Introduction
Multiple organ dysfunction caused by sepsis and
septic shock is an important issue in acute and critical care.
Sepsis-induced myocardial injury is the most common complication of
organ dysfunction (1). Among adult
patients with sepsis, 25-50% of them present with myocardial
injury, which is an indicator of poor prognosis of sepsis and can
increase the mortality rate to 70% (2). Therefore, active treatment of
sepsis-induced myocardial injury is necessary to improve the
survival rate and prognosis of patients.
Propofol is the most commonly used intravenous
anesthetic in clinical practice (3). Previous studies have reported that
propofol has a protective effect on the myocardium (4-6).
Propofol mediates cardioprotective effects against myocardial
ischemia/reperfusion injury in a microRNA-451/high mobility group
box 1 (HMGB1)-dependent manner (4), alleviates doxorubicin-induced toxic
injury through activation of Nrf2/GPx4 signaling (5), and protects the myocardium from
ischemia-reperfusion injury by inhibiting ferroptosis through the
AKT/p53 signaling pathway (6).
These results suggested that propofol may have important protective
effects on cardiac function. In addition, a previous study revealed
that propofol significantly improves the survival rate of rats with
sepsis, and has protective effects on the liver, kidneys and heart
(7). Moreover, a recent study
demonstrated that propofol inhibits inflammation and apoptosis
through the PPARγ/HMGB1/NLRP3 axis to improve endotoxin-induced
cardiomyocyte injury (8). These
results indicated that propofol may participate in the protective
effects against septic injury through multiple pathways.
It has been shown that propofol effectively
activates sirtuin 1 (SIRT1), and reduces lung injury, liver injury
and human umbilical vein endothelial cell injury induced by high
glucose (9-11).
SIRT1 is a key regulator of autophagy, which has recently been
recognized as a new selective substrate for nuclear autophagy in
senescence and aging (12). SIRT1
promotes autophagy through AMPK activation, thereby protecting
cardiomyocytes from hypoxia (13).
Melatonin regulates apoptosis and autophagy by activating SIRT1 in
mice to prevent heart dysfunction induced by sepsis (14). In addition, it has been shown that
propofol has an important regulatory effect on autophagy, thus
affecting cellular homeostasis (15).
The present study hypothesized that propofol may
activate SIRT1-mediated autophagy in lipopolysaccharide
(LPS)-induced myocardial injury and aimed to investigate this
hypothesis in vitro.
Materials and methods
Cell culture
The rat cardiomyocyte H9C2 cells (MilliporeSigma)
were cultured in DMEM supplemented with 10% FBS (both from Gibco;
Thermo Fisher Scientific, Inc.) at 37˚C in an atmosphere containing
5% CO2. Cells were sub-cultured when they reached 90%
confluence. For the in vitro sepsis-induced myocardial
injury model, H9C2 cells were sub-cultured for 12 h and then
induced with LPS (1 µg/ml; cat. no. HY-D1056; MedChemExpress) for
24 h at 37˚C (16). Concentrations
of 12.5, 25 and 50 µM propofol (cat. no. HY-B0649; MedChemExpress)
were selected to pretreat H9C2 cells at 37˚C for 24 h prior to LPS
treatment (8,17). In the inhibition experiments, cell
were pre-treated for 30 min with the autophagy inhibitor
3-methyladenine (3-MA; 10 µM; cat. no. HY-19312; MedChemExpress)
and the SIRT1 inhibitor EX527 (100 nM; cat. no. HY-15452;
MedChemExpress) at 37˚C, followed by LPS induction and propofol
treatment (18,19). Untreated H9C2 cells were regarded
as the control group.
Cell Counting Kit 8 (CCK8) assay
The viability of H9C2 cells was measured using the
CCK8 assay (Nanjing Jiancheng Bioengineering Institute), according
to the manufacturer's instructions. The absorbance was measured at
a wavelength of 450 nm using a microplate reader (Thermo Fisher
Scientific, Inc.).
Immunofluorescence (IF) analysis
After treatment, H9C2 cells were fixed with 4%
paraformaldehyde in PBS for 30 min at room temperature.
Subsequently, cells were washed and incubated with 0.2% Triton
X-100 (MilliporeSigma) for 10 min at room temperature. The cells
were then incubated with 5% goat serum (MilliporeSigma) for 30 min
at room temperature and with a primary antibody against LC3 (cat.
no. ab192890; 1:2,000; Abcam) overnight at 4˚C. Following primary
antibody incubation, cells were incubated with goat anti-rabbit IgG
H&L (Alexa Fluor® 488; cat. no. ab150077; 1:200;
Abcam) at room temperature for 1 h. DAPI was used to stain the cell
nuclei for 5 min at room temperature and images were captured using
a confocal laser scanning microscopy (Zeiss AG).
Western blotting
Proteins were extracted from cells on ice with RIPA
buffer (Beyotime Institute of Biotechnology) and protein
concentrations were determined using a BCA kit (MilliporeSigma).
Total proteins (~30 µg/lane) were separated by SDS-PAGE on a 10%
gel and were transferred onto a PVDF membrane (Bio-Rad
Laboratories, Inc.). After blocking for 1 h in 5% skim milk, the
membranes were incubated with the primary antibodies against SIRT1
(cat. no. ab189494; 1:1,000; Abcam), LC3 (cat. no. ab192890;
1:2,000; Abcam), Beclin-1 (cat. no. ab207612; 1:2,000; Abcam), p62
(cat. no. ab109012; 1:10,000; Abcam), Bcl-2 (cat. no. ab196495;
1:1,000; Abcam), Bax (cat. no. ab32503; 1:1,000; Abcam), cleaved
caspase-3 (cat. no. #9661; 1:1,000; Cell Signaling Technology,
Inc.), caspase-3 (cat. no. ab184787; 1:2,000; Abcam),
phosphorylated (p)-p65 (cat. no. ab76302; 1:1,000; Abcam), p65
(cat. no. #8242; 1:1,000; Cell Signaling Technology, Inc.), iNOS
(cat. no. ab178945; 1:1,000; Abcam), COX-2 (cat. no. ab179800;
1:1,000; Abcam) and GAPDH (cat. no. ab181602 1:10,000; Abcam)
overnight at 4˚C. Following primary antibody incubation, the
membranes were incubated with the appropriate horseradish
peroxidase-conjugated secondary antibody (cat. no. ab6721; 1:2,000;
Abcam) at room temperature for 2 h and visualized using the Pierce™
ECL Western Blotting Substrate (Thermo Fisher Scientific, Inc.).
Protein bands were analyzed with Quantity One® software
version 4.5 (Bio-Rad Laboratories, Inc.).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's instructions. RNA was
reverse-transcribed into cDNA using a Primescript™ RT reagent kit
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. The synthesized cDNA was amplified and
quantified using the Real-time PCR Master Mix (SYBR Green) kit
(cat.no. KGA1339-1; Nanjing KeyGen Biotech Co., Ltd.). GAPDH was
used as an internal reference for evaluating the relative
expression of SIRT1. The thermocycling conditions were as follows:
Denaturation at 94˚C for 2 min; followed by 30 cycles at 94˚C for
30 sec, 56˚C for 30 sec and 72˚C for 30 sec; and a final elongation
step at 72˚C for 5 min. Relative expression levels were calculated
using the 2-ΔΔCq method (20). The following primer pairs were used
for qPCR: SIRT1 forward, 5'-ATCTCCCAGATCCTCAAGCCA-3' and reverse,
5'-CTTCCACTGCACAGGCACAT-3'; and GAPDH forward,
5'-GCATCTTCTTGTGCAGTGCC-3' and reverse,
5'-GATGGTGATGGGTTTCCCGT-3'.
