1. Introduction
The kidney has excretory, metabolic and endocrine
functions. Among all the organs of the human body, the kidney has
one of the highest blood flow rates per gram of organ tissue. Based
on its excretory function, it intervenes in maintaining the
hydro-electrolytic balance of the body, by forming and eliminating
urine in the adequate amounts and with the adequate concentrations
of ions.
To perform such roles, the kidney requires its
normal blood flow, which is ~20-25% of the resting cardiac output.
Physiologically, the distribution of renal blood flow is not
uniform. From the entire renal blood flow, the cortex receives
85-90%, the external medulla receives 10%, and the internal medulla
receives only 2%. The kidney has multiple, complex, and
interrelated mechanisms for the autoregulation of blood flow
(1). It is well known that, in the
human kidney, autoregulation occurs for values of the systemic
arterial blood pressure (ABP) approximately between 60 and 180 mm
Hg. Thus, the renal blood flow remains fairly constant despite ABP
changes between these values. There is a linear association between
ABP and renal blood flow beyond the respective pressure interval.
As a result, variations in systemic blood pressure within the
mentioned ABP range are accompanied only by transient changes in
the glomerular filtration rate (GFR).
Stimulation of the sympathetic nervous system causes
intrarenal vasomotor changes. It is well known that sympathetic
hyperstimulation causes more pronounced vasoconstriction of the
afferent arteriole, with reduced glomerular blood flow and reduced
hydrostatic pressure in the glomerular capillaries. Consequently,
the rate of glomerular filtration is markedly reduced. If blood
pressure values remain high, renal dysfunction occurs, with the
onset of chronic kidney disease (CKD).
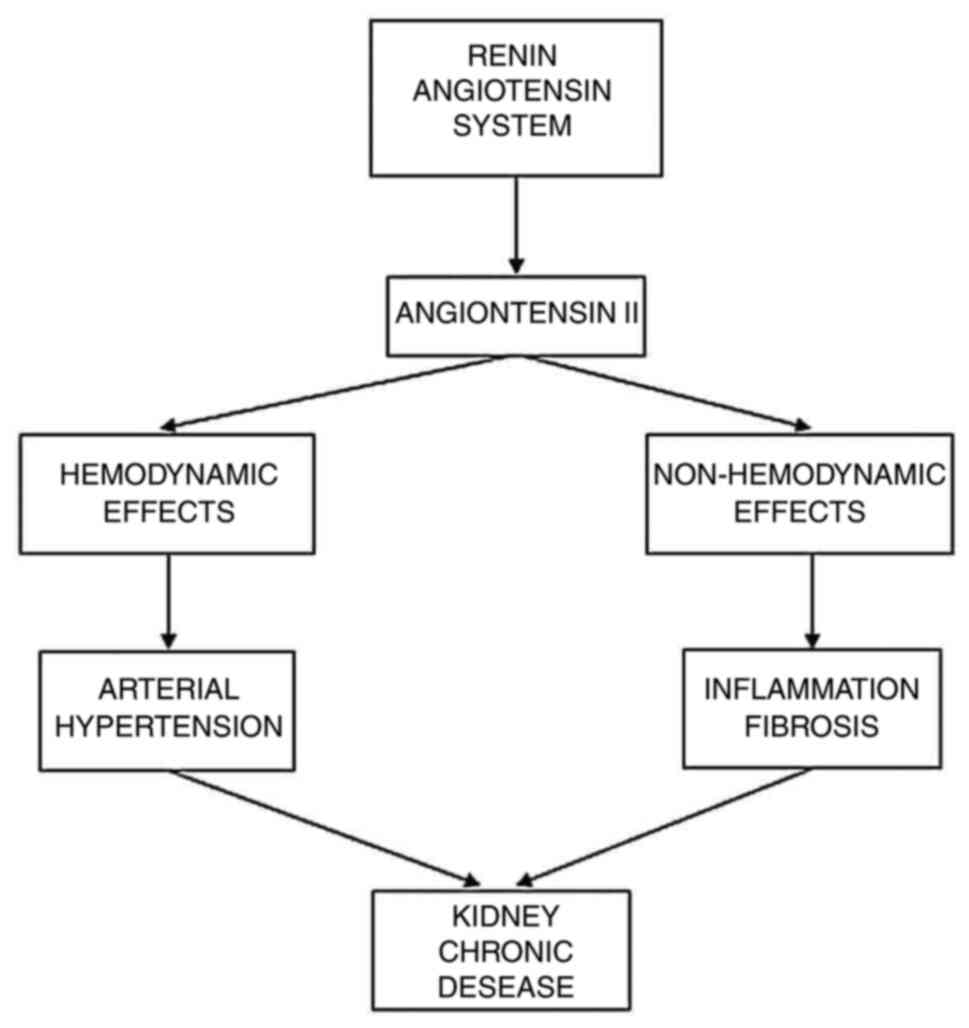
One of the pathophysiological mechanisms involved in
the increase of blood pressure and in the development of arterial
hypertension is represented by the increase of the circulating
level of angiotensin II (Ang II), due to the excessive activation
of the renin angiotensin system (RAS). Damage of the target organs,
which occurs as a result of increased ABP, is accompanied by a
higher risk of cardiovascular events. In addition, Ang II
stimulates inflammation and atherosclerosis in the target organs
(2).
Arterial hypertension is closely related to CKD,
while CKD is promoted and perpetuated by the chronic kidney
inflammation that involves the increased RAS activity (both the
circulating one and local, renal one) (Fig. 1).
2. Chronic kidney disease
