Introduction
Macrophages serve a key role in host defense against
infections by viruses and microorganisms (1). However, activated macrophages can
also contribute to tissue damage and repair by secreting a variety
of growth factors, cytokines and proteolytic enzymes (1,2).
Overproduction of these inflammatory mediators may promote the
development of inflammatory diseases, including atherosclerosis,
septic shock and even cancer (1).
Atherosclerosis has been defined to be a chronic inflammatory
disease and is one of the leading causes of cardiovascular
diseases, such as myocardial infarctions and coronary artery
disease. As an important constituent of atherosclerotic lesions,
macrophages serve an important part in the progression of
atherosclerosis by forming the foam cells and regulating
inflammation (1,3). Macrophages treated with
lipopolysaccharide (LPS) has been frequently used as a model of
activated macrophages experimentally (1,4). LPS
can activate a number of intracellular signaling pathways,
including ERK1/2, c-JNK and p38, which belongs to the MAPKs family
of signaling cascades (1). In
addition, LPS can activate PI3K/AKT, which ultimately leads to the
activation of NF-κB and activator protein-1 (AP-1) to release
various inflammatory factors, including TNFα, IL-1β and IL-6
(5-7).
Activated MAPKs can also regulate the activity of other
transcription factors such as Sp1 to control the expression of
downstream targets such as TRIM59 (1,8).
Tripartite motif-containing (TRIM) 65 belongs to a
member of the TRIM protein family (9,10).
Structurally, it contains a RING-finger domain, a B-box domain and a
coiled-coil domain in the N-terminus (9,10).
The presence of a RING domain can mediate the conjugation of
proteins with ubiquitin. The B-box domains have been shown to
contribute to innate resistance to HIV, and the coiled-coil domains
mediate homomeric and heteromeric interactions among TRIM family
members and other proteins, in particular self-association
(11). Previous studies found
TRIM65 to serve a key role in tumor progression by promoting
hepatocellular carcinoma via ubiquitylation of Axin1 and supporting
bladder urothelial carcinoma cell aggressiveness by promoting ANXA2
ubiquitination and degradation (12-15),
melanoma differentiation-associated protein 5-mediated antiviral
innate immunity by promoting K63 polyubiquitination of MDA5
(16,17) and modulating the miRNA pathway by
ubiquitination of TNRC6 (13,18).
Furthermore, TRIM65 has been found to suppress LPS-induced lung
inflammation by targeting vascular cell adhesion molecule
1(19). TRIM65 has also been
observed to function as an endogenous negative regulator of NACHT
domain-, leucine-rich repeat- and PYD-containing protein 3
inflammasome activation in macrophages (20). However, the molecular mechanism
that controls its expression and physiological function in
activated macrophages remain poorly understood.
Therefore, the present study aimed to investigate
the possible effect and mechanism of LPS on TRIM65 expression in
vitro and in vivo and further to evaluate the potential
role of TRIM65 on macrophages activation.
Materials and methods
Cell culture
The murine macrophage cell line RAW264.7 (cat. no.
SCSP-5036) were obtained from The Cell Bank of Type Culture
Collection of the Chinese Academy of Sciences. They were cultured
in complete RPMI-1640 medium (Gibco; Thermo Fisher Scientific,
Inc.) containing 10% FBS (Shanghai ExCell Biology, Inc.), 100 U/ml
penicillin and 100 µg/ml streptomycin (cat. no. P1400; Beijing
Solarbio Science & Technology Co., Ltd.) at 37˚C in a
humidified incubator with 5% CO2.
Human umbilical vein endothelial cells (HUVECs)
(cat. no. PCS-100-013; passage 3), human aortic endothelial cells
(HAECs; cat. no. PCS-100-011, passage 3) and human aortic smooth
muscle cells (HASMCs; cat. no. QC470, passage 2) were gifts from
Professor Yuanli Chen (College of Food and Biological Engineering,
Hefei University of Technology, Hefei), human cerebral
microvascular endothelial cells (HCMEC/D3) were purchased from BeNa
Culture Collection: Beijing Beina Chunglian Biotechnology Research
Institute (cat. no. BNCC337728, passage 3), mouse cerebral
microvascular endothelial cells (bEnd.3; cat. no. TCM40) and THP-1
cells (a human monocytic cell line; cat. no. SCSP-567, passage 5)
were also obtained from The Cell Bank of Type Culture Collection of
the Chinese Academy of Sciences.
HUVECs, HAECs and HCMEC/D3 were cultured in
Endothelial Cell Medium (cat. no. 1001; ScienCell Research
Laboratories, Inc.) supplemented with 5% FBS (Shanghai ExCell
Biology, Inc.) and 1% penicillin/streptomycin at 37˚C in a
humidified incubator with 5% CO2 . HASMC and bEnd.3
cells were cultured in complete DMEM (Gibco; Thermo Fisher
Scientific) containing 10% FBS (Shanghai ExCell Biology, Inc.), 100
U/ml penicillin and 100 µg/ml streptomycin (cat. no. P1400; Beijing
Solarbio Science & Technology Co., Ltd.) at 37˚C in a
humidified incubator with 5% CO2.
