientific, Inc.), PVDF membranes were blocked with
5% BSA (cat. no. ST025; Beyotime Institute of Biotechnology) for 1
h at room temperature. Following blocking, membranes were incubated
with a primary antibody overnight in a cold room at 4˚C and with a
secondary antibody for 1 h at room temperature. The following
primary antibodies were used: Rabbit anti-total-Ras homolog family
member A (RhoA; cat. no. 2117; 1:2,000; Cell Signaling Technology,
Inc.); rabbit anti-total-Rac Family Small GTPase 1 (Rac1; cat. no.
2465; 1:2,000; Cell Signaling Technology, Inc.); rabbit
anti-total-cell division cycle 42 (CDC42; cat. no. 2466; 1:1,000;
Cell Signaling Technology, Inc.), rabbit anti-β Tubulin (cat. no.
2416; 1:5,000, Cell Signaling Technology, Inc.), and rabbit
anti-PKCε (cat. no. 2683; 1:1,000; Cell Signaling Technology,
Inc.). The secondary antibodies were horseradish peroxidase
anti-rabbit IgG (cat. no. 7074; 1:5,000; Cell Signaling Technology,
Inc.). Protein bands were visualized using Pierce™ ECL
Western Blotting Substrate (cat no. 32209; Thermo Fisher
Scientific, Inc.) and developed in a dark room using X-OMAT BT film
(cat. no. FF057; Beyotime Institute of Biotechnology). The
quantification of the bands was performed using ImageJ software
(version 1.50i; National Institutes of Health).
Statistical analysis
Results were analyzed using GraphPad Prism Software
Version 6.0 (GraphPad Software, Inc.; Dotmatics). All data are
presented as the mean ± standard deviation, with replicates from 3
to 10. Statistical comparisons were performed using unpaired
Student's t test (for comparisons between two groups), one-way
ANOVA followed by Tukey's post hoc test (for comparisons between 3
groups) or two-way ANOVA followed by Tukey's post hoc test (for