The renal function is assessed by evaluation of GFR,
which is normally ~125 ml/min. Structural and/or functional changes
of the kidneys may correspond to various extents of true renal
injury/lesion. If renal injury is acute, then the kidney has the
ability to recover, but if the damage is chronic, then the ability
of the kidney to regenerate is affected/lost and CKD tends to
develop and progress (3). This
irreversible disease, which affects 5-7% of the population of the
world, thus progresses to the final stage, a situation that
requires the replacement of the kidney function by dialysis or
transplantation (4).
CKD is a clinical syndrome characterized by GFR
decrease below 60 ml/min/1.73 m2, lasting at least three
months. Arterial hypertension and diabetes are the main etiological
causes of CKD, but CKD can also be a complication of other diseases
such as infectious glomerulonephritis, urethral obstruction,
genetic damage, vasculitis, autoimmune diseases, and drugs
(5). CKD is a huge public health
problem, due to the increasing incidence of arterial hypertension,
diabetes (6), obesity, and life
expectancy and it is the 18th leading cause of death worldwide.
Some 30-40% of the individuals diagnosed with nephropathy develop
kidney failure in 10-15 years and these individuals need to improve
their kidney function, which involves huge costs (7).
Regardless of the cause of CKD, the common elements
encountered are inflammation, followed by the development of
fibrosis (8) and the progressive
loss of kidney function. Over time, these renal changes lead to the
progression of the final stage of CKD. Inflammation of low
intensity, but persistent, is characterized by the presence of
leukocyte infiltration and the secretion of proinflammatory
cytokines. Inflammatory phenomena at the renal level occur a few
years before the start of specific clinical symptoms (9) and represent a protective response to
potential endogenous and/or exogenous aggressors (10).
Progressive renal sclerosis, which develops after
the uncontrolled inflammatory response, is a major risk factor, by
increasing the pressure in the glomerular capillaries (11). Several changes occur: A decrease in
the number of podocytes, loss of the electrical charge of the
glomerular basement membrane, and mesangial distension. All these,
together with the endothelial dysfunction, lead to
glomerulosclerosis.
Fibrosis, which develops after kidney inflammation,
involves tissue repair, and is characterized by excessive
accumulation of extracellular matrix components, which gradually
affects the cellular tissue architecture (12,13).