Peritoneal macrophages (PMs) were collected from the
abdomen of C57BL/6 mice (WT or TRIM65 knockout, male, n=4 per
group) by rinsing with PBS as described previously (21). In brief, 8-week-old C57BL/6 male
mice were injected intraperitoneally with 3 ml 4% thioglycolate
solution (cat. no. 211716; BD Biosciences) at day 0 and maintained
with access to water and normal chow for 5 days in SPF units of the
Animal Center at Nanchang University (with a 12-h light cycle from
8 a.m. to 8 p.m. and at 23±3˚C with 60-70% humidity). The mice were
sacrificed in a CO2 chamber with the flow displacement
rate of 30% container volume per minute on day 5 and the PMs were
collected from mouse abdomen by lavage with 10 ml PBS. After
removal of erythrocytes with Red Blood Cell Lysis Buffer (cat. no.
R1010; Beijing Solarbio Science & Technology Co., Ltd.)
according to the manufacturer's protocol, PMs were collected by
centrifugation (500 x g for 5 min) at 4˚C, and re-suspended and
cultured in complete RPMI-1640 medium for 2 days at 37˚C in a
humidified incubator with 5% CO2 followed by treatment
with LPS after the 2 days.
Mouse aortic SMCs (MASMCs) were isolated from the
aortas of male C57BL/6 mice (8 weeks) as described previously
(22,23). Briefly, aortas were carefully
dissected and cleaned of connective tissue, and then were cut into
1-2 mm pieces followed by digestion for 6 h at 37˚C in DMEM
containing 200 U/ml type-II collagenase. After 6 h, MASMCs were
harvested by centrifugation at 200 x g for 5 min at room
temperature and cultured in DMEM containing 20% FBS, 100 U/ml
penicillin, and 100 U/ml streptomycin at 37˚C in a humidified
incubator with 5% CO2.
Bone marrow cells were isolated from mouse tibias
and femurs and the erythrocytes were removed with Red Blood Cell
Lysis Buffer (cat. no. R1010; Beijing Solarbio Science &
Technology Co., Ltd.). The remaining cells, termed BM were
collected by centrifugation (500 x g for 5 min at 4˚C) and then
re-suspended in PBS twice to wash out the Red Blood Cell Lysis
Buffer and this was followed by extraction of RNA and protein. Bone
marrow-derived macrophages (BMDMs) were derived from BM in DMEM
containing 10% FBS and 50 ng/ml macrophage-colony stimulating
factor (cat. no. HY-P7085A; MedChemExpress) for 7 days at 37˚C in a
humidified incubator with 5% CO2 (1).
Human blood monocyte-derived macrophages (hBMDMs)
were acquired as described previously with 10 ml blood from a
healthy volunteer (female aged 35) (1). Briefly, after the removal of red
blood cells by incubating the blood with a dextran sedimentation
mixture (cat. no. D2870; Beijing Solarbio Science & Technology
Co., Ltd.), the remaining cells were collected by centrifugation
(250 x g for 5 min at 4˚C) and re-suspended in serum-free RPMI1640
medium. The suspension was then overlaid onto the top of a Ficoll
solution (cat. no. P8610; Beijing Solarbio Science & Technology
Co., Ltd.). After gradient centrifugation for 20 min at 1,000 x g
at room temperature, monocytes in the interface layer were
collected and washed 3 times with serum-free RPMI-1640 medium
followed by culture in complete RPMI-1640 medium containing 50
ng/ml macrophage-colony stimulating factor (cat. no. HY-P7050A;
MedChemExpress) for 7 days at 37˚C in a humidified incubator with
5% CO2 to differentiate into macrophages. Studies with
human samples were approved by the Ethics Committee of Second
Affiliated Hospital of Nanchang University (Nanchang, China) and
complied strictly with the 2008 Declaration of Helsinki Principle.
Written informed consents were signed by the participant prior to
the collection of samples in the present study.
Cell treatment
To determine the effect of LPS (cat. no. L3129;
MilliporeSigma) on TRIM65 expression in macrophages, RAW264.7, PMs,
THP-1 derived macrophages and hBMDMs were treated with 0.1, 0.2,
0.5 and 1 µg/ml LPS or with 0.2 µg/ml LPS for 3, 6, 12 and 24 h in
RPMI-1640 or DMEM serum-free medium at 37˚C in a humidified
incubator with 5% CO2 followed by the extraction of
protein or RNA. LPS has been recognized to be a viable reagent for
inducing macrophage activation and all the doses of LPS in study
used exerted no toxicity on the macrophages (1,2).
Further to our previously reported results (1) and the maximum inhibition of 0.2 µg/ml
LPS on TRIM65 expression in primary PMs, 0.2 µg/ml LPS was chosen
for treating all the macrophages for 24 h at 37˚C instead of the
lowest concentration of 0.1 µg/ml and the highest concentration 1
µg/ml.
To identify the signaling pathways by which
LPS-inhibited TRIM65 expression, RAW264.7 cells were pretreated
with U0126 (cat. no. HY-12031; MedChemExpress), SB203580 (cat. no.
HY-10256; MedChemExpress), SP600125 (cat. no. HY-12041;
MedChemExpress), LY294002 (cat. no. HY-10108; MedChemExpress) and
Wedelolactone (cat. no. W4016; MilliporeSigma) for 2 h at 37˚C and
then were treated with 0.2 µg/ml LPS for 12 h at 37˚C in a
humidified incubator with 5% CO2 followed by the
extraction of protein or RNA.
To determine the role of TRIM65 in macrophage
activation, the PMs isolated from WT and TRIM65 knockout
(TRIM65-/-) mice were treated with 10 ng/ml LPS for 4 h
at 37˚C in a humidified incubator with 5% CO2 followed
by extraction of RNA.