quantification of spine length and width). All electrophysiological
data were analyzed using Clampfit (Molecular Device, LLC).
P<0.05 was considered to indicate a statistically significant
difference.
Results
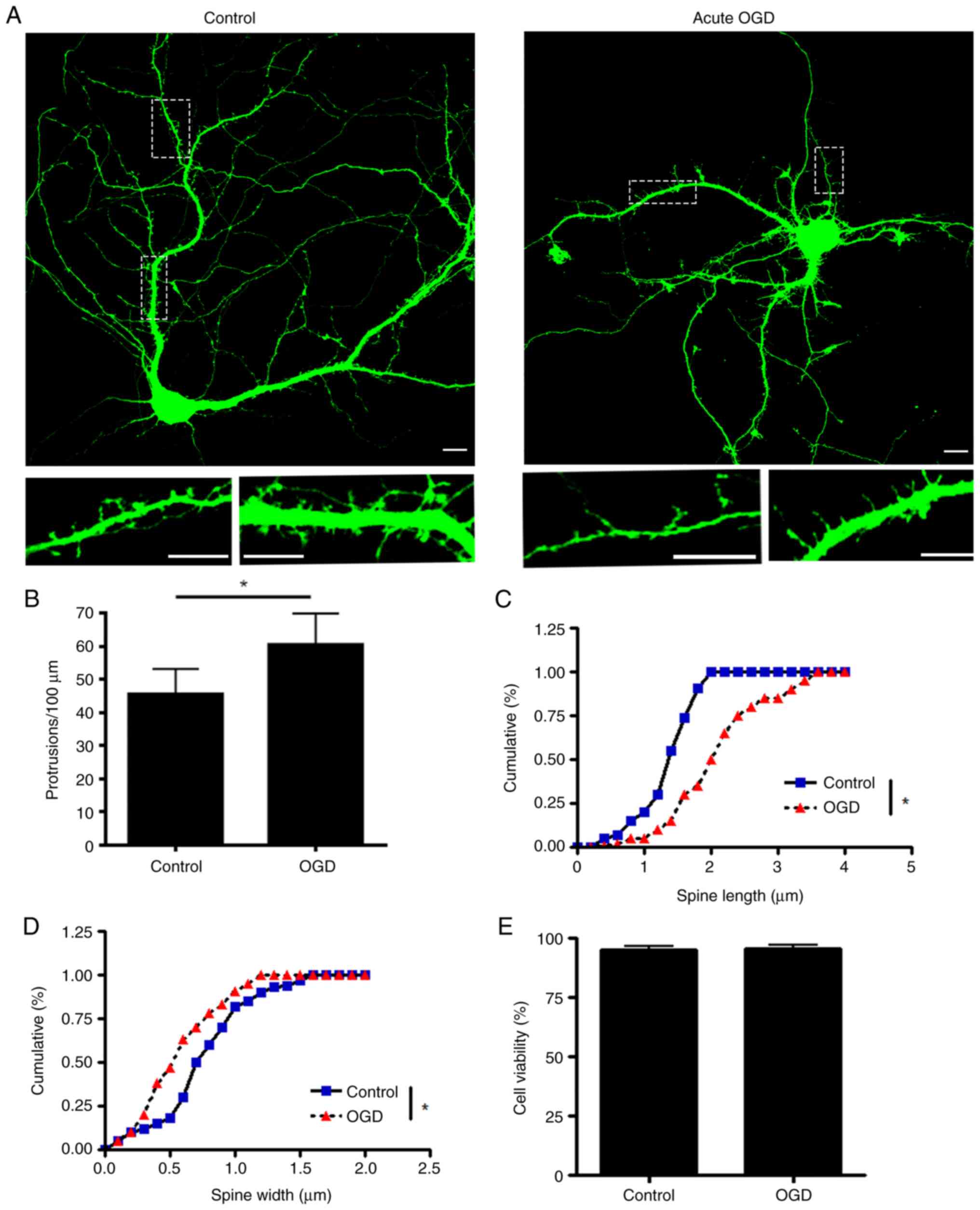
OGD damages the morphology and
function of the dendritic spine in primary hippocampus neuronal
culture
To investigate how ischemia interrupts dendritic
spine morphology and function, an acute OGD model was induced in
cultured primary hippocampus neurons. The OGD treatment is an
established in vitro method to simulate severe hypoxic and
ischemic conditions by withdrawing glucose in culture media while
limiting the oxygen supply to 1%. Primary hippocampus neurons were
transfected with eGFP plasmid for visualization at DIV6 and
transferred into a hypoxia chamber at DIV17 with DMEM without
glucose. According to a previous study (19), most of the dendritic spines in
cultured hippocampus neurons mature at DIV17. After 2 h treatment,
hippocampus neurons were fixed and imaged using a confocal
microscope to analyse the morphology of dendritic spines. Following
acute phase OGD, total protrusions in the dendrites were
significantly increased, while the number of mature dendritic
spines was decreased. The mean width of the spine head was
decreased (Fig. 1A-D), which
indicated that the dendritic spine underwent shrinking.
Furthermore, acute OGD treatment did not induce significant
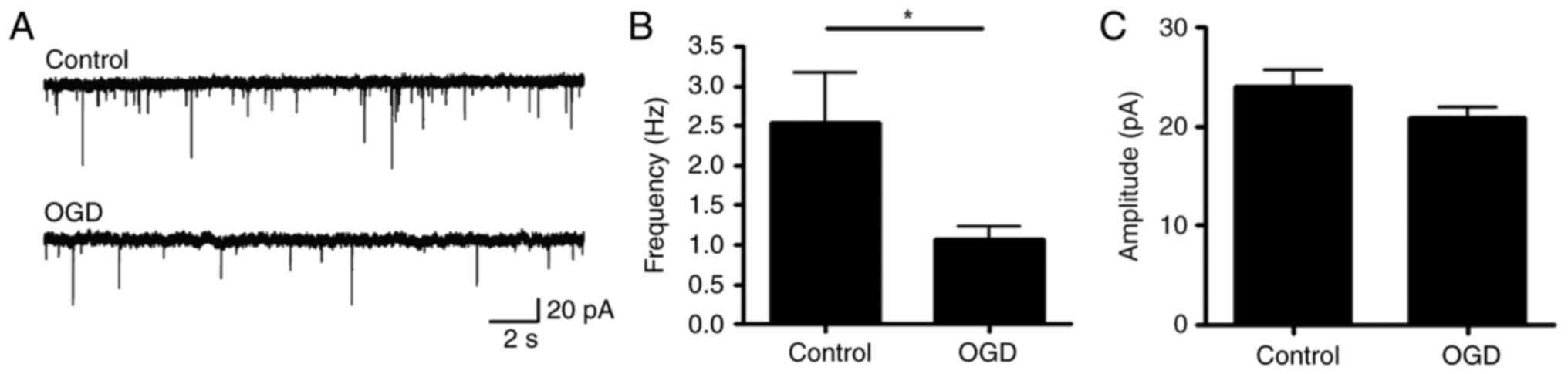
neuronal death (Fig. 1E). Finally,
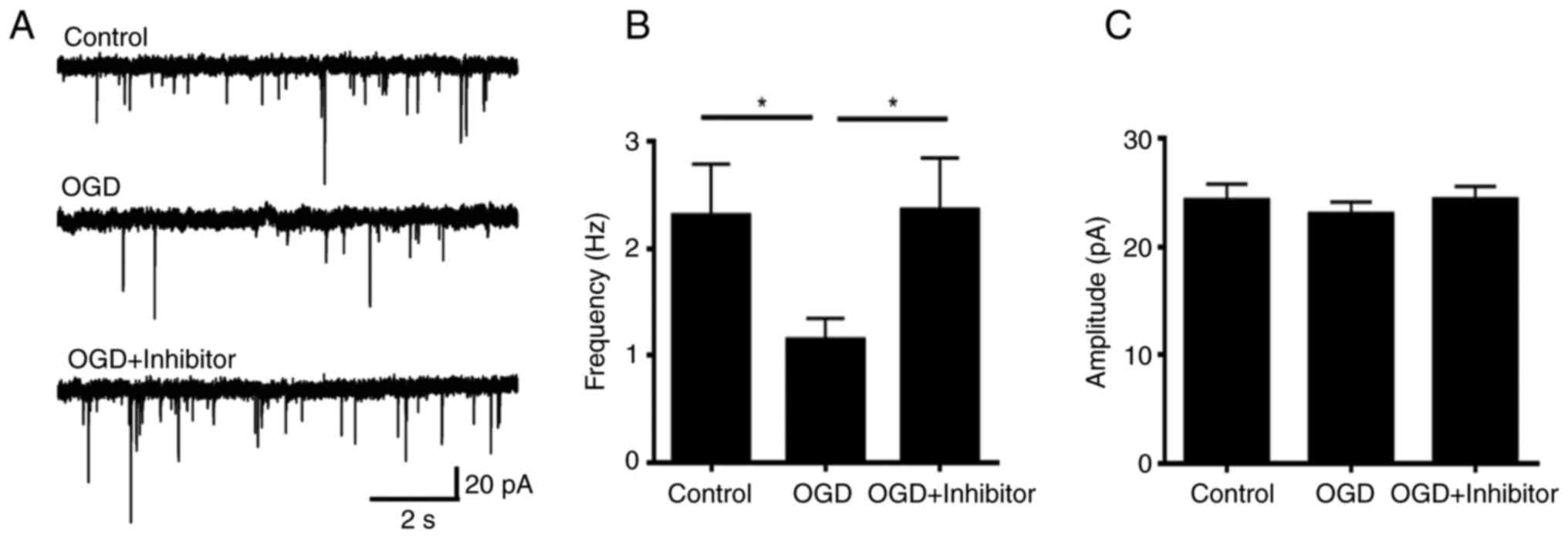
electrophysiology was performed to measure the function of the
synapses. The frequency of mEPSC was significantly decreased
following acute OGD treatment (Fig.