TUNEL assay
H9C2 cells (2x106/ml) were fixed in 4%
paraformaldehyde for 15 min at room temperature, and were then
washed and incubated with 0.2% Triton X-100 (Sigma-Aldrich; Merck
KGaA) for 20 min at room temperature. Cells were incubated with 50
µl TUNEL reaction mixture (cat. no. 11684817910; MilliporeSigma) at
37˚C for 60 min in the dark. The cells were then incubated with
DAPI at room temperature for 10 min, washed with PBS, mounted in
glycerol and visualized with a fluorescence microscope (five
fields).
2,7-dichlorohydrofluorescein diacetate
(DCFH-DA) assay
Reactive oxygen species (ROS) levels were measured
by staining with the fluorescent probe DCFH-DA (Beyotime Institute
of Biotechnology), according to the manufacturer's instructions.
Following drug treatments, cells were stained in the dark with
DCFH-DA (10 µM) in DMEM at 37˚C for 30 min. Images of the stained
cells were captured under a fluorescence microscope (Olympus
Corporation) using excitation and emission wavelengths of 488 and
525 nm, respectively.
Glutathione peroxidase (GSH-Px)
activity, malondialdehyde (MDA) content and superoxide dismutase
(SOD) activity detection
H9C2 cells maintained in PBS were centrifuged at
1,500 x g for 10 min at 4˚C and the cell supernatants were then
collected. The GSH-Px activity, MDA contents and SOD activity in
the cell supernatants were measured using the GSH-Px assay kit
(cat. no. A005-1-2), MDA assay kit (cat. no. A003-1-2) and SOD
assay kit (cat. no. A001-3-2), respectively, according to the
manufacturer's instructions (Nanjing Jiancheng Bioengineering
Institute).
ELISA
H9C2 cells maintained in PBS were centrifuged at
1,500 x g for 10 min at 4˚C and the cell supernatants were then
collected. The levels of TNF-α, IL-6, IL-1β and lactate
dehydrogenase (LDH) in the supernatants of H9C2 cells after
different treatments were measured using the TNF-α assay kit (cat.
no. H052-1-2), IL-6 assay kit (cat. no. H007-1-1), IL-1β assay kit
(cat. no. H002-1-2) and LDH assay kit (cat. no. A020-2-2),
respectively, according to the manufacturer's protocols (Nanjing
Jiancheng Bioengineering Institute).
Cell transfection
H9C2 cells seeded into 6-well plates at a density of
1x106 cells/well were transfected with 100 nM
non-targeting small interfering RNA (siRNA)-negative control
(si-NC), si-SIRT1-1 or si-SIRT2-2 (all purchased from Shanghai
GenePharma Co., Ltd.) for 48 h at 37˚C with
Lipofectamine® RNAiMAX (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol.
Post-transfection, cells were used for subsequent functional
experiments 48 h post-transfection. The siRNA sequences were as
follows: si-SIRT1-1 sense, 5'-AAAACUUAACUCUAACGACAA-3' and
antisense, 5'-GUCGUUAGAGUUAAGUUUUUC-3'; si-SIRT1-2 sense,
5'-UUUAGGAAUAGCUCUUUCCUU-3' and antisense,
5'-GGAAAGAGCUAUUCCUAAAAC-3'; and si-NC sense,
5'-UUCUCCGAACGUGUCACGUTT-3' and antisense,
5'-ACGUGACACGUUCGGAGAATT-3'.
Statistical analysis
Data are presented as the mean ± standard
derivation. Statistical analyses were performed using one-way ANOVA
followed by Tukey's post hoc test in GraphPad Prism 8 (GraphPad
Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
Propofol activates autophagy in
LPS-induced cardiomyocyte injury
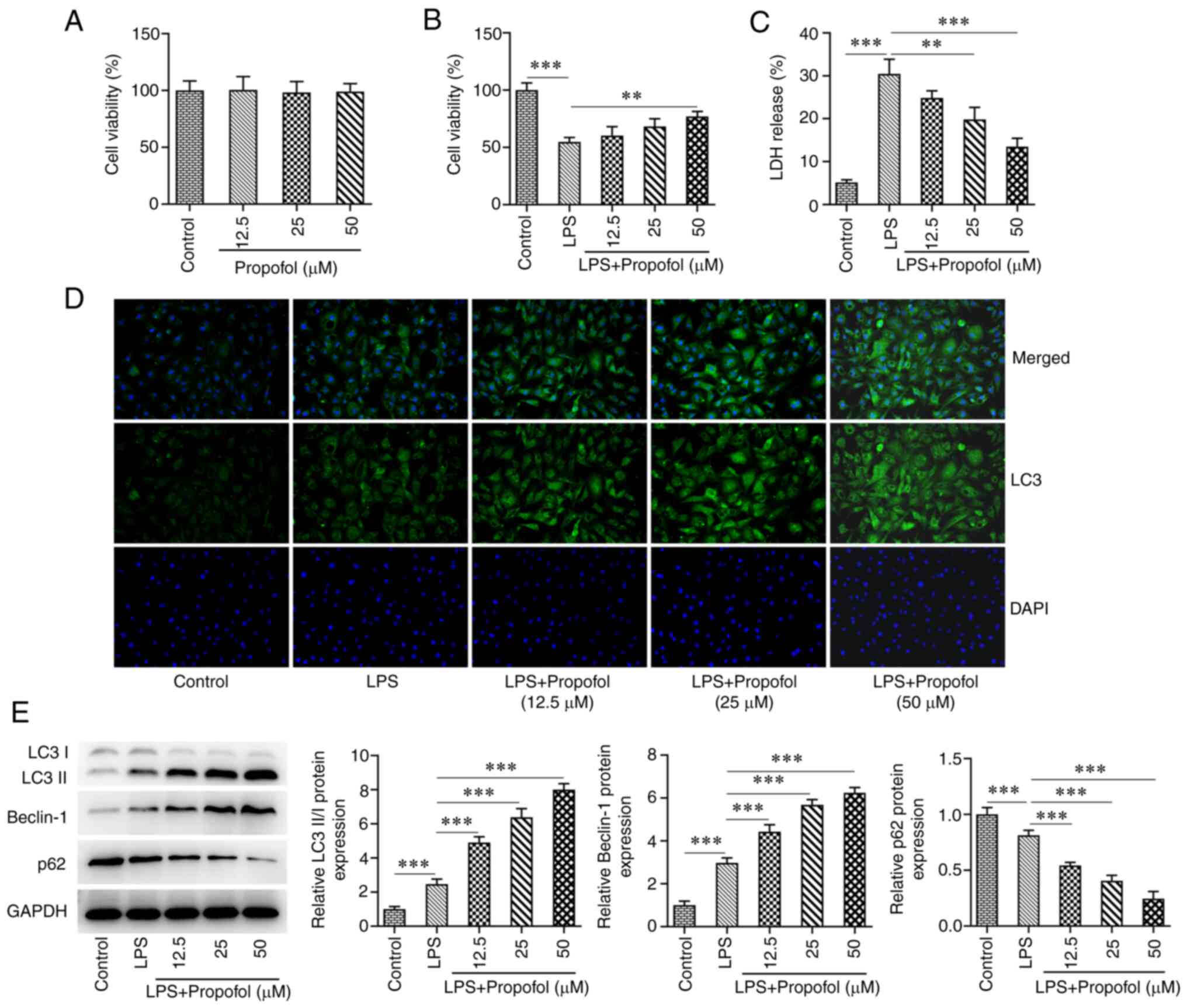
H9C2 cells were treated with 12.5, 25 and 50 µM
propofol, and the CCK8 assay showed no significant difference in
the viability of H9C2 cells at this concentration range compared
with in the control group (Fig.