To induce monocyte/macrophage differentiation, THP-1
cells at a density of 2.5x105 cells/cm2 were
treated with 100 nM PMA (cat. no. 19-144; MilliporeSigma) and
cultured overnight in complete RPMI-1640 medium. PMA was then
removed by aspiration and the adhesive cells were washed twice with
PBS. The derived THP-1/PMA macrophages were cultured in complete
RPMI- 1640 medium at 37˚C in a humidified incubator with 5%
CO2 for another 2 days followed by treatment by LPS.
Animals and in vivo study
WT mice with C57BL/6 background (male, 7-week-old,
n=20) were purchased from Hunan SJA Laboratory Animal Co., Ltd.
(Changsha, China). Apolipoprotein E-knockout (ApoE-/-)
mice on a C57BL/6 J background (8-week-old, two female and 1 male)
were obtained from Charles River Laboratories, Inc. A total of 8
male ApoE-/- mice were produced by interbreeding and
used in the present study. Heterozygous TRIM65-knockout
(TRIM65+/-) mice on a C57BL/6 background (8-week-old,
two female and 1 male) were purchased from Cyagen Biosciences,
Inc., whereas TRIM65 knockout homozygous (TRIM65-/-)
mice (male, 8-week-old, n=4) were generated by interbreeding and
used in this study. All mice were housed in SPF units of the Animal
Center at Nanchang University (with a 12-h light cycle from 8 a.m.
to 8 p.m. at 23±3˚C with 60-70% humidity) and maintained on a
standard rodent diet with free access to water in plastic bottles.
Animal health and behavior were monitored every day. The protocols
for animal experiments were approved by the Ethics Committee of
Nanchang University (approval nos. 20X049 and X20200703) and
complied with the Guide for the Care and Use of Laboratory Animals
published by NIH (1). All the mice
were sacrificed at the end of the experiments or upon reaching
humane endpoints. The humane endpoints set for the present study
included loss of appetite, persistent weight loss, dyspnea,
persistent convulsions and severe hypothermia that could not be
recovered by warming. Mortality in mice was verified by the loss of
heartbeat and dilation of pupils.
The male WT mice were allowed to acclimatize to
their housing environment for ≥7 days before the experiments, where
≤ five mice were kept per plastic cage with corn cob bedding
material. The mice were then divided into two groups at random
(n=6), before they underwent the following treatment scheme at 8
a.m. on day 0: Mice in the control group were fed with normal chow
and injected intraperitoneally with saline (100 µl/mice); whereas
mice in the LPS group were fed with normal diet and injected
intraperitoneally with LPS (20 mg/kg). After treatment for 12 h,
the mice were sacrificed in a CO2 chamber with the flow
displacement rate of 30% container volume per minute at 8 p.m. on
day 0. Subsequently, the spleen, lung, aorta and bone marrow of
mice were all collected. Protein and RNA samples were then
extracted, which were used for the determination of TRIM65 protein
and mRNA expression by western blotting and RT-qPCR.
To determine the expression level of TRIM65 in the
aorta during the progress of atherosclerosis, ApoE-/-
mice (~12-week-old, male) were randomly divided into two groups
(four mice/group) and were fed with either Normal diet (ND) or
western diet (WD: 0.5% cholesterol and 21% fat) at week 0. On week
16, all of the mice were anesthetized and euthanized in a
CO2 chamber with the flow displacement rate of 30%
container volume per minute, followed by the collection of mouse
aortas and the measurement of TRIM65 mRNA expression by
RT-qPCR.
Identification of the
TRIM65-/- mice genotype by PCR
DNA were extracted from 0.5 cm mouse tails from mice
with mouse tissue lysis buffer (cat. no. 19697ES70; YEASEN
Biotechnology (Shanghai) Co., Ltd.), before PCR was performed using
2X Rapid Taq Master mix (cat. no. P222-01; Vazyme Biotech Co.,
Ltd.). The primers were as follows: Mouse TRIM65 forward,
5'-CTGGAACTCCCATCTGCTCTGCT-3' and reverse,
5'-GGGAGGAGTGTGGACAGGACAGTT-3' and mouse TRIM 65-Wt/He forward,
5'-CAGGAGATTCAGTAGCCTGCTTCAGG-3' and reverse,
5'-GGGAGGAGTGTGGACAGGACAGTT-3'. The following thermocycling
conditions were used: Initial denaturation at 94˚C for 5 min,
followed by 35 cycles at 94˚C for 30 sec, 60˚C for 30 sec, 72˚C for
1 min and then 72˚C for 10 min. The gel electrophoresis was
performed with 1X TAE (cat. no. T1061; Beijing Solarbio Science
& Technology Co., Ltd.) in 1% agarose gel with YeaRed Nucleic
Acid Gel stain (cat. no 10202ES76; Shanghai Yeasen Biotechnology
Co., Ltd.). If the PCR revealed one band at 660 bp, then the mice
would be considered to be of the TRIM65-/- genotype. By
contrast, if the result of PCR revealed two bands with lengths of
660 and 909 bp, then the mice would be identified to be of the
TRIM65+/- genotype. If the result of PCR showed one band
at 909 bp, then the mouse would be considered as WT.