2), which indicated that the function of the dendritic spine
was damaged.
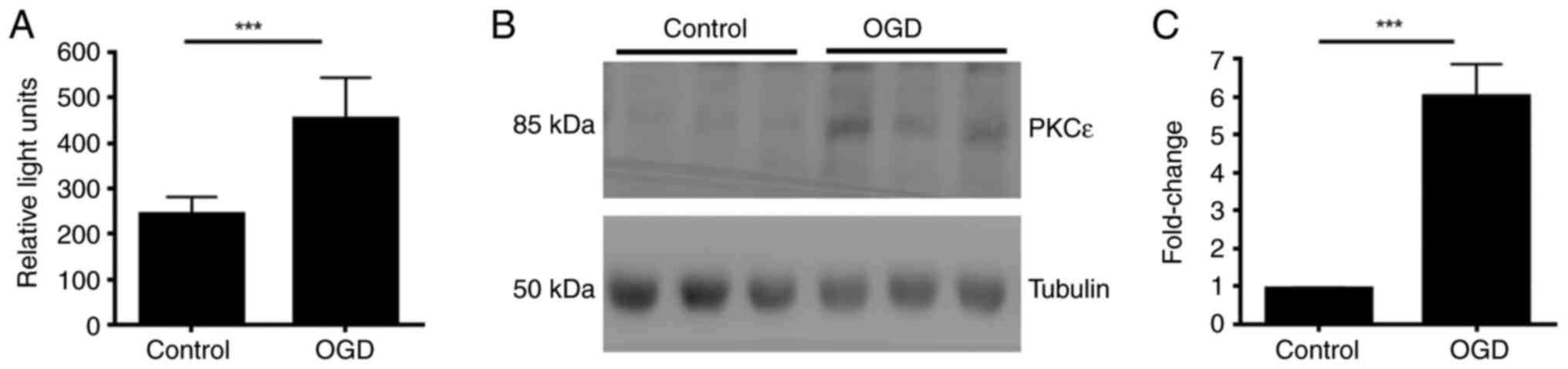
Acute OGD increases PKCε kinase
activity and protein expression levels in primary neuronal
culture
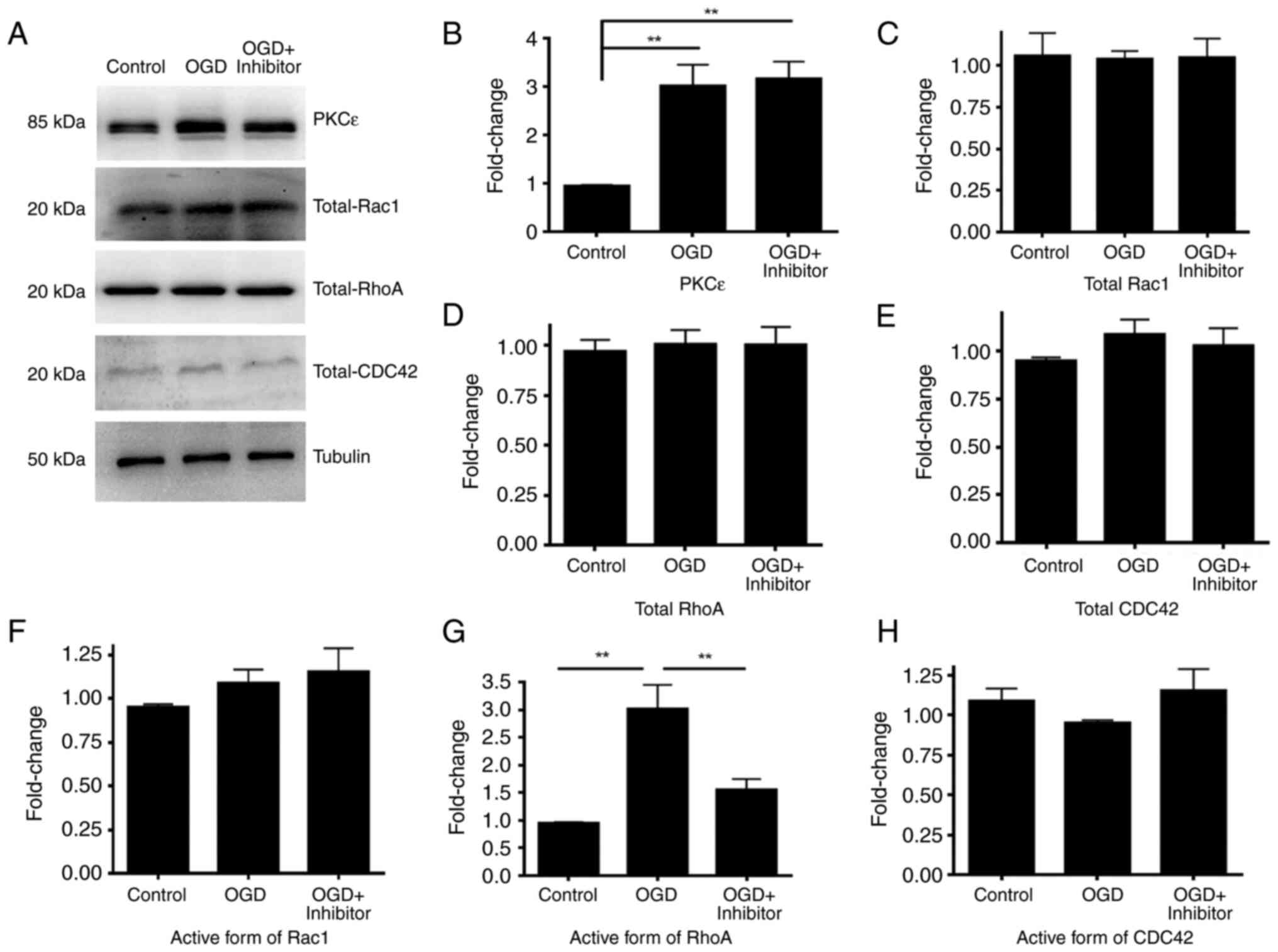
The present study investigated how acute OGD
treatment regulates PKCε. High-density cortical neurons at DIV17
were treated with OGD for 2 h before measuring PKCε protein level
and activity, respectively by western blotting and kinase activity
assay Following 2 h treatment, the kinase activity of PKCε
significantly increased compared with that in the control (Fig. 3A). In addition, protein expression
of PKCε was also significantly increased after 2 h OGD treatment
(Fig. 3B and C).
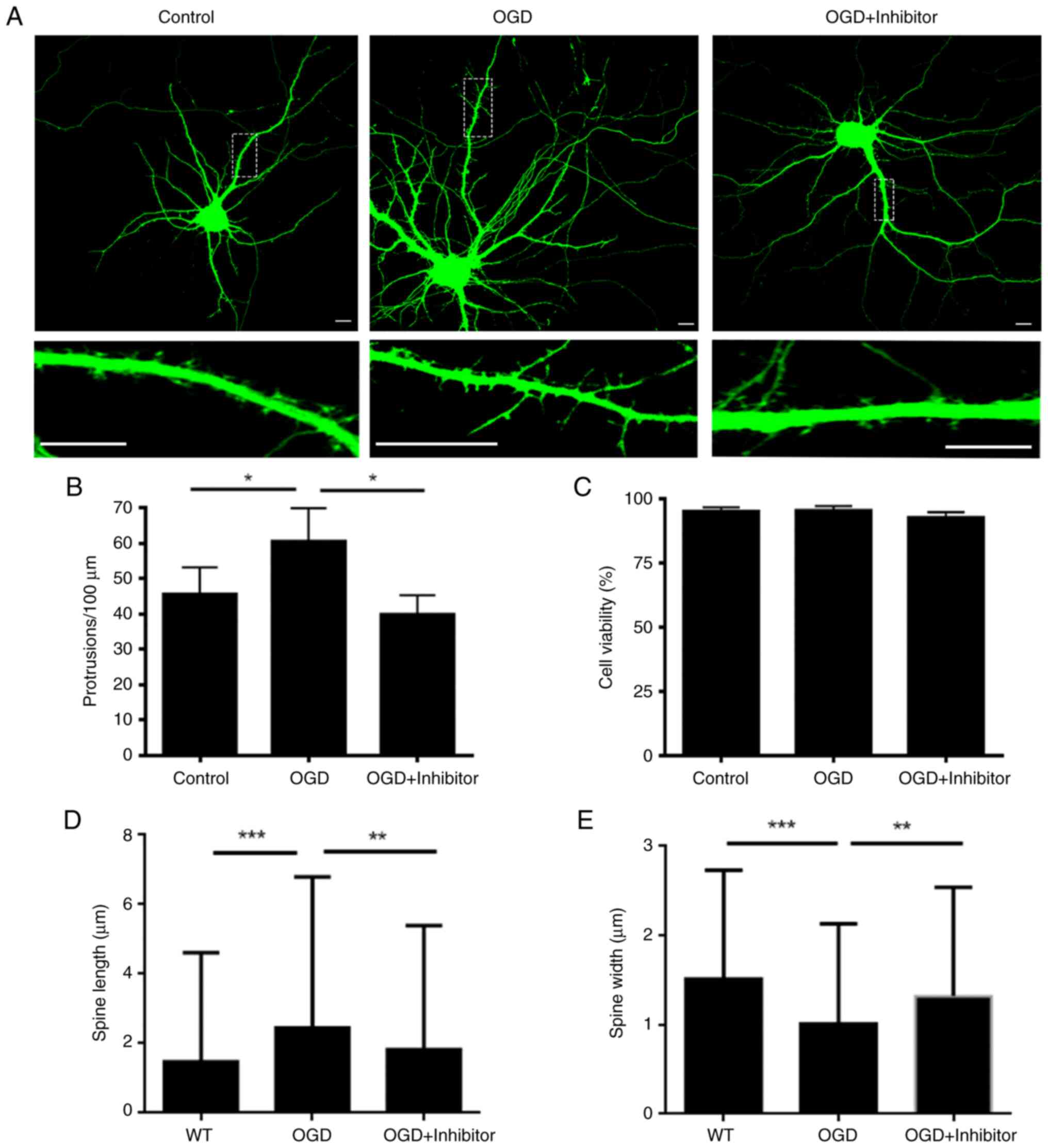
Inhibition of PKCε activity following
acute OGD rescues dendritic spine dysfunction
To investigate whether functional change of PKCε
induced dendritic spine dysfunction following OGD, we applied
PKCε-specific inhibiting peptide, EAVSLKPT. This inhibiting peptide
specifically blocks binding of PKCε with its downstream effecter,
Rho-associated coiled-coil containing protein kinase 2, and thus
inhibits PKCε function without interfering with its ATP-binding
motif (20). Confocal microscopy
results showed that the number of dendritic spines was
significantly rescued by inhibitor pre-treatment (Fig. 4A and B). MTT assay showed that the cell
viability did not change significantly following different
treatments (Fig. 4C). Dendritic
spine length and width were quantified in each group. OGD treatment
significantly increased the mean spine length whereas the mean
spine width was significantly decreased, indicating that the number