The progression of CKD to the final stage is the consequence of
losing the renal cells and their replacement with extracellular
matrix, in the glomeruli and in other renal tissue structures
(11). With a role in preserving
renal function, the processes of inflammation and fibrosis affect
the specialized renal cells (tubular epithelial cells, podocytes,
and glomerular mesangial cells), as well as the vascular cells
(13). Glomerulosclerosis,
vascular sclerosis and tubulo-interstitial fibrosis appear in the
final stage of CKD (3).
3. Role of hypertension in chronic kidney
disease
Arterial hypertension, the leading modifiable risk
factor for a variety of cardiovascular diseases, continues to be
underdiagnosed and undertreated and/or untreated worldwide. The
kidneys, heart, brain, blood vessels, and eyes are the main target
organs that are affected by increased ABP. Over 90% of hypertension
in CKD patients is closely related to endothelial dysfunction and
it is either an etiological factor or a complication of CKD. It is
not known exactly whether impaired renal function in CKD is the
cause and/or consequence of increased blood pressure. Being
surpassed only by diabetic nephropathy, hypertensive nephropathy is
the second cause of CKD progression to the final stage (14) and it is considered an independent
risk factor for the decline of the GFR (15).
The GFR physiologically has a linear association
with ABP, classically designated pressure diuresis. Furthermore,
chronically high ABP produces renal effects at multiple levels
including vascular, glomerular, interstitial, tubular, and also
that of the renal immune system. High ABP, the cause and effect of
CKD, promotes and perpetuates inflammation and fibrosis at the
glomerular, interstitial, and tubular levels and in addition causes
important changes in renal microcirculation. Renal disease followed
by increased ABP, hypertensive nephropathy or hypertensive
nephrosclerosis, is associated with histopathological changes in
the pre- and intra-glomerular microvascularization and with
tubulo-interstitial changes (16-18).
From a pathophysiological point of view, nephrosclerosis can set in
slowly, with progressive changes in the intima of the arteries and
arterioles of the renal parenchyma, or rapidly, with histological
changes in fibrinoid necrosis and/or proliferation of myointimal
cells.
In addition, atherosclerotic changes (hyalinization)
occur in the afferent arterioles. Both processes contribute to the
occurrence of pulsating blood flow in the renal arterioles
(19) which causes ischemic
glomerular changes (focal and segmental glomerular sclerosis) and
tubular atrophy, accompanied by interstitial fibrosis.
Tubulo-interstitial hypoxia is instated and, together with hormonal
vasoconstriction (including RAS and/or Ang II), it progresses CKD
to the final stage (20). The
immune system can also be an important etiopathogenic contributor
to the development and maintenance of high ABP, as further
discussed in the next section.
4. Renin-angiotensin system
RAS is an important regulator of systemic ABP, but
other hormones are also involved, such as catecholamines, thyroid
hormones, corticosteroids, and sex hormones (12).
Classically, the biologically active product of RAS,
which is Ang II, is formed by a cascade of enzymatic reactions
(Fig. 2), which involves two
important enzymes: Renin and angiotensin-converting enzyme (ACE).
Secreted by exocytosis from the cells of the renal juxtaglomerular
apparatus (RJA), renin acts upon angiotensinogen (a serpentine
polypeptide, synthesized mainly in the liver) and forms angiotensin
I (Ang I) (apparently an inactive decapeptide). Low ABP,
hypovolemia, hyponatremia, and sympathetic stimulation increase
renin synthesis in RJA.
Under the action of ACE, released by the lung
tissue, Ang II is formed from Ang I. ACE2 is a homologous enzyme of
ACE, which generates angiotensin 1-9 (Ang 1-9) from Ang I and
angiotensin 1-7 (Ang 1-7) from Ang II. Due to its vasodilating and
Ang II-antagonizing effects, Ang 1-7 is considered a mechanism of
counter-regulation of the classical RAS. Ang II can also be
generated from Ang I, through the action of an enzyme called
chymase.