Determination of TRIM65 protein
expression by western blotting
After treatment, total protein samples were
extracted from tissues or cells using a protein lysis buffer (RIPA;
cat. no. R0010; Beijing Solarbio Science & Technology Co.,
Ltd.) containing PMSF and cocktail (10 µg/ml), before being
centrifuged at 10,000 x g for 10 min at 4˚C. After detection of
protein content using a BCA assay kit (cat. no. 23227; Thermo
Fisher Scientific, Inc.), TRIM65 protein level was examined in 30
µg total protein of each sample using western blotting as described
previously (2,21). Briefly, proteins from each sample
were separated by SDS/PAGE (10% gel) and transferred onto a
nylon-enhanced nitrocellulose membrane. The membrane was then
blocked with a solution of 5% skimmed milk in PBS for 1 h at room
temperature, before being incubated with primary antibodies against
TRIM65 (1:1,000; cat. no. HPA021578; Sigma-Aldrich; Merck KGaA),
p-ERK (1:1,000; cat. no. 4370S; Cell Signaling, Inc.), ERK
(1:1,000; cat. no. 4695S; Cell Signaling, Inc.) and GAPDH (1:5,000,
cat. no. 60004-1-Ig; ProteinTech Group, Inc.) or β-actin (1:5,000;
cat. no. 66009-1-Ig; ProteinTech Group, Inc.) at 4˚C overnight.
This was followed by washing three times for 10 min each with a
solution of 0.5% Tween-20 in PBS (PBS-T). The membranes were then
exposed to HRP-conjugated goat anti-rabbit (1:5,000; cat. no.
31460; Thermo Fisher Scientific, Inc.) or anti-mouse (1:5,000; cat.
no. 31430; Thermo Fisher Scientific, Inc.) IgG secondary antibodies
and incubated for 1 h at room temperature. After three washes with
PBS-T (10 min each), the membrane was incubated for 5 min in a
mixture of equal volumes of Western blot chemiluminescence reagents
1 and 2 of the Ultra High Sensitivity ECL Kit (cat. no. GK10008;
GlpBio Technology). The protein bands were visualized using an ECL
detection system (Tanon Science and Technology Co., Ltd.) and
analyzed using the Image J software (version 1.46r; National
Institutes of Health).
Examination of TRIM65, TNFα, IL-1β and
IL-6 mRNA expression by RT-qPCR
After treatment, tissues or cells were lysed before
RNA was extracted using the M5 HiPer Total RNA Extraction Reagent
(cat. no. MF034-01; JHM IT Group) as described previously (1). After quantification, total RNA (2 µg)
was used to synthesize cDNA with HiScript Q RT SuperMix for qPCR
(+gDNA wiper; cat. no. R123-01; Vazyme Biotech Co., Ltd.). The
following conditions were used: Initial wiper gDNA at 42˚C for 2
min, followed by 50˚C for 15 min and 85˚C for 5 sec. TRIM65, TNFα,
IL-1β and IL-6 mRNA expression was examined by RT-qPCR using a 2x
Universal Blue SYBR Green qPCR Master Mix (cat. no. G3326; Wuhan
Servicebio Technology Co., Ltd.) with primers listed in Table I. The following thermocycling
conditions were used: Initial denaturation at 95˚C for 30 sec,
followed by 40 cycles at 95˚C for 15 sec, 60˚C for 30 sec, before
the melting curve was made according to the default Settings of the
instrument. GAPDH was used as the internal reference and the
2-ΔΔCq method (24) was
used to calculate the relative expression.
 | Table ISequences of primers for reverse
transcription-quantitative PCR. |
Table I
Sequences of primers for reverse
transcription-quantitative PCR.
| Gene | Forward
(5'-3') | Reverse
(5'-3') |
|---|
| Mouse TRIM65 |
AAGAGAAGAGCCTCCCCAAG |
GGTCTCTGGGTCAAAGGTCA |
| Mouse TNFα |
ATGGCCTCCCTCTCATCAGT |
TTTGCTACGACGTGGGCTAC |
| Mouse IL-1β |
AACCTGCTGGTGTGTGACGTTC |
CAGCACGAGGCTTTTTTGTTGT |
| Mouse IL-6 |
GGGACTGATGCTGGTGACAA |
TCCACGATTTCCCAGAGAACA |
| Mouse GAPDH |
ACCCAGAAGACTGTGGATGG |
ACACATTGGGGGTAGGAACA |
| Human TRIM65 |
CCTGGAAATACAGCACACGA |
AAGGTCTGCTCATCCACCTG |
| Human GAPDH |
GGTGGTCTCCTCTGACTTCAACA |
GTTGCTGTAGCCAAATTCGTTGT |
Statistical analysis
All data are presented as the mean ± standard error
of mean. All statistical analyses were conducted with the GraphPad
Prism 7 software (Dotmatics) and the statistical significance of
differences was assessed using the unpaired Student's t-test
between two groups or one-way analysis of variance followed by
Tukey's post hoc test among > two groups (Fig. 1, Fig.
2, Fig. 3 and Fig. 4). Statistical significance of
differences in Fig. 5 was analyzed
using two-way ANOVA followed by Sidak's post hoc tests. P<0.05
was considered to indicate a statistically significant
difference.