of mature spines was significantly decreased in the OGD treatment
group. On the other hand, dendritic spine defects caused by OGD
treatment were decreased in cells pre-treated with PKCε inhibitor
compared with those in the OGD treatment group (Fig. 4D and E). Electrophysiological analysis, which
was performed to measure the function of dendritic spines from
hippocampus neurons, showed that the decrease in mEPSC frequency
due to the OGD treatment was abolished by the PKCε inhibitor
pre-treatment, indicating that the synapse function was largely
preserved following OGD with PKCε inhibitor pre-treatment (Fig. 5).
PKCε-induced RhoA activation mediates
dendritic spine impairment following OGD
The morphology and function of dendritic spines are
regulated by the cytoskeleton network and Rho-GTPases are key
regulators of actin cytoskeleton rearrangement, which is the major
structure maintaining dendritic spine morphology (21). To investigate if Rho-GTPases are
involved in OGD, the activity of RhoA, Rac1 and CDC42 were measured
following OGD. High-density cortical neurons at DIV17 were treated
with OGD for 2 h. Cells were lysed and the same amount of protein
was used to measure the active forms of RhoA, Rac1 and CDC42. Total
RhoA, Rac1 and CDC42 protein levels were measured by western
blotting as a control. Following OGD treatment, the activity of
RhoA was significantly increased while the activity of Rac1 and
CDC42 was not significantly different compared with that in the
control (Fig. 6). Furthermore, the
present study investigated if RhoA was downstream of PKCε by
pre-treating cortical neurons with PKCε inhibitor peptide.
Following 24 h PKCε inhibitor pre-treatment, activation of RhoA was