In order to exert its effects, Ang II acts on
specific receptors: The angiotensin receptor type 1 (AT1) and the
angiotensin receptor type 2 (AT2) (Fig. 2). Ang II binding to AT1 mediates
the following: Sodium retention, vasoconstriction (preferentially
in the renal efferent arteriole), thirst stimulation, increased
sympathetic activity, and increased release of aldosterone from the
glomerular area of the adrenal gland. In the fetus, Ang II binding
to the AT2 receptor counteracts the effects of AT1 receptor
activation by Ang II and has vasodilator, anti-inflammatory and
antifibrotic effects, but in adults these effects are
irrelevant.
All RAS components can be produced locally, in
tissues and organs, and act independently of systemic RAS. The
kidney possesses all the components of RAS, and the amount of Ang
II produced in the kidney is 1,000 times higher than the
circulating one (21). Ang II
influences both renal hemodynamics and tubular function. Ang II
decreases renal blood flow and decreases the GFR, exerting effects
both on renal microvasculature and on the glomerular mesangium. Ang
II predominantly contracts preglomerular arterioles. When the pre-
and post-glomerular resistance increases in parallel, the outcome
is an increased GFR. Regarding tubular function, Ang II has the
following effects: It causes sodium and water retention (mediated
by Ang II-stimulated aldosterone secretion and modulated by
hemodynamic changes in the peritubular capillaries); it causes
cellular hypertrophy; and it induces oxidative stress. Local Ang II
production directly causes podocyte impairment via AT1 receptor
activation, regardless of hemodynamic changes (22,23).
Decreased blood pressure in response to Ang II
inhibition is more pronounced during a low sodium diet. Normally,
in hypertensive patients with kidney disease, ACE-I and sartans are
used with other drugs, generally diuretics. It has been observed
that ACE-I and sartans lead to a marked decrease in systemic ABP,
accompanied by decreased GFR, when the volume of extracellular
fluid is reduced. In patients with severe kidney disease, the
related arterioles become less responsive to ACE-I and sartans
(24).
Beyond the hemodynamic effects it possesses, Ang II
behaves similar to a cytokine, with proinflammatory and profibrotic
properties. Ang II-induced fibrosis is achieved by a dual
mechanism: i) By a direct effect on the process of
synthesis/degradation of the extracellular matrix; and ii)
indirectly, by increasing the expression of profibrotic factors
with a crucial role in mesangial proliferation (25), such as transforming growth factor β
(TGF-β) and platelet-derived growth factor (PDGF) (26). Renin and aldosterone also possess
profibrotic effects by increasing TGF-β expression. Both the renal
cells (especially the glomerular ones) and the macrophages
recruited in the kidneys express TGF-β (27). TGF-β is involved in modulating the
renal immune response by regulating the proliferation,
differentiation and migration of inflammatory cells (23), while stimulating the proliferation
of fibroblasts (28). Together
with Ang II, TGF-β induces oxidative stress and stimulates
extracellular matrix synthesis, in parallel with inhibition of
proteases that degrade the extracellular matrix. Ang II exerts
these effects through AT1 receptors. Furthermore, following renal
injury, an increase in apoptosis is observed, a phenomenon induced
by Ang II through AT2 receptors. Animal studies have shown the
possible involvement of the AT2 receptor in the regression of renal
fibrosis, by reducing post-aggression vascular damage. Renal
fibrosis, associated with glomerulosclerosis and interstitial renal
fibrosis is characterized by atrophy and tubular dilation, and
increased fibrogenesis and collagen deposits in the extracellular
matrix (29,30).
In previous studies by the authors some systemic and
renal aspects of the proinflammatory effects of Ang II were
investigated, using the Ang II-induced hypertension rat model
(17,18,31).
In fact, in these studies an attempt was made to gain insight into
the associations among Ang II, hypertension, and inflammation. This
trilateral association could be relevant to the topic of the
present review, particularly regarding the disease stages just
before and/or immediately after the true onset of CKD (as defined
in terms of diminished GFR for at least three months).