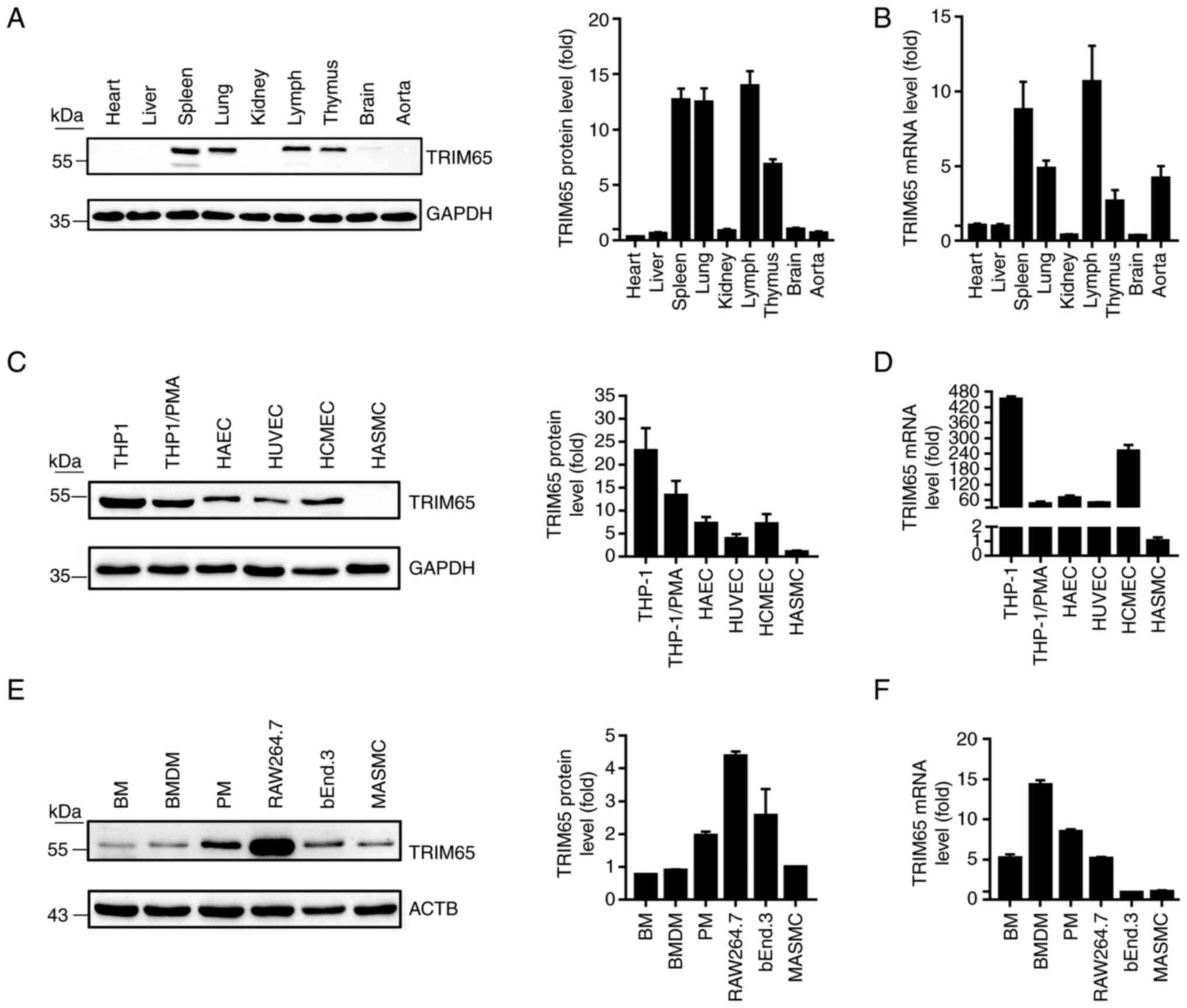 | Figure 1TRIM65 expression levels in mouse
tissues and cell lines. Total protein or RNA samples were extracted
from tissues or cell lines, and TRIM65 expression was examined by
western blotting and RT-qPCR. (A) TRIM65 protein and (B) mRNA
expression in mouse heart, liver, spleen, lung, kidney, lymph,
thymus, brain and aorta tissues. (C) TRIM65 protein and (D) mRNA
expression in a panel of human cell lines, namely THP1, THP1/PMA,
HAEC, HUVEC, HCMEC and HASMC. (E) TRIM65 protein and (F) mRNA
expression in a panel of mouse cell lines, namely BM, BMDM, PM,
RAW264.7, bEnd.3 and MASMC. HUVEC, human umbilical vein endothelial
cells; TRIM65, Tripartite motif-containing protein 65; PMA, phorbol
12-myristate 13-acetate; HAEC, human aortic endothelial cells;
HCMEC, human cerebral microvascular endothelial cells; HASMC, human
aortic smooth muscle cells; BM, bone marrow; BMDM, bone
marrow-derived macrophages; PM, peritoneal macrophages; MASMCs,
mouse aortic smooth muscle cells. |
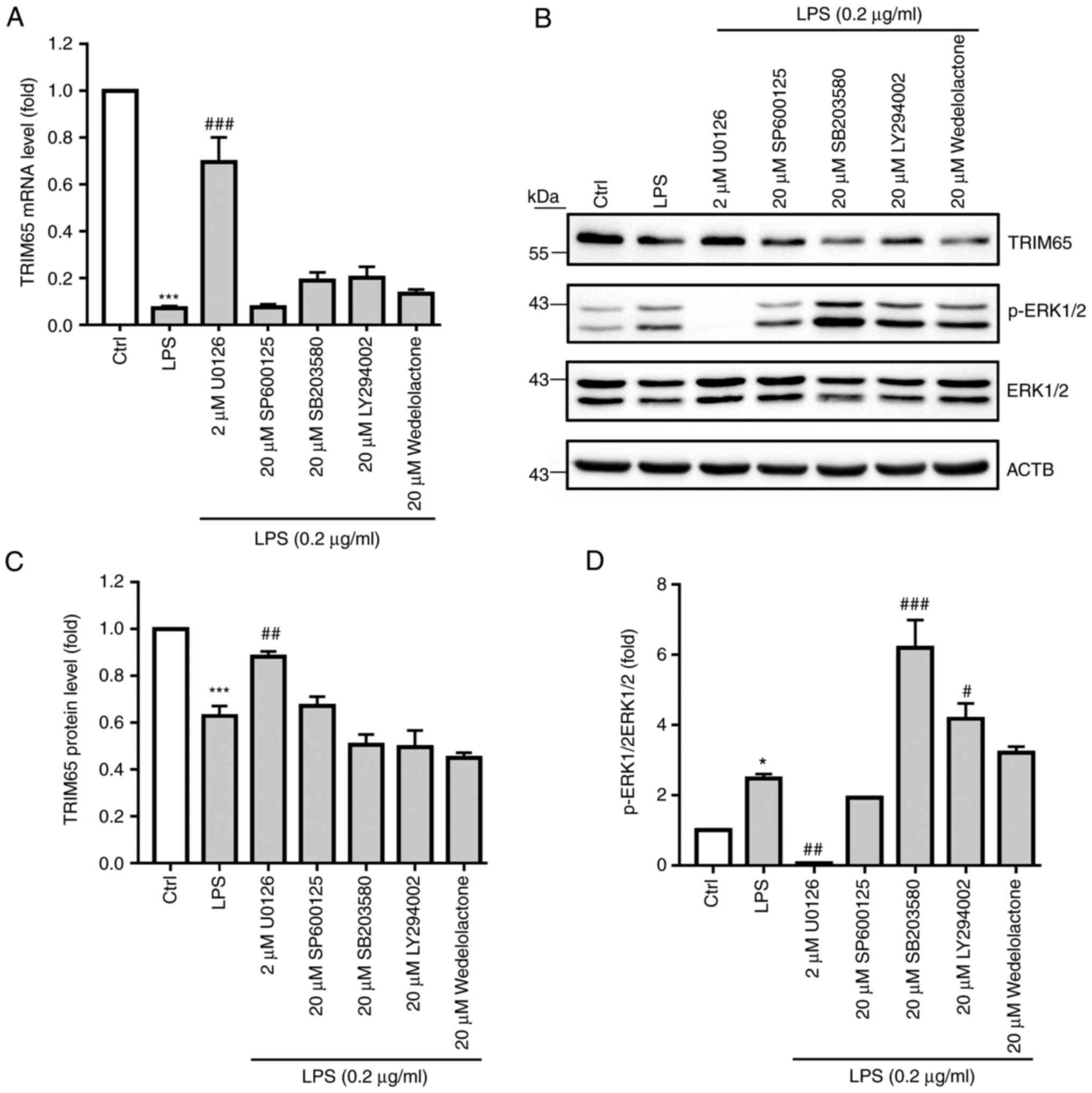 | Figure 4LPS decreases macrophage TRIM65
expression by activating the ERK1/2 signaling pathway. RAW264.7
macrophages were pre-treated with a variety of inhibitors, namely
the c-JNK inhibitor SP600125, the p38 inhibitor SB203580, the
ERK1/2 inhibitor U0126, the PI3K inhibitor LY294002 and the
inhibitor of NF-κB kinase/NF-κB inhibitor Wedelolactone, for 2 h