1A). By contrast, cell viability was significantly decreased
following LPS induction compared with in the control group.
Compared with in the LPS group, cell viability in the LPS +
propofol groups was increased, with a significant increase detected
in response to 50 µM propofol (Fig.
1B). The levels of LDH in the cell supernatants were
significantly increased in the LPS group compared with those in the
control group, whereas LDH levels were significantly decreased in
the LPS + 25 µM propofol and LPS + 50 µM propofol groups compared
with those in the LPS group (Fig.
1C). IF assay revealed that LC3 expression was markedly
increased following LPS induction. Compared with in the LPS group,
LC3 expression was further increased in all of the LPS + propofol
groups in a concentration-dependent manner (Fig. 1D). Western blot analysis
demonstrated that the expression levels of LC3II/I and Beclin-1
were increased in the LPS group, whereas the expression of p62 was
decreased compared with that in the control group. Compared with in
the LPS group, LC3II/I and Beclin-1 expression levels were further
increased and p62 expression was further decreased in all of the
LPS + propofol groups (Fig. 1E).
Propofol (50 µM) was selected for subsequent experiments since it
exhibited a greater effect on the expression of autophagy-related
markers.
Propofol reduces LPS-induced apoptosis
and decreases in cardiomyocyte viability by activating
autophagy
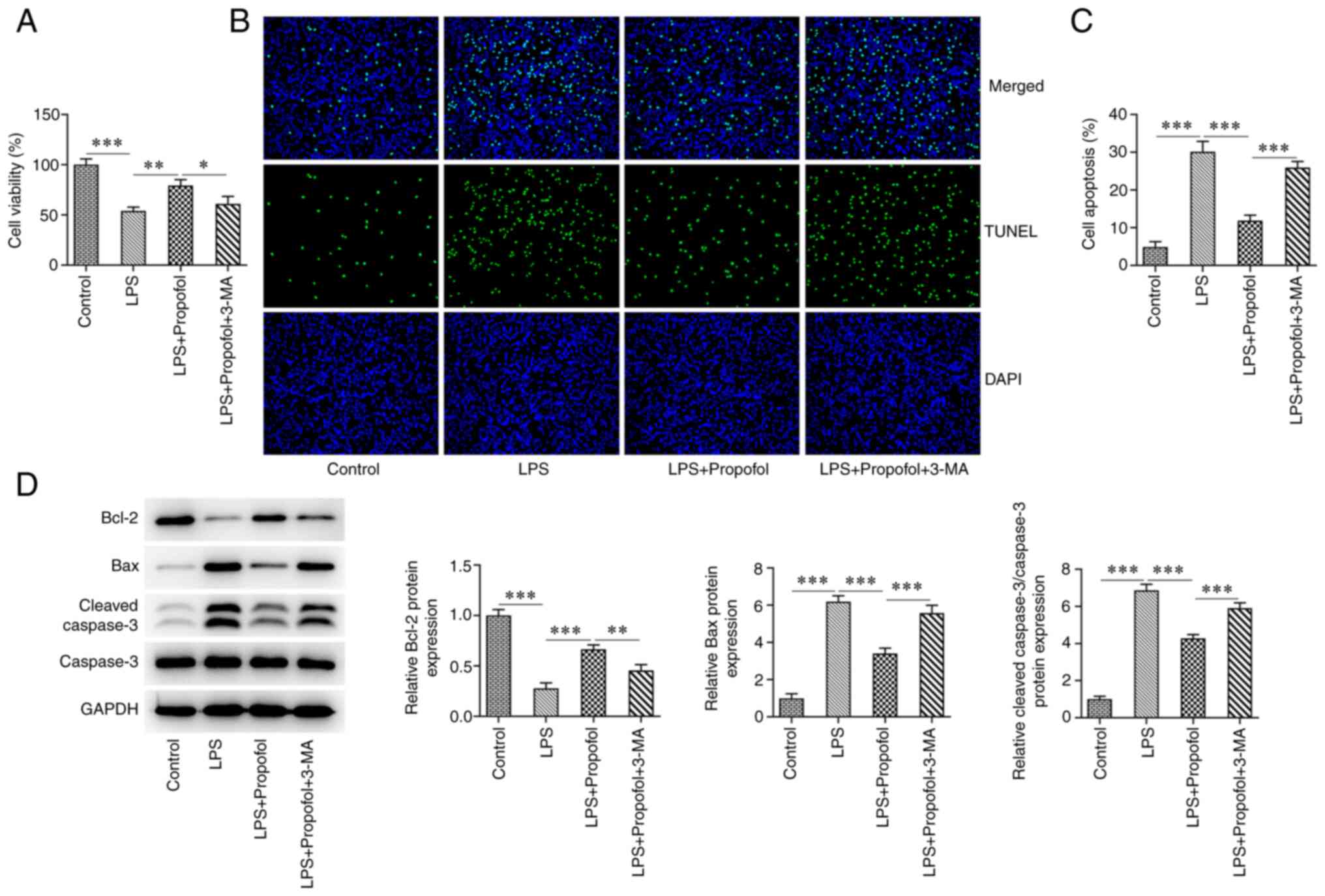
To further investigate whether 50 µM propofol
regulated myocardial cell injury by activating autophagy, cells
were pre-treated with the autophagy inhibitor 3-MA. Cell viability
was significantly decreased in the LPS + propofol + 3-MA group
compared with that in the LPS + 50 µM propofol group (Fig. 2A). The TUNEL assay indicated that
LPS exposure significantly potentiated H9C2 cell apoptosis, whereas
the apoptosis in the LPS + 50 µM propofol + 3-MA group was
significantly increased compared with that in the LPS + 50 µM
propofol group (Fig. 2B and
C). Furthermore, western blotting
revealed that LPS treatment significantly reduced Bcl-2 expression,
and elevated Bax and cleaved caspase-3 expression. Also, the
expression levels of Bax and cleaved caspase-3 were increased,
whereas the expression levels of Bcl-2 were decreased in the LPS +
50 µM propofol + 3-MA group compared with those in the LPS + 50 µM
propofol group (Fig. 2D).
Propofol reduces LPS-induced oxidative
stress and inflammatory damage in cardiomyocytes by activating
autophagy
ROS levels were detected by DCFH-DA staining.
Notably, DCFH-DA staining, and thus ROS generation, was markedly
increased in the LPS group compared with that in the control group,
whereas it was decreased in the LPS + 50 µM propofol group. By
contrast, ROS generation was higher in the LPS + 50 µM propofol +
3-MA group than that in the LPS + 50 µM propofol group (Fig. 3A). Subsequently, the levels of
oxidative stress markers, MDA, SOD and GSH-Px, were assessed. The
MDA levels were significantly increased in the LPS group compared
with those in the control group, whereas SOD and GSH-Px activities
were significantly decreased. Compared with in the LPS group, the
levels of MDA were significantly decreased in the LPS + 50 µM
propofol group, whereas the activities of SOD and GSH-Px were
significantly increased. Compared with in the LPS + 50 µM propofol
group, the MDA levels were significantly increased in the LPS + 50
µM propofol + 3-MA group, whereas the SOD and GSH-Px activities
were significantly decreased (Fig.