blocked, indicating that RhoA is a downstream molecule of PKCε that
mediates OGD-induced dendritic spine impairment (Fig. 6).
Discussion
Brain ischemia is a severe disorder that damages
brain tissue and leads to neuronal death (19), triggering cognitive impairment or
dementia that includes alterations in learning, memory and function
needed to perform basic daily life activities (22). AD and brain ischemia are common
pathologies of ageing and their frequent co-occurrence has been
recognized (23,24). A previous study reported that
patients with AD reporting cerebrovascular events exhibit more
severe cognitive function decline compared with those without
cerebrovascular events (25).
Preclinical and clinical evidence has indicated that AD and brain
ischemia may share common pathological pathways (26-29).
However, to the best of our knowledge, the molecular mechanisms
that connect these two diseases remain unclear.
The majority (>95%) of all excitatory synapses
are located at dendritic spines and loss of excitatory synapses is
a key characteristic of AD (30).
Synapse dysfunction and dendritic spine loss are key early-stage
symptoms of cognitive function impairments (31). Contrary to irreversible neuronal
death, synapse loss is a reversible process if treated at an early
stage (32). The present study
investigated dendritic spine morphology and synapse function
following acute OGD, which distinguished this study from other
studies that focused on ischemia-induced cell death (33-36).
Members of the PKC family are involved in basic
cellular functions, such as cell metabolism and movements (37). Several studies found that several
members of the PKC family are involved in the occurrence of AD
(38-40).