before they were treated with 0.2 µg/ml LPS for 12 h. After
treatment, (A) TRIM65 mRNA expression was examined by RT-qPCR. (B)
TRIM65 and ERK1/2 expression, in addition to ERK1/2
phosphorylation, were examined by western blot analysis. (C) TRIM65
expression and (D) p-ERK/ERK ratio were then semi-quantified from
(B). *P<0.05 and ***P<0.001 vs. Ctrl,
#P<0.05, ##P<0.01 and
###P<0.001 vs. LPS. LPS, lipopolysaccharide; p-,
phosphorylated; Ctrl, control. |
Results
Expression profile of TRIM65 in
tissues and cells
Mouse tissues were first isolated to examine their
TRIM65 expression profile by western blotting and RT-qPCR in
vivo. As shown in Fig. 1A and
B, TRIM65 is highly expressed in
spleen, axillary lymph node, lung, thymus and aorta, but is
expressed at low levels in the heart, liver, brain and kidneys.
Subsequently, TRIM65 expression in macrophages,
endothelial cells (ECs) and smooth muscle cells (SMCs) was measured
because they have all been reported to serve key roles in the
progression of atherosclerosis (1,25).
The results of Fig. 1C-F suggest
that TRIM65 is highly expressed not only in monocytes or
macrophages (THP1, THP1/PMA, RAW264.7, PM, BMDM and BM), but also
in ECs (HAEC, HUVEC, HCMEC and bEnd.3). However, the expression
level of TRIM65 in SMCs appeared to be species-dependent, with
moderate expression in mice and negligible expression in human.