3B). The levels of TNF-α, IL-6 and IL-1β, which were measured
via ELISA, were significantly increased following LPS induction
compared with those in the control group. Compared with in the LPS
group, TNF-α, IL-6 and IL-1β levels were significantly decreased in
the LPS + 50 µM propofol group. By contrast, a significant increase
in TNF-α, IL-6 and IL-1β levels was observed in the LPS + 50 µM
propofol + 3-MA group compared with in the LPS + 50 µM propofol
group (Fig. 3C). Western blot
analysis showed a significant increase in the expression levels of
inflammatory enzymes, including iNOS, p-p65 and COX-2, in the LPS
group compared with those in the control group, whereas the
expression levels of these proteins were decreased in the LPS + 50
µM propofol group. Conversely, a significant increase in the
expression levels of iNOS, p-p65 and COX-2 was observed in the LPS
+ 50 µM propofol + 3-MA group compared with in the LPS + 50 µM
propofol group (Fig. 3D).
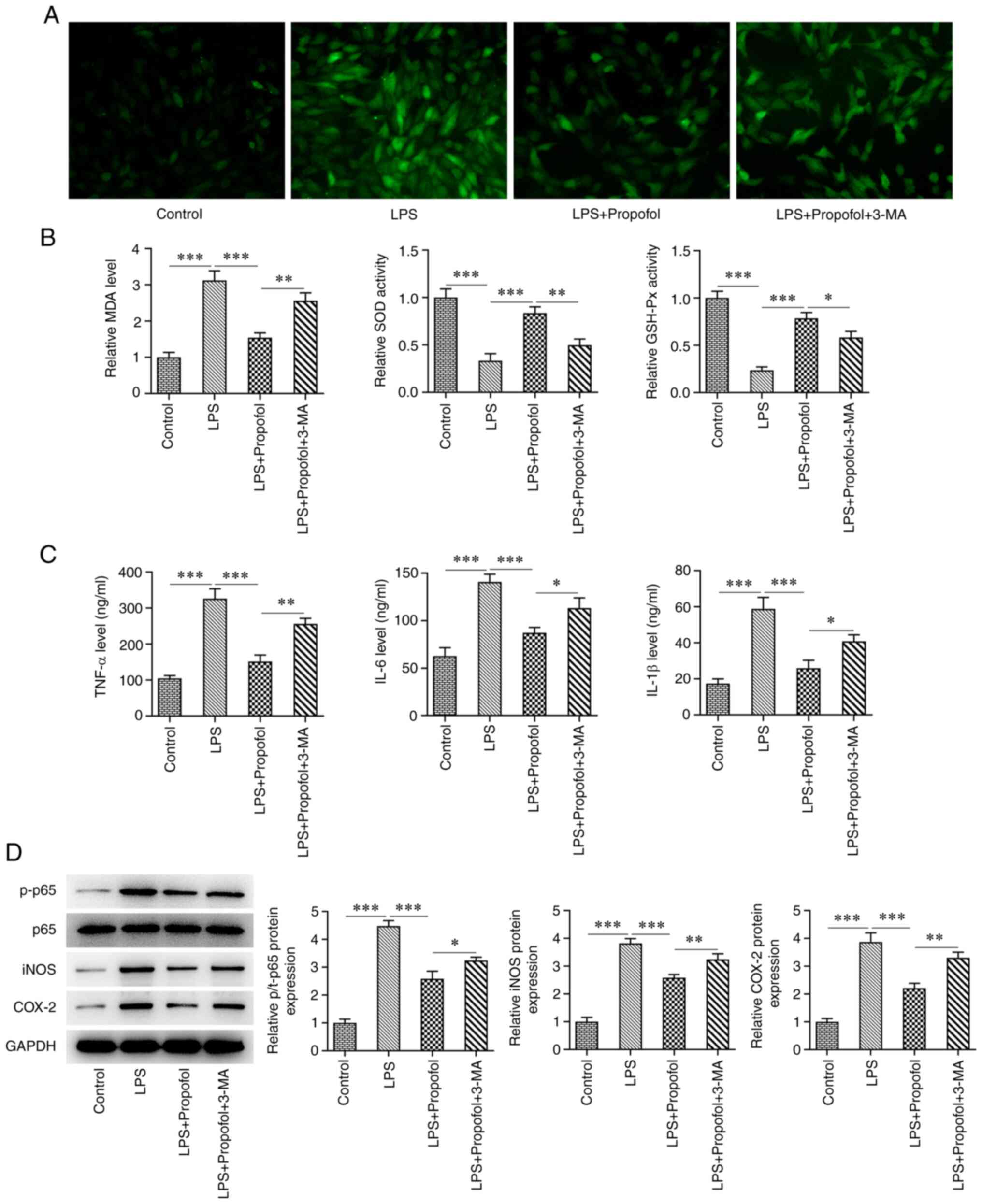 | Figure 3Propofol reduces LPS-induced
oxidative stress and inflammatory damage in cardiomyocytes by
activating autophagy. (A) 2,7-dichlorohydrofluorescein diacetate
staining assay for the detection of reactive oxygen species
generation. Magnification, x200. (B) Levels of MDA content, SOD and
GSH-Px activities. (C) ELISA of the levels of TNF-α, IL-6 and
IL-1β. (D) Western blot analysis of the expression levels of p65,
p-p65, iNOS and COX-2. *P<0.05,
**P<0.01, ***P<0.001. LPS,
lipopolysaccharide; GSH-Px, glutathione peroxidase; MDA,
malondialdehyde; SOD superoxide dismutase; p-, phosphorylated;
3-MA, 3-methyladenine. |
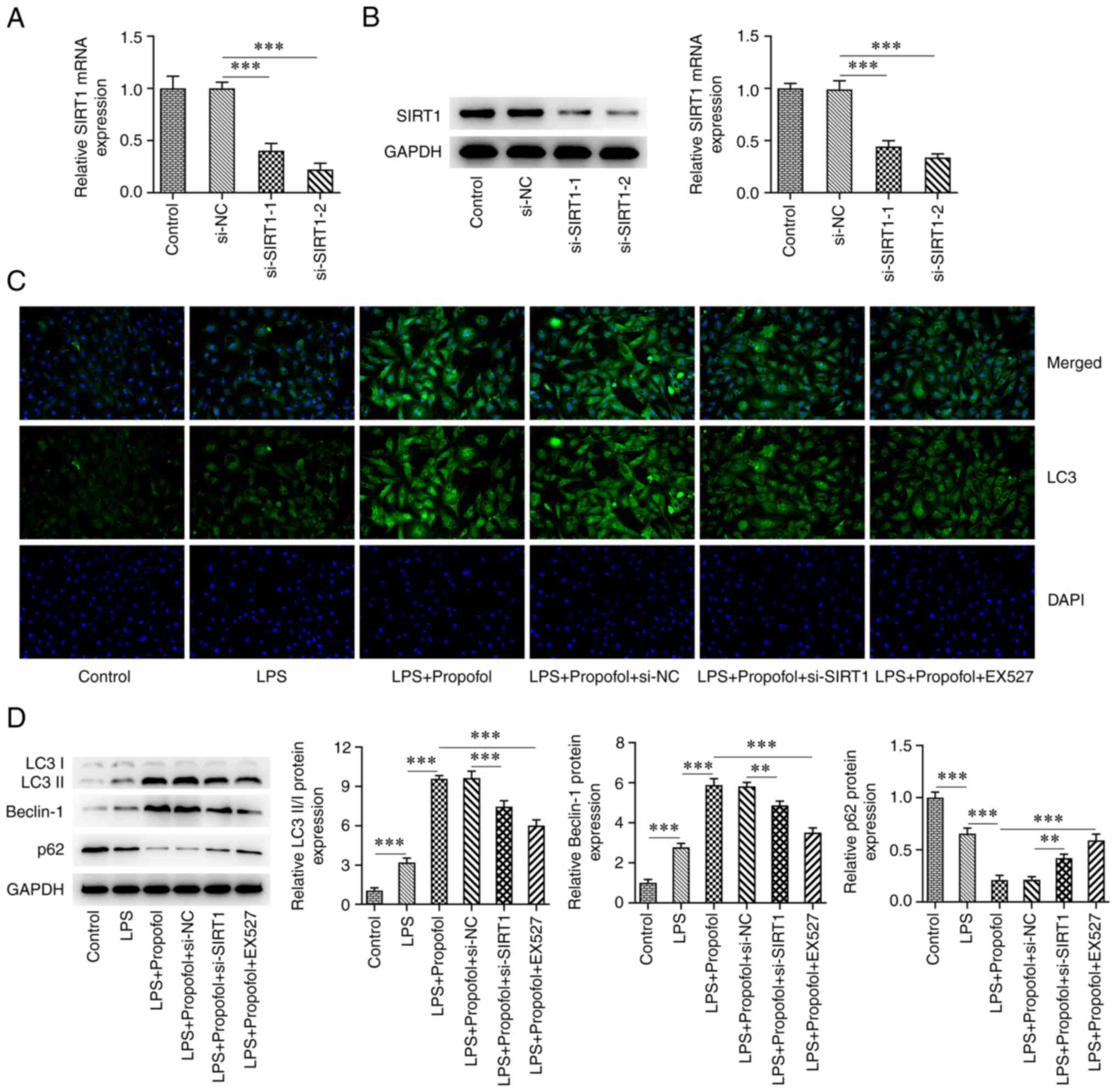
Inhibition of SIRT1 reduces
propofol-activated autophagy in LPS-induced cardiomyocytes
Cells were transfected with either si-SIRT1-1 or
si-SIRT1-2, and exhibited a significant decrease in the mRNA and
protein expression levels of SIRT1 (Fig. 4A and B). Notably, si-SIRT1-2 exerted the
greatest effect on SIRT1 expression and was thus chosen for
subsequent experiments. In addition, the SIRT1 inhibitor EX527 was
also used to inhibit SIRT1. The IF assay revealed that the
expression levels of LC3 in the LPS + 50 µM propofol + si-SIRT1
group were markedly decreased compared with those in the LPS + 50
µM propofol + si-NC group. Compared with in the LPS + 50 µM
propofol group, the expression levels of LC3 were also markedly
decreased in the LPS + 50 µM propofol + EX527 group (Fig. 4C). Western blot analysis revealed a
significant decrease in the expression levels of LC3II/I and
Beclin-1 in the LPS + 50 µM propofol + si-SIRT1 group compared with
those in the LPS + 50 µM propofol + si-NC group, whereas the
expression of p62 was significantly increased. In addition,