5. Conclusions
In conclusion, Ang II acts on the kidney through a
dual mechanism: Indirectly, by increasing systemic ABP and
directly, by stimulating the inflammatory and fibrotic processes in
the vascular wall and in the renal tissue. Kidney inflammation
associated with high ABP is an area open to research due to
insufficient available data. Expanding pathophysiological knowledge
with regard to hypertension and kidney disease has an applicative
potential, opening up new directions in the therapeutic approach of
these diseases.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
All the authors substantially contributed to each of
the following aspects of this review: conception and design (mainly
MAM, AC, ACP, DMT, DNS and ILS) and selection, analysis and
interpretation of cited references (all the authors, but mainly
MAM, AC, ACP, DMT, CS, RAS and BH). Moreover, all the authors were
involved in drafting of the manuscript (mainly MAM, AC, ACP, DCB,
DEB and DMT) and in revising it critically for important
intellectual content (mainly DNS, CS, RAS and ILS). All the authors
have read and approved the final version of the manuscript to be
published. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Burke M, Pabbidi MR, Farley J and Roman
RJ: Molecular mechanisms of renal blood flow autoregulation. Curr
Vasc Pharmacol. 12:845–858. 2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Touyz RM: Molecular and cellular
mechanisms in vascular injury in hypertension: Role of angiotensin
II. Curr Opin Nephrol Hypertens. 14:125–131. 2005.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Nogueira A, Piresi MJ and Oliveira PA:
Pathophysiological mechanisms of renal fibrosis: A review of animal
models and therapeutic strategies. In vivo. 31:1–22.
2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Hewitson TD: Renal tubulointerstitial
fibrosis: Common but never simple. Am J Physiol Renal Physiol.
296:F1239–F1244. 2009.PubMed/NCBI View Article : Google Scholar
|
|
5
|
López-Novoa JM, Martínez-Salgado C,
Rodríguez-Peña AB and López-Hernández FJ: Common pathophysiological
mechanisms of chronic kidney disease: Therapeutic perspectives.
Pharmacol Ther. 128:61–81. 2010.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Tanase DM, Gosav EM, Costea CF, Ciocoiu M,
Lacatusu CM, Maranduca MA, Ouatu A and Floria M: The intricate
relationship between type 2 diabetes mellitus (T2DM), insulin
resistance (IR), and nonalcoholic fatty liver disease (NAFLD). J
Diabetes Res. 2020(3920196)2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Remuzzi G, Benigni A and Remuzzi A:
Mechanisms of progression and regression of renal lesions of
chronic nephropathies and diabetes. J Clin Invest. 116:288–296.
2006.PubMed/NCBI View
Article : Google Scholar
|
|
8
|
Owens EP, Vesey DA, Kassianos AJ, Healy H,
Hoy WE and Gobe GC: Biomarkers and the role of mast cells as
facilitators of inflammation and fibrosis in chronic kidney
disease. Transl Androl Urol. 8 (Suppl 2):S175–S183. 2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Fan J, Xie K, Wang L, Zheng N and Yu X:
Roles of inflammasomes in inflammatory kidney diseases. Mediators
Inflamm. 2019(2923072)2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Meng XM, Nikolic-Paterson DJ and Lan HY:
Inflammatory processes in renal fibrosis. Nat Rev Nephrol.
10:493–503. 2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Fogo AB: Progression and potential
regression of glomerulosclerosis. Kidney Int. 59:804–819.
2001.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Gupta J, Mitra N, Kanetsky PA, Devaney J,
Wing MR, Reilly M, Shah VO, Balakrishnan VS, Guzman NJ, Girndt M,
et al: Association between albuminuria, kidney function, and
inflammatory biomarker profile in CKD in CRIC. Clin J Am Soc
Nephrol. 7:1938–1946. 2012.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wynn TA and Ramalingam TR: Mechanisms of
fibrosis: Therapeutic translation for fibrotic disease. Nat Med.
18:1028–1040. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Sievers LK and Eckardt K: Molecular
mechanisms of kidney injury and repair in arterial hypertension.
Int J Mol Sci. 20(2138)2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Gabriele MM and Nogueira PCK: Management
of hypertension in CAKUT: Protective factor for CKD. Front Pediatr.