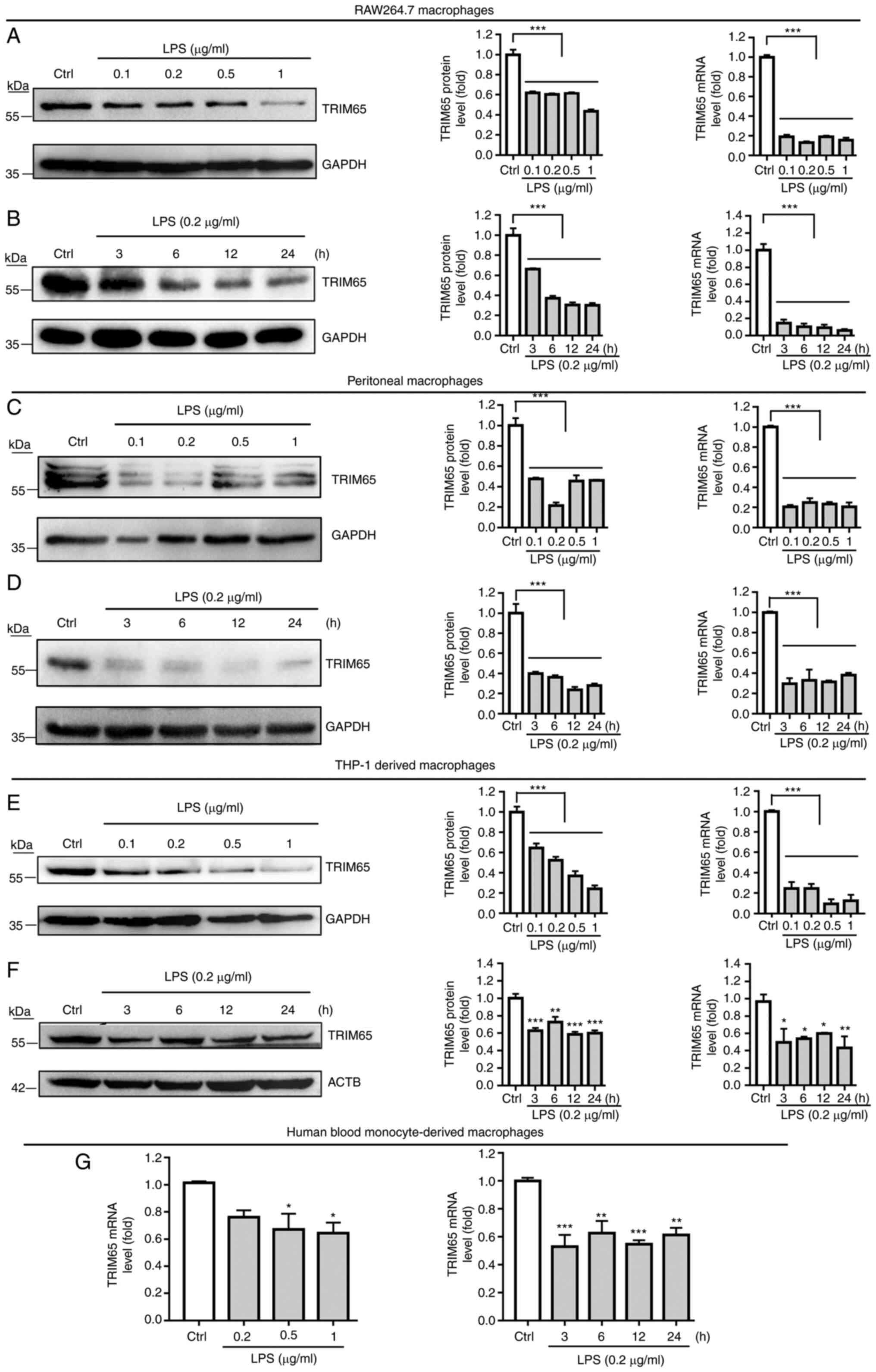
LPS reduces the expression of TRIM65
in macrophages in vitro
The effect of LPS on macrophage TRIM65 expression
was then examined. Both RAW264.7 macrophages and PMs were treated
with LPS at different doses for 24 h or at 0.2 µg/ml for the
indicated duration. TRIM65 protein and mRNA expression was then
examined by western blotting or RT-qPCR. The results in Fig. 2A and C revealed that different doses of LPS all
markedly decreased TRIM65 protein and mRNA expression, in both cell
types. As shown in Fig. 2B and
D, the inhibition of LPS on TRIM65
expression occurred at 3 h, minimized at 12 h, and sustained until
24 h of treatment, in both cell types.
Furthermore, to define if this LPS-mediated
inhibition on TRIM65 expression is dependent on species, LPS was
used to treat the THP-1-derived macrophages. Consistent with
results observed on mouse macrophages, LPS significantly decreased
TRIM65 protein and mRNA expression in THP-1 derived macrophages
(Fig. 2E and F). To further examine whether LPS can
alter TRIM65 expression in primary macrophages, hBMDMs were also
treated with LPS followed by the examination of TRIM65 mRNA
expression with RT-qPCR. The results showed that LPS also markedly
reduced TRIM65 mRNA expression in the hBMDMs (Fig. 2G). Taken together, these results
suggest that LPS can inhibit TRIM65 expression in macrophages in
vitro.