compared with in the LPS + 50 µM propofol group, the expression
levels of LC3II/I and Beclin-1 were significantly decreased,
whereas the expression of p62 was significantly increased in the
LPS + 50 µM Propofol + EX527 group (Fig. 4D).
Inhibition of SIRT1 reduces the
protective effect of propofol on LPS-induced cardiomyocytes
The CCK8 assay showed that cell viability in the LPS
+ 50 µM propofol + si-SIRT1 group was significantly decreased
compared with that in the LPS + 50 µM propofol + si-NC group.
Compared with in the LPS + 50 µM propofol group, cell viability in
the LPS + 50 µM propofol + EX527 group was also significantly
decreased (Fig. 5A). The TUNEL
assay revealed that apoptosis was significantly increased in the
LPS + 50 µM propofol + si-SIRT1 group compared with that in the LPS
+ 50 µM propofol + si-NC group. Compared with in the LPS + 50 µM
propofol group, apoptosis was also significantly increased in the
LPS + 50 µM propofol + EX527 group (Fig. 5C). The results of western blotting
showed that the expression levels of Bax and cleaved caspase-3 were
significantly increased in the LPS + 50 µM propofol + si-SIRT1
group compared with in the LPS + 50 µM propofol + si-NC group,
whereas Bcl-2 expression was significantly decreased. Similar
results were obtained in the LPS + 50 µM propofol + EX257 group
compared with in the LPS + 50 µM propofol group (Fig. 5D). ROS generation in the LPS + 50
µM propofol + si-SIRT1 group was markedly higher than that in the
LPS + 50 µM propofol + si-NC group (Fig. 5E). Compared with in the LPS + 50 µM
propofol + si-NC group, the MDA levels in the LPS + 50 µM propofol
+ si-SIRT1 group were significantly increased, whereas a
significant decrease in the SOD and GSH-Px activities was observed
(Fig. 5F). The trend of oxidative
stress factors in the LPS + 50 µM propofol + EX527 group was
consistent with that in the LPS + 50 µM propofol + si-SIRT1 group
(Fig. 5E and F). ELISA showed that, compared with those
in the LPS + 50 µM propofol + si-NC groups, the levels of TNF-α,
IL-6 and IL-1β were significantly increased in the LPS + 50 µM
propofol + si-SIRT1 group. Similarly, TNF-α, IL-6 and IL-1β levels
were significantly increased in the LPS + 50 µM propofol + EX527
group compared with those in the LPS + 50 µM propofol group
(Fig. 5G). Furthermore, western
blot analysis indicated that, compared with those in the LPS + 50
µM propofol + si-NC group, the expression levels of iNOS, p-p65 and
COX-2 were significantly increased in the LPS + 50 µM propofol +
si-SIRT1 group. Similarly, iNOS, p-p65 and COX-2 expression levels
were significantly increased in the LPS + 50 µM propofol + EX527
group compared with those in the LPS + 50 µM propofol group
(Fig. 5E).