For example, PKCε was found to be downregulated in patients with AD
and a chemical agonist of PKCε, bryostatin 1, is now under clinical
trial for AD treatment (41). A
previous study also found that PKCε negatively regulates dendritic
spine function through RhoA and Ephexin5 activation in the
embryonic development stage (42).
The present study used cultured neurons at DIV17, which were
considered mature in vitro (43). PKCε activity was found to be
significantly upregulated following acute OGD, which may underlie
the association between PKCε and brain ischemia-induced cognitive
function impairment.
Previous studies have indicated that following heart
ischemia, the hypoxia-inducible factor-1α upregulates PKCε through
epigenetic modifications and then impairs mitochondrial function in
cardiocytes (44). Furthermore,
studies also showed that a calcium signal in neurons is upregulated
following hypoxia, which may also regulate the protein expression
of PKCε in neurons (45-47).
Considering that the changes in protein generation usually occur
days after stimulation, it was hypothesized that the increase in
the PKCε protein levels at acute phase after 2 h OGD treatment was
due to inhibition of PKCε degradation. This should be further
investigated in future studies.
The present study used the term ‘protrusion’ to
represent all types of dendritic spine dynamics, which include a
mature spine with a typical mushroom shape, an immature spine with
a typical thin shape, a newly formed spine with stubby shape, as
well as the non-functional filopodia and all other atypical shapes
(48). Due to the limited image
resolution, further separation into sub-groups was not possible.
Because all the aforementioned types of protrusions represent a
certain moment of the spine dynamics, the present study calculated
the number of matured spines vs. all protrusions to quantified the
percentage of mature spine.
In conclusion, an OGD cell model in primary neuronal
culture was established in the present study to simulate adult
brain ischemia in vitro. Dendritic spine of the hippocampus
and cortical neurons was impaired following acute phase of OGD
treatment. Concomitantly, PKCε protein levels and activity were
also increased. Furthermore, inhibiting the function of PKCε
ameliorated excitatory synapse dysfunction caused by OGD treatment.
Finally, activity of RhoA, but not that of Rac1 or CDC42, was
increased following PKCε activation. The findings of the present
study may suggest novel therapeutic targets for synapse dysfunction
and cognitive loss following brain ischemia.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Huzhou Science
And Technology Grant (grant no. 2020GZ42).
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SW designed the study and drafted the manuscript. SW
and YW confirm the authenticity of all the raw data. XW designed
the study and performed cell culture, electrophysiology recording
and imaging. CG performed imaging studies. LL performed the primary
cell culture and blinded dendritic spine numbering. MQ performed
the statistical analysis. GS performed blinded dendritic spine
numbering. YW designed the electrophysiology study. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
Procedures involving animals were approved by the
Institutional Animal Care and Use Committee of WuXi AppTec Co.,
Ltd. (Qidong site, Jiangsu, China; approval no.
GP01-QD089-2022v1.0).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kalaria RN, Akinyemi R and Ihara M: Stroke
injury, cognitive impairment and vascular dementia. Biochim Biophys
Acta. 1862:915–925. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kalaria RN: The role of cerebral ischemia
in Alzheimer's disease. Neurobiol Aging. 21:321–330.
2000.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Pluta R, Januszewski S and Czuczwar SJ:
Brain ischemia as a prelude to Alzheimer's disease. Front Aging
Neurosci. 13(636653)2021.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Pluta R, Jabłoński M, Ułamek-Kozioł M,
Kocki J, Brzozowska J, Januszewski S, Furmaga-Jabłońska W,
Bogucka-Kocka A, Maciejewski R and Czuczwar SJ: Sporadic
Alzheimer's disease begins as episodes of brain ischemia and
ischemically dysregulated Alzheimer's disease genes. Mol Neurobiol.
48:500–515. 2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Mungas D, Jagust WJ, Reed BR, Kramer JH,
Weiner MW, Schuff N, Norman D, Mack WJ, Willis L and Chui HC: MRI
predictors of cognition in subcortical ischemic vascular disease
and Alzheimer's disease. Neurology. 57:2229–2235. 2001.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Qi JP, Wu H, Yang Y, Wang DD, Chen YX, Gu
YH and Liu T: Cerebral ischemia and Alzheimer's disease: The
expression of amyloid-beta and apolipoprotein E in human
hippocampus. J Alzheimers Dis. 12:335–341. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Pluta R, Ułamek-Kozioł M and Czuczwar SJ:
Neuroprotective and neurological/cognitive enhancement effects of
curcumin after brain ischemia injury with Alzheimer's disease
phenotype. Int J Mol Sci. 19(4002)2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Jiang T, Handley E, Brizuela M, Dawkins E,
Lewis KEA, Clark RM, Dickson TC and Blizzard CA: Amyotrophic
lateral sclerosis mutant TDP-43 may cause synaptic dysfunction
through altered dendritic spine function. Dis Model Mech.
12(dmm038109)2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Reza-Zaldivar EE, Hernández-Sápiens MA,
Minjarez B, Gómez-Pinedo U, Sánchez-González VJ, Márquez-Aguirre AL
and Canales-Aguirre AA: Dendritic spine and synaptic plasticity in
Alzheimer's disease: A focus on MicroRNA. Front Cell Dev Biol.