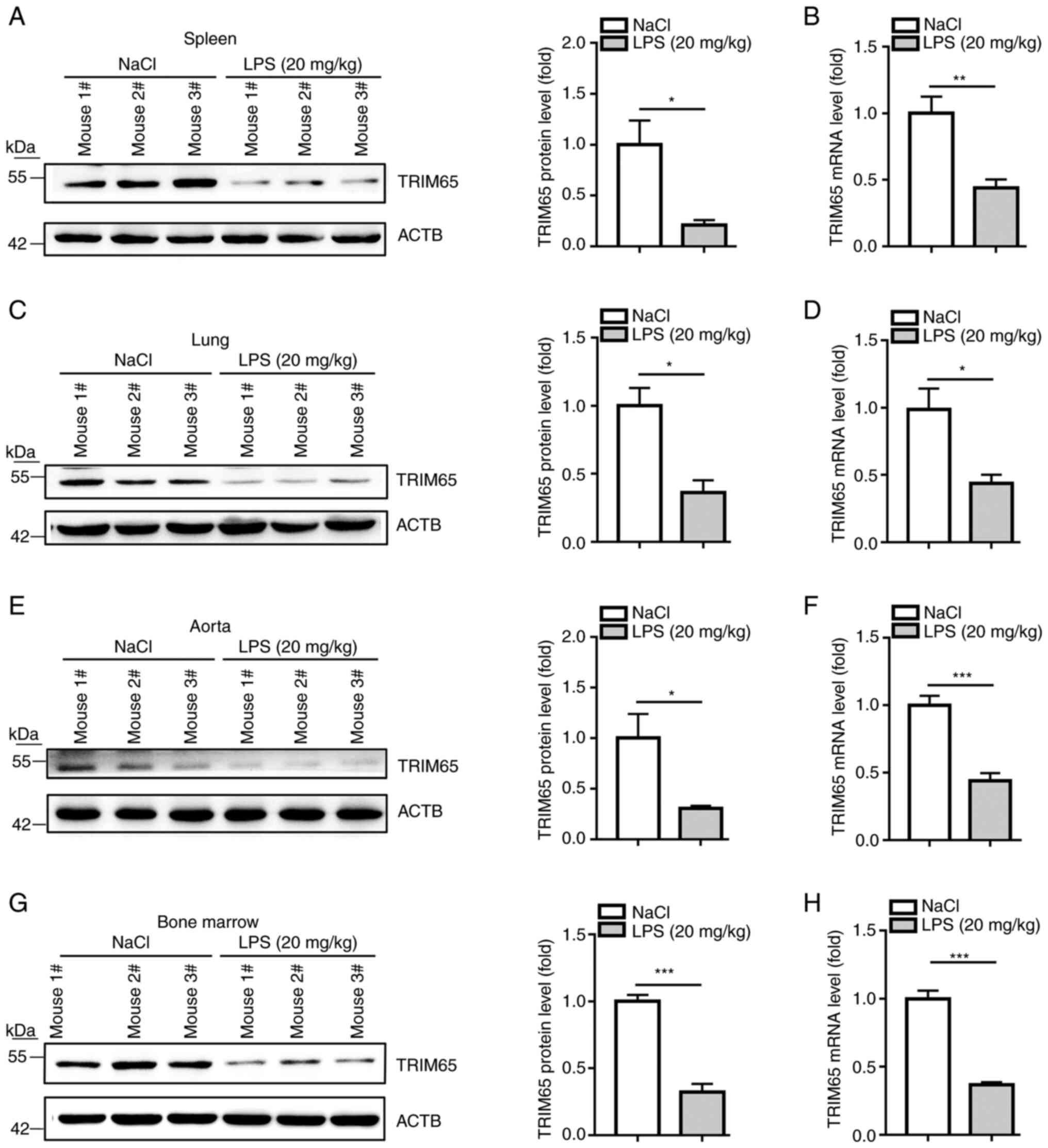
TRIM65 can be reduced by LPS in
vivo
To determine if LPS can inhibit TRIM65 expression
in vivo, protein and mRNA samples were extracted from the
spleen, lung, aorta and bone marrow of mice injected with LPS (20
mg/kg). The results demonstrate that LPS significantly decreased
TRIM65 protein expression in all tissues tested (Fig. 3A, C, E and
G). Consistent with the protein
expression changes, RT-qPCR results (Fig. 3B, D, F and
H) indicate that TRIM65 mRNA
expression in these tissues was likewise significantly decreased by
LPS treatment. Taken together, these observations suggest that
TRIM65 expression is reduced by LPS in vivo.
ERK1/2 signaling serve a key role in
the LPS-decreased TRIM65 expression in macrophages
MAPK (p38, ERK1/2, c-JNK), PI3K and NF-κB have been
reported to be the primary signaling pathways activated by
LPS/toll-like receptor 4 (TLR4) signaling. To clarify if any of
these pathways are responsible for LPS-decreased TRIM65 expression,
RAW264.7 macrophages were pre-treated with U0126 (ERK1/2
inhibitor), SP600125 (c-JNK inhibitor), SB203580 (p38 inhibitor),
LY294002 (PI3K inhibitor) or Wedelolactone (inhibitor of NF-κB
kinase/NF-κB inhibitor) for 2 h, followed by LPS treatment for 12
h. LPS-reduced TRIM65 mRNA expression inhibition was significantly
reversed by U0126, but not by other inhibitors (Fig. 4A). Consistent with mRNA expression,
LPS-reduced expression of TRIM65 protein was likewise significantly
reversed by U0126, but not by other inhibitors (Fig. 4B and C). In addition, U0126 significantly
reversed, whilst SB203580 and LY294002 significantly potentiated
the phosphorylation of ERK1/2 induced by LPS (Fig. 4B and D). In conclusion, these results indicate
that the ERK1/2 pathway serves a crucial role in LPS-induced
suppression of TRIM65 expression.