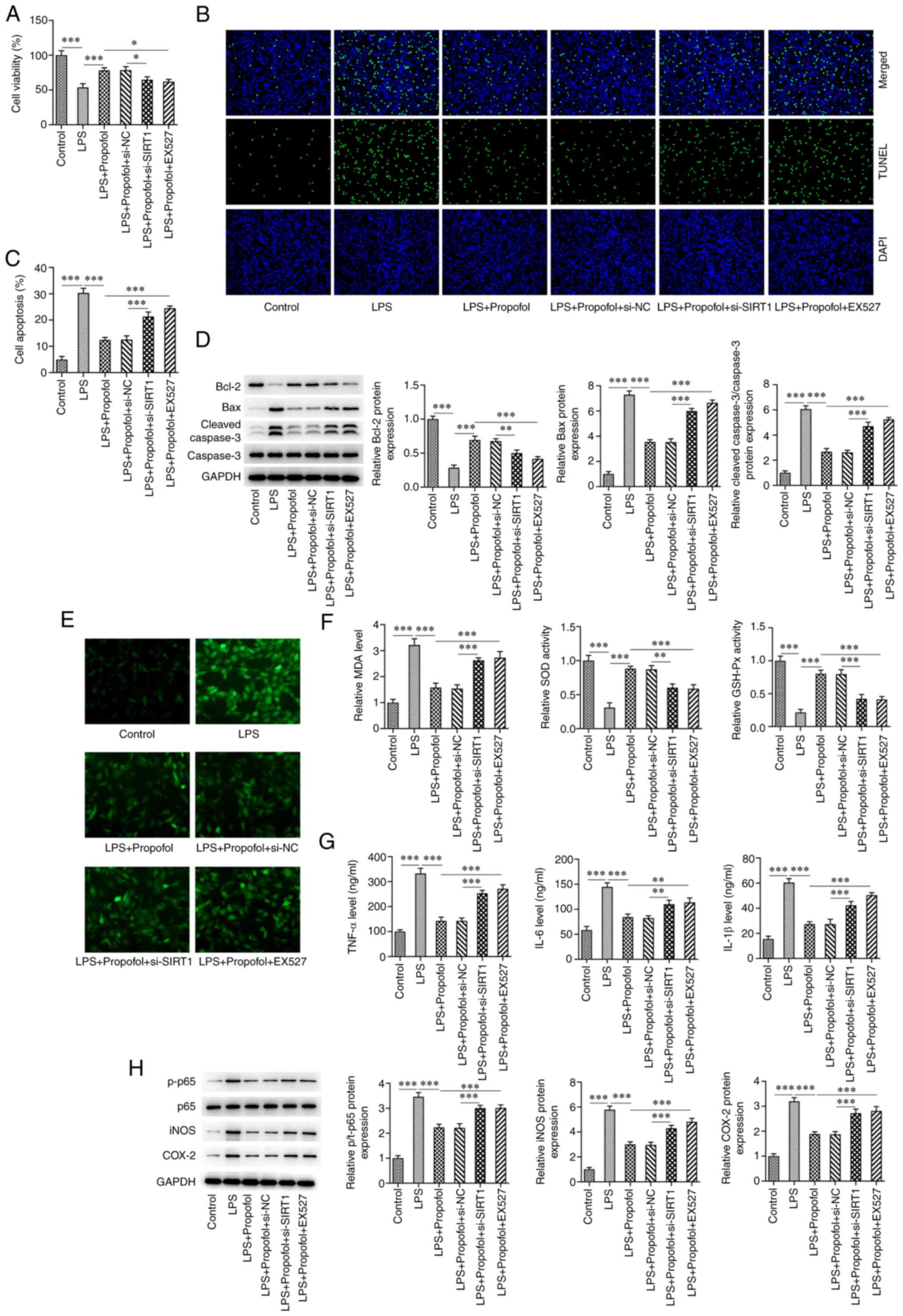 | Figure 5Inhibition of SIRT1 reduces the
protective effect of propofol on LPS-induced cardiomyocytes. (A)
Cell viability was measured using the Cell Counting Kit 8 assay.
(B) Cell apoptosis was measured using the TUNEL assay.
Magnification, x200. (C) Percentage of apoptotic cells. (D) Western
blot analysis of the expression levels of Bcl-2, Bax, cleaved
caspase-3 and caspase-3. (E) 2,7-dichlorohydrofluorescein diacetate
staining assay for the detection of reactive oxygen species
generation. Magnification, x200. (F) Levels of MDA content, SOD and
GSH-Px activities. (G) ELISA of the levels of TNF-α, IL-6 and
IL-1β. (H) Western blot analysis of the expression levels of p65,
p-p65, iNOS and COX-2. *P<0.05,
**P<0.01, ***P<0.001. LPS,
lipopolysaccharide; GSH-Px, glutathione peroxidase; MDA,
malondialdehyde; SOD superoxide dismutase; p-, phosphorylated;
SIRT1, sirtuin 1; NC, negative control; si, small interfering. |
Discussion
Severe sepsis can lead to organ dysfunction, with
heart dysfunction being the most common type (21). Therefore, ameliorating cardiac
dysfunction and myocardial injury after sepsis can improve the
prognosis of patients. Endotoxin is a toxic component of LPS, which
can significantly promote inflammation and the immune response
(22). Previous studies have shown
that when LPS enters the body, free LPS can interact with immune
cells and induce the release of inflammatory mediators,
pro-apoptotic factors and pro-fibrotic factors, thus leading to
abnormalities in coronary arteries, directly damaging myocardial
cells, affecting cardiac function (23), and eventually resulting in
myocardial fibrosis and cardiac disease, such as myocardial
infarction (24,25). In the present study, H9C2 cells
were induced by LPS treatment to create a model of myocardial cell
injury in vitro. The present results showed that H9C2 cells
underwent oxidative stress and inflammatory injury following LPS
induction, with decreased cell viability and increased
apoptosis.
The Increased generation of ROS activates the
antioxidant system (26). MDA is a
lipid peroxidation product that is widely detected as an oxidative
stress marker (27). The
antioxidant GSH can prevent ROS-induced cell damage; notably, all
cells in the human body are able to synthesize GSH, which is
essential for protection against oxidative stress (28). A decrease in SOD activity can
result in the accumulation of peroxides, which can also lead to
oxidative stress (29,30). Therefore, the present study
investigated the levels of oxidative stress markers, ROS, MDA, SOD
and GSH-Px, in H9C2 cells and revealed that, following LPS
induction, the levels of ROS and MDA were increased, whereas the
activities of SOD and GSH-Px were decreased, indicating that the
level of oxidative stress was increased after LPS induction. TNF-α
is considered a key regulator of the inflammatory response that is
released when the body is injured (31). Moreover, IL-1β and IL-6 are
important pro-inflammatory cytokines that are related to several
autoimmune diseases and have a key role following infection
(32). In the present study,
following LPS induction, the levels of TNF-α, IL-6 and IL-1β were
increased, indicating the activation of an inflammatory response.
Autophagy is an important self-protection mechanism of cells, which
aims to decompose intracellular protein macromolecules and
organelles into small molecules for recycling, in order to maintain
the metabolic process of cells upon exposure to various stimulating
factors, including oxidative stress and toxin stimulation, and
improve cell survival (33). The
level of autophagy is low under normal physiological conditions and
autophagy can be activated by several factors, such as nutrient
deficiency, hypoxia, infection, tumorigenesis, tissue damage,
protein misfolding, DNA damage, radiotherapy and chemotherapy
(34). Autophagy can be observed
in the early stage of sepsis via detection of the aggregation of
autophagy-related proteins (LC3II/I, Beclin1 and p62) and the
formation of autophagosomes (35).
MicroRNA-155 has been reported to alleviate septic lung injury by
inhibiting transforming growth factor-β-activated binding protein 2
and thus inducing autophagy (36).
The present study indicated that autophagy may be activated to
protect cells from damage following LPS stimulation.
Drugs that can improve the autophagy of
cardiomyocytes may ameliorate cardiac injury in sepsis (35,37).