8(255)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Pedrazzoli M, Losurdo M, Paolone G,
Medelin M, Jaupaj L, Cisterna B, Slanzi A, Malatesta M, Coco S and
Buffelli M: Glucocorticoid receptors modulate dendritic spine
plasticity and microglia activity in an animal model of Alzheimer's
disease. Neurobiol Dis. 132(104568)2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Niftullayev S and Lamarche-Vane N:
Regulators of Rho GTPases in the nervous system: Molecular
implication in axon guidance and neurological disorders. Int J Mol
Sci. 20(1497)2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Gao K, Liu M, Ding Y, Yao M, Zhu Y, Zhao
J, Cheng L, Bai J, Wang F, Cao J, et al: A phenolic amide (LyA)
isolated from the fruits of Lycium barbarum protects against
cerebral ischemia-reperfusion injury via PKCε/Nrf2/HO-1 pathway.
Aging (Albany NY). 11:12361–12374. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Lancaster TS, Jefferson SJ and Korzick DH:
Local delivery of a PKCε-activating peptide limits ischemia
reperfusion injury in the aged female rat heart. Am J Physiol Regul
Integr Comp Physiol. 301:R1242–R1249. 2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Akaike A: Preclinical evidence of
neuroprotection by cholinesterase inhibitors. Alzheimer Dis Assoc
Disord. 20 (2 Suppl 1):S8–S11. 2006.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Villalba HAlbekairi T, Vaidya B and
Abbruscato TJ: Role of Myo-inositol in ischemic stroke outcome in a
preclinical tobacco smoke exposed mouse model. FASEB J. 33
(S1)(S500.2)2019.
|
|
16
|
Babu M, Singh N and Datta A: In vitro
oxygen glucose deprivation model of ischemic stroke: A
proteomics-driven systems biological perspective. Mol Neurobiol.
59:2363–2377. 2022.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Sun M, Bernard LP, Dibona VL, Wu Q and
Zhang H: Calcium phosphate transfection of primary hippocampal
neurons. J Vis Exp. (e50808)2013.PubMed/NCBI View
Article : Google Scholar
|
|
18
|
Juntunen M, Hagman S, Moisan A, Narkilahti
S and Miettinen S: In vitro oxygen-glucose deprivation-induced
stroke models with human neuroblastoma cell- and induced
pluripotent stem cell-derived neurons. Stem Cells Int.
2020(8841026)2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Niizuma K, Yoshioka H, Chen H, Kim GS,
Jung JE, Katsu M, Okami N and Chan PH: Mitochondrial and apoptotic
neuronal death signaling pathways in cerebral ischemia. Biochim
Biophys Acta. 1802:92–99. 2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Rechfeld F, Gruber P, Kirchmair J, Boehler
M, Hauser N, Hechenberger G, Garczarczyk D, Lapa GB,
Preobrazhenskaya MN, Goekjian P, et al: Thienoquinolines as novel
disruptors of the PKCε/RACK2 protein-protein interaction. J Med
Chem. 57:3235–3246. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Nayak RC, Chang KH, Vaitinadin NS and
Cancelas JA: Rho GTPases control specific cytoskeleton-dependent
functions of hematopoietic stem cells. Immunol Rev. 256:255–268.
2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kawabori M and Yenari MA: Inflammatory
responses in brain ischemia. Curr Med Chem. 22:1258–1277.
2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Vijayan M and Reddy PH: Stroke, vascular
dementia, and Alzheimer's disease: Molecular links. J Alzheimers
Dis. 54:427–443. 2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Takizawa C, Gemmell E, Kenworthy J and
Speyer R: A systematic review of the prevalence of oropharyngeal
dysphagia in stroke, Parkinson's disease, Alzheimer's disease, head
injury, and pneumonia. Dysphagia. 31:434–441. 2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhang X, Zhou K, Wang R, Cui J, Lipton SA,
Liao FF, Xu H and Zhang YW: Hypoxia-inducible factor 1alpha
(HIF-1alpha)-mediated hypoxia increases BACE1 expression and
beta-amyloid generation. J Biol Chem. 282:10873–10880.
2007.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zlokovic BV and Griffin JH: Cytoprotective
protein C pathways and implications for stroke and neurological
disorders. Trends Neurosci. 34:198–209. 2011.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Hook V, Yoon M, Mosier C, Ito G, Podvin S,
Head BP, Rissman R, O'Donoghue AJ and Hook G: Cathepsin B in
neurodegeneration of Alzheimer's disease, traumatic brain injury,
and related brain disorders. Biochim Biophys Acta Proteins Proteom.
1868(140428)2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Fu Z, Caprihan A, Chen J, Du Y, Adair JC,
Sui J, Rosenberg GA and Calhoun VD: Altered static and dynamic
functional network connectivity in Alzheimer's disease and
subcortical ischemic vascular disease: Shared and specific brain
connectivity abnormalities. Hum Brain Mapp. 40:3203–3221.
2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Manda-Handzlik A and Demkow U: The brain
entangled: The contribution of neutrophil extracellular traps to
the diseases of the central nervous system. Cells.
8(1477)2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Rochefort NL and Konnerth A: Dendritic
spines: From structure to in vivo function. EMBO Rep. 13:699–708.