7(222)2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Liang S, Le W, Liang D, Chen H, Xu F, Chen
H, Liu Z and Zeng C: Clinico-pathological characteristics and
outcomes of patients with biopsy-proven hypertensive
nephrosclerosis: A retrospective cohort study. BMC Nephrol.
17(42)2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Maranduca MA, Tarniceriu CC, Carasevici E
and Cojocaru E: Interaction between angiotensin II, hypertension
and inflammation in rat kidney. Rev Med Soc Nat. 119:791–797.
2015.
|
|
18
|
Mărănducă MA, Cojocaru E and Carasevici E:
Correlations between arterial hypertension induced by angiotensin
II and systemic inflammation in rat. Rev Med Chir Soc Med Nat Iasi.
120:77–82. 2016.PubMed/NCBI
|
|
19
|
Hill GS: Hypertensive nephrosclerosis.
Curr Opin Nephrol Hypertens. 17:266–270. 2008.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Jordan J: Device-based approaches for the
treatment of arterial hypertension. Curr Hypertens Rep.
19(59)2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
VanDeVoorde RG and Mitsnefes MM:
Hypertension and CKD. Adv Chronic Kidney Dis. 18:355–361.
2011.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Schaefer B and Wühl E: Educational paper:
Progression in chronic kidney disease and prevention strategies.
Eur J Pediatr. 171:1579–1588. 2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Klahr S and Morrissey JJ: The role of
vasoactive compounds, growth factors and cytokines in the
progression of renal disease. Kidney Int Suppl. 57:S7–S14.
2000.PubMed/NCBI
|
|
24
|
Liang XB, Ma LJ, Naito T, Wang Y, Madaio
M, Zent R, Pozzi A and Fogo AB: Angiotensin type 1 receptor blocker
restores podocyte potential to promote glomerular endothelial cell
growth. J Am Soc Nephrol. 17:1886–1895. 2006.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Liebau MC, Lang D, Böhm J, Endlich N, Bek
MJ, Witherden I, Mathieson PW, Saleem MA, Pavenstädt H and Fischer
KG: Functional expression of the renin-angiotensin system in human
podocytes. Am J Physiol Renal Physiol. 290:F710–F719.
2006.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kobori H, Ozawa Y, Suzaki Y,
Prieto-Carrasquero MC, Nishiyama A, Shoji T, Cohen EP and Navar LG:
Young Scholars Award Lecture: Intratubular angiotensinogen in
hypertension and kidney diseases. Am J Hypertens. 19:541–550.
2006.PubMed/NCBI View Article : Google Scholar
|
|
27
|
el Nahas AM, Muchaneta-Kubara EC, Essawy M
and Soylemezoglu O: Renal fibrosis: Insights into pathogenesis and
treatment. Int J Biochem Cell Biol. 29:55–62. 1997.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Rodríguez-Peña A, Prieto M, Duwel A, Rivas
JV, Eleno N, Pérez-Barriocanal F, Arévalo M, Smith JD, Vary CP,
Bernabeu C and López-Novoa JM: Up-regulation of endoglin, a
TGF-beta-binding protein, in rats with experimental renal fibrosis
induced by renal mass reduction. Nephrol Dial Transplant. 16 (Suppl
1):S34–S39. 2001.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Cattran DC and Rao P: Long-term outcome in
children and adults with classic focal segmental
glomerulosclerosis. Am J Kidney Dis. 32:72–79. 1998.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Pei Y, Cattran D, Delmore T, Katz A, Lang
A and Rance P: Evidence suggesting under-treatment in adults with
idiopathic focal segmental glomerulosclerosis. Regional
glomerulonephritis registry study. Am J Med. 82:938–944.
1987.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Maranduca MA, Tanase DM, Branisteanu DC,
Serban DN, Branisteanu DE and Serban IL: Involvement of
proinflammatory cytokines in angiotensin II-induced hypertension in
rat. Exp Ther Med. 20:3541–3545. 2020.PubMed/NCBI View Article : Google Scholar
|