A previous study reported that valproic acid can accelerate
autophagy in rats through the PTEN/AKT/mTOR pathway and relieve
cardiac dysfunction caused by sepsis (38). Pan et al (39) demonstrated that melatonin can
regulate mitochondrial uncoupling protein, improve LPS-induced
cardiac autophagy levels, protect the structure and function of
mitochondria, and alleviate LPS-induced cardiac injury. Unuma et
al (40) revealed that cobalt
protoporphyrin can activate transcription factor EB and
lysosomal-associated membrane protein in cardiomyocytes to promote
autophagosome formation, protect cardiomyocytes and alleviate
LPS-induced myocardial injury. It has been demonstrated that
propofol can inhibit oxygen glucose deprivation and
reperfusion-induced neuronal injury by inhibiting autophagy
(41). The present study also
hypothesized that propofol may regulate autophagy and serve a role
in LPS-induced myocardial cell injury. Conversely, the experimental
data demonstrated that after propofol treatment in LPS-induced
cardiomyocytes, the expression levels of autophagy markers were
increased, suggesting that propofol increased autophagy, which was
consistent with a previous study, which suggested that propofol may
serve a protective role in myocardial ischemia-reperfusion injury
by inducing autophagy of cardiomyocytes through the
stress-activated protein kinases/JNK pathway (42). Additionally, the present study
demonstrated that, after propofol treatment in LPS-induced
cardiomyocytes, the cell viability was increased, whereas the
expression of oxidative stress markers and inflammatory factors
were significantly decreased. Apoptosis is a process by which cells
organize self-destruction. The mitochondrial apoptosis pathway is
regulated by Bcl-2 family member proteins, including pro-apoptotic
Bax, and anti-apoptotic Bcl-2 and Bcl-xL. Bax is able to enhance
mitochondrial outer membrane permeability, thus leading to
excessive release of cytochrome c and caspase-3 activation.
Subsequently, caspase-3 activation participates in various
apoptotic pathways, finally resulting in apoptosis (43). The present study detected the
expression levels of Bcl-2, Bax and caspase-3 through western
blotting and measured apoptosis through TUNEL assay, and
demonstrated that propofol inhibited LPS-induced apoptosis of
cardiomyocytes. These results suggested that propofol may improve
myocardial cell injury by regulating autophagy. Subsequently, to
further explore the mechanism of action, the effect of the
autophagy inhibitor 3-MA was investigated and the results showed
that 3-MA could significantly reverse the protective effect of
propofol on myocardial cell injury.
SIRT1 is a key regulator of autophagy. A recent
study reported that propofol can relieve the oxidative stress
response of cerebral ischemia-reperfusion injury through the SIRT1
signaling pathway (44). In
addition, SIRT1 inhibitors can decrease the protective effect of
propofol on renal ischemia-reperfusion-mediated acute lung injury
(45). Moreover, daming capsule, a
hypolipidemic drug, promotes mitochondrial autophagy through the
SIRT1/AMPK signaling pathway, thereby inhibiting myocardial cell
injury and protecting against myocardial infarction (46). Quercetin has been shown to improve
cardiomyocyte susceptibility to hypoxia by regulating
SIRT1/TMBIM6-related mitochondrial autophagy and endoplasmic
reticulum stress (47). The
present study thus aimed to investigate whether propofol regulated
myocardial cell injury by regulating SIRT1-mediated autophagy. In
the present study, SIRT1 was silenced though siRNA transfection and
inhibited using the SIRT1 inhibitor EX527. It was revealed that
both silencing and inhibition of SIRT1 decreased propofol-induced
autophagy and protected LPS-induced cardiomyocytes from injury.
While normal autophagy protects cells, an excessive activation
leads to cell damage; therefore, further investigation on the
appropriate propofol dosage is required.
In conclusion, the present study indicated that
propofol may reduce LPS-induced cardiomyocyte injury by activating
SIRT1-mediated autophagy, implying that propofol may serve as a
potential therapeutic agent for myocardial injury. However, the
lack of detection of autophagosome formation is a limitation of the
present study; therefore, further exploration is required to
confirm the association between propofol and SIRT1-mediated
autophagy in LPS-exposed cardiomyocytes.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JD and YZ designed the study and performed the
experiments. YZ revised the manuscript for important intellectual
content. JD collected and analyzed the data. JD and YZ confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Merx MW and Weber C: Sepsis and the heart.
Circulation. 116:793–802. 2007.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Romero-Bermejo FJ, Ruiz-Bailen M,
Gil-Cebrian J and Huertos-Ranchal MJ: Sepsis-induced
cardiomyopathy. Curr Cardiol Rev. 7:163–183. 2011.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Fan W, Zhu X, Wu L, Wu Z, Li D, Huang F
and He H: Propofol: An anesthetic possessing neuroprotective
effects. Eur Rev Med Pharmacol Sci. 19:1520–1529. 2015.PubMed/NCBI
|
|
4
|
Li YM, Sun JG, Hu LH, Ma XC, Zhou G and
Huang XZ: Propofol-mediated cardioprotection dependent of
microRNA-451/HMGB1 against myocardial ischemia-reperfusion injury.
J Cell Physiol. 234:23289–23301. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Lai HC, Yeh YC, Wang LC, Ting CT, Lee WL,
Lee HW, Wang KY, Wu A, Su CS and Liu TJ: Propofol ameliorates
doxorubicin-induced oxidative stress and cellular apoptosis in rat
cardiomyocytes. Toxicol Appl Pharmacol. 257:437–448.
2011.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lu Z, Liu Z and Fang B: Propofol protects
cardiomyocytes from doxorubicin-induced toxic injury by activating
the nuclear factor erythroid 2-related factor 2/glutathione
peroxidase 4 signaling pathways. Bioengineered. 13:9145–9155.
2022.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Bao HG and Li S: Effects of propofol on
the outcomes of rats with sepsis. J Surg Res. 168:e111–e115.
2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zhao H, Gu Y and Chen H: Propofol
ameliorates endotoxininduced myocardial cell injury by inhibiting
inflammation and apoptosis via the PPARgamma/HMGB1/NLRP3 axis. Mol
Med Rep. 23(176)2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Liu Z, Meng Y, Miao Y, Yu L and Yu Q:
Propofol reduces renal ischemia/reperfusion-induced acute lung
injury by stimulating sirtuin 1 and inhibiting pyroptosis. Aging
(Albany NY). 13:865–876. 2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Liu Y, Du X, Zhang S, Liu X and Xu G:
Propofol alleviates hepatic ischemia/reperfusion injury via the
activation of the Sirt1 pathway. Int J Clin Exp Pathol.
10:10959–10968. 2017.PubMed/NCBI
|
|
11
|
Wang J, Qi J, Wu Q, Jiang H, Yin Y, Huan
Y, Zhao Y and Zhu M: Propofol attenuates high glucose-induced
P66shc expression in human umbilical vein endothelial cells through
Sirt1. Acta Biochim Biophys Sin (Shanghai). 51:197–203.
2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wang L, Xu C, Johansen T, Berger SL and
Dou Z: SIRT1-a new mammalian substrate of nuclear autophagy.
Autophagy. 17:593–595. 2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Luo G, Jian Z, Zhu Y, Zhu Y, Chen B, Ma R,
Tang F and Xiao Y: Sirt1 promotes autophagy and inhibits apoptosis
to protect cardiomyocytes from hypoxic stress. Int J Mol Med.
43:2033–2043. 2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zhang WX, He BM, Wu Y, Qiao JF and Peng
ZY: Melatonin protects against sepsis-induced cardiac dysfunction
by regulating apoptosis and autophagy via activation of SIRT1 in
mice. Life Sci. 217:8–15. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Guo XN and Ma X: The effects of propofol
on autophagy. DNA Cell Biol. 39:197–209. 2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wang Y, Yu W, Shi C and Hu P: Crocetin
attenuates sepsis-induced cardiac dysfunction via regulation of
inflammatory response and mitochondrial function. Front Physiol.