2012.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Counts SE, Nadeem M, Lad SP, Wuu J and
Mufson EJ: Differential expression of synaptic proteins in the
frontal and temporal cortex of elderly subjects with mild cognitive
impairment. J Neuropathol Exp Neurol. 65:592–601. 2006.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Qi Y, Yu S, Du Z, Qu T, He L, Xiong W, Wei
W, Liu K and Gong S: Long-term conductive auditory deprivation
during early development causes irreversible hearing impairment and
cochlear synaptic disruption. Neuroscience. 406:345–355.
2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Forouzanfar F, Asadpour E, Hosseinzadeh H,
Boroushaki MT, Adab A, Dastpeiman SH and Sadeghnia HR: Safranal
protects against ischemia-induced PC12 cell injury through
inhibiting oxidative stress and apoptosis. Naunyn Schmiedebergs
Arch Pharmacol. 394:707–716. 2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Li C, Sun G, Chen B, Xu L, Ye Y, He J, Bao
Z, Zhao P, Miao Z, Zhao L, et al: Nuclear receptor coactivator
4-mediated ferritinophagy contributes to cerebral ischemia-induced
ferroptosis in ischemic stroke. Pharmacol Res.
174(105933)2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Lee ML, Sulistyowati E, Hsu JH, Huang BY,
Dai ZK, Wu BN, Chao YY and Yeh JL: KMUP-1 ameliorates
ischemia-induced cardiomyocyte apoptosis through the NO-cGMP-MAPK
signaling pathways. Molecules. 24(1376)2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Dietz RM, Cruz-Torres I, Orfila JE, Patsos
OP, Shimizu K, Chalmers N, Deng G, Tiemeier E, Quillinan N and
Herson PS: Reversal of global ischemia-induced cognitive
dysfunction by delayed inhibition of TRPM2 ion channels. Transl
Stroke Res. 11:254–266. 2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Rosse C, Linch M, Kermorgant S, Cameron
AJ, Boeckeler K and Parker PJ: PKC and the control of localized
signal dynamics. Nat Rev Mol Cell Biol. 11:103–112. 2010.PubMed/NCBI View
Article : Google Scholar
|
|
38
|
Ortiz-Sanz C, Balantzategi U,
Quintela-López T, Ruiz A, Luchena C, Zuazo-Ibarra J,
Capetillo-Zarate E, Matute C, Zugaza JL and Alberdi E: Amyloid
β/PKC-dependent alterations in NMDA receptor composition are
detected in early stages of Alzheimer's disease. Cell Death Dis.
13(253)2022.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Sasahara T, Satomura K, Tada M, Kakita A
and Hoshi M: Alzheimer's Aβ assembly binds sodium pump and blocks
endothelial NOS activity via ROS-PKC pathway in brain vascular
endothelial cells. iScience. 24(102936)2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Sajan MP, Braun U, Leitges M, Park C,
Diamond DM, Wu J, Hansen BC, Duncan MA, Apostolatos CA, Apostolatos
AH, et al: Atypical PKC controls β-secretase expression and thereby
regulates production of Alzheimer plaque precursor Aβ in brain and
insulin receptor degradation in liver. Metab Clin Exper. 104
(Suppl)(S154112)2020.
|
|
41
|
Schrott LM, Jackson K, Yi P, Dietz F,
Johnson GS, Basting TF, Purdum G, Tyler T, Rios JD, Castor TP and
Alexander JS: Acute oral bryostatin-1 administration improves
learning deficits in the APP/PS1 transgenic mouse model of
Alzheimer's disease. Curr Alzheimer Res. 12:22–31. 2015.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Schaffer TB, Smith JE, Cook EK, Phan T and
Margolis SS: PKCε inhibits neuronal dendritic spine development
through dual phosphorylation of Ephexin5. Cell Rep.
25:2470–2483.e8. 2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Liao MH, Xiang YC, Huang JY, Tao RR, Tian
Y, Ye WF, Zhang GS, Lu YM, Ahmed MM, Liu ZR, et al: The disturbance
of hippocampal CaMKII/PKA/PKC phosphorylation in early experimental
diabetes mellitus. CNS Neurosci Ther. 19:329–336. 2013.PubMed/NCBI View Article : Google Scholar
|
|
44
|
McCarthy J, Lochner A, Opie LH, Sack MN
and Essop MF: PKCε promotes cardiac mitochondrial and metabolic
adaptation to chronic hypobaric hypoxia by GSK3β inhibition. J Cell
Physiol. 226:2457–2468. 2011.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Sommer N, Strielkov I, Pak O and Weissmann
N: Oxygen sensing and signal transduction in hypoxic pulmonary
vasoconstriction. Eur Respir J. 47:288–303. 2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Mungai PT, Waypa GB, Jairaman A, Prakriya
M, Dokic D, Ball MK and Schumacker PT: Hypoxia triggers AMPK
activation through reactive oxygen species-mediated activation of
calcium release-activated calcium channels. Mol Cell Biol.
31:3531–3545. 2011.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Shimoda LA and Undem C: Interactions
between calcium and reactive oxygen species in pulmonary arterial
smooth muscle responses to hypoxia. Respir Physiol Neurobiol.
174:221–229. 2010.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Sanchez-Arias JC, Candlish RC, van der
Slagt E and Swayne LA: Pannexin 1 regulates dendritic protrusion
dynamics in immature cortical neurons. eNeuro.
7(ENEURO.0079-20.2020)2020.PubMed/NCBI View Article : Google Scholar
|