11(514)2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Tang J, Hu JJ, Lu CH, Liang JN, Xiao JF,
Liu YT, Lin CS and Qin ZS: Propofol inhibits
lipopolysaccharide-induced tumor necrosis factor-alpha expression
and myocardial depression through decreasing the generation of
superoxide anion in cardiomyocytes. Oxid Med Cell Longev.
2014(157376)2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yuan X, Chen G, Guo D, Xu L and Gu Y:
Polydatin alleviates septic myocardial injury by promoting
SIRT6-mediated autophagy. Inflammation. 43:785–795. 2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Xu C, Xiao Z, Wu H, Zhou G, He D, Chang Y,
Li Y, Wang G and Xie M: BDMC protects AD in vitro via AMPK and
SIRT1. Transl Neurosci. 11:319–327. 2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Lv X and Wang H: Pathophysiology of
sepsis-induced myocardial dysfunction. Mil Med Res.
3(30)2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Giordano NP, Cian MB and Dalebroux ZD:
Outer membrane lipid secretion and the innate immune response to
gram-negative bacteria. Infect Immun. 88:e00920–19. 2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Abel FL: Myocardial function in sepsis and
endotoxin shock. Am J Physiol. 257:R1265–R1281. 1989.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Norouzi F, Abareshi A, Asgharzadeh F,
Beheshti F, Hosseini M, Farzadnia M and Khazaei M: The effect of
Nigella sativa on inflammation-induced myocardial fibrosis in male
rats. Res Pharm Sci. 12:74–81. 2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Tung CL, Ju DT, Velmurugan BK, Ban B, Dung
TD, Hsieh DJ, P Viswanadha V, Day CH, Lin YM and Huang CY:
Carthamus tinctorius L. extract activates insulin-like growth
factor-I receptor signaling to inhibit FAS-death receptor pathway
and suppress lipopolysaccharides-induced H9c2 cardiomyoblast cell
apoptosis. Environ Toxicol. 34:1320–1328. 2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Snezhkina AV, Kudryavtseva AV, Kardymon
OL, Savvateeva MV, Melnikova NV, Krasnov GS and Dmitriev AA: ROS
Generation and antioxidant defense systems in normal and malignant
cells. Oxid Med Cell Longev. 2019(6175804)2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Tsikas D: Assessment of lipid peroxidation
by measuring malondialdehyde (MDA) and relatives in biological
samples: Analytical and biological challenges. Anal Biochem.
524:13–30. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Diaz-Vivancos P, de Simone A, Kiddle G and
Foyer CH: Glutathione-linking cell proliferation to oxidative
stress. Free Radic Biol Med. 89:1154–1164. 2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Han B, Lv Z, Zhang X, Lv Y, Li S, Wu P,
Yang Q, Li J, Qu B and Zhang Z: Deltamethrin induces liver fibrosis
in quails via activation of the TGF-β1/Smad signaling pathway.
Environ Pollut. 259(113870)2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Yang X, Fang Y, Hou J, Wang X, Li J, Li S,
Zheng X, Liu Y and Zhang Z: The heart as a target for deltamethrin
toxicity: Inhibition of Nrf2/HO-1 pathway induces oxidative stress
and results in inflammation and apoptosis. Chemosphere.
300(134479)2022.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Jang DI, Lee AH, Shin HY, Song HR, Park
JH, Kang TB, Lee SR and Yang SH: The role of tumor necrosis factor
alpha (TNF-α) in autoimmune disease and current TNF-α inhibitors in
therapeutics. Int J Mol Sci. 22(2719)2021.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Li J, Yu Z, Han B, Li S, Lv Y, Wang X,
Yang Q, Wu P, Liao Y, Qu B and Zhang Z: Activation of the GPX4/TLR4
signaling pathway participates in the alleviation of selenium yeast
on deltamethrin-provoked cerebrum injury in quails. Mol Neurobiol.
59:2946–2961. 2022.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Galluzzi L and Green DR:
Autophagy-independent functions of the autophagy machinery. Cell.
177:1682–1699. 2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Onorati AV, Dyczynski M, Ojha R and
Amaravadi RK: Targeting autophagy in cancer. Cancer. 124:3307–3318.
2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Yin X, Xin H, Mao S, Wu G and Guo L: The
role of autophagy in sepsis: Protection and injury to organs. Front
Physiol. 10(1071)2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Liu F, Nie C, Zhao N, Wang Y, Liu Y, Li Y,
Zeng Z, Ding C, Shao Q, Qing C, et al: MiR-155 alleviates septic
lung injury by inducing autophagy via inhibition of transforming
growth factor-β-activated binding protein 2. Shock. 48:61–68.
2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Xie M, Morales CR, Lavandero S and Hill
JA: Tuning flux: Autophagy as a target of heart disease therapy.
Curr Opin Cardiol. 26:216–222. 2011.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Shi X, Liu Y, Zhang D and Xiao D: Valproic
acid attenuates sepsis-induced myocardial dysfunction in rats by
accelerating autophagy through the PTEN/AKT/mTOR pathway. Life Sci.
232(116613)2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Pan P, Zhang H, Su L, Wang X and Liu D:
Melatonin balance the autophagy and apoptosis by regulating UCP2 in
the LPS-induced cardiomyopathy. Molecules. 23(675)2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Unuma K, Aki T, Funakoshi T, Yoshida K and
Uemura K: Cobalt protoporphyrin accelerates TFEB activation and
lysosome reformation during LPS-induced septic insults in the rat
heart. PLoS One. 8(e56526)2013.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Sun B, Ou H, Ren F, Huan Y, Zhong T, Gao M
and Cai H: Propofol inhibited autophagy through
Ca2+/CaMKKβ/AMPK/mTOR pathway in OGD/R-induced neuron
injury. Mol Med. 24(58)2018.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Li H, Zhang X, Tan J, Sun L, Xu LH, Jiang
YG, Lou JS, Shi XY and Mi WD: Propofol postconditioning protects
H9c2 cells from hypoxia/reoxygenation injury by inducing autophagy
via the SAPK/JNK pathway. Mol Med Rep. 17:4573–4580.
2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Li S, Wu P, Han B, Yang Q, Wang X, Li J,
Deng N, Han B, Liao Y, Liu Y and Zhang Z: Deltamethrin induces
apoptosis in cerebrum neurons of quail via promoting endoplasmic
reticulum stress and mitochondrial dysfunction. Environ Toxicol.
37:2033–2043. 2022.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Liu XB, Xia H, Wang G, Zhang W, Hu Y and
Zhang J: Propofol relieves oxidative stress response of cerebral
ischemiareperfusion injury through SIRT1 signaling pathway. J Biol
Regul Homeost Agents. 34:435–443. 2020.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Liu Z, Li C, Li Y, Yu L and Qu M: Propofol
reduces renal ischemia reperfusion-mediated necroptosis by
up-regulation of SIRT1 in rats. Inflammation. 45:2038–2051.
2022.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Sun X, Han Y, Dong C, Qu H, Yu Y, Ju J,
Bai Y and Yang B: Daming capsule protects against myocardial
infarction by promoting mitophagy via the SIRT1/AMPK signaling
pathway. Biomed Pharmacother. 151(113162)2022.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Chang X, Zhang T, Meng Q, ShiyuanWang Yan
P, Wang X, Luo D, Zhou X and Ji R: Quercetin improves cardiomyocyte
vulnerability to hypoxia by regulating SIRT1/TMBIM6-related
mitophagy and endoplasmic reticulum stress. Oxid Med Cell Longev.
2021(5529913)2021.PubMed/NCBI View Article : Google Scholar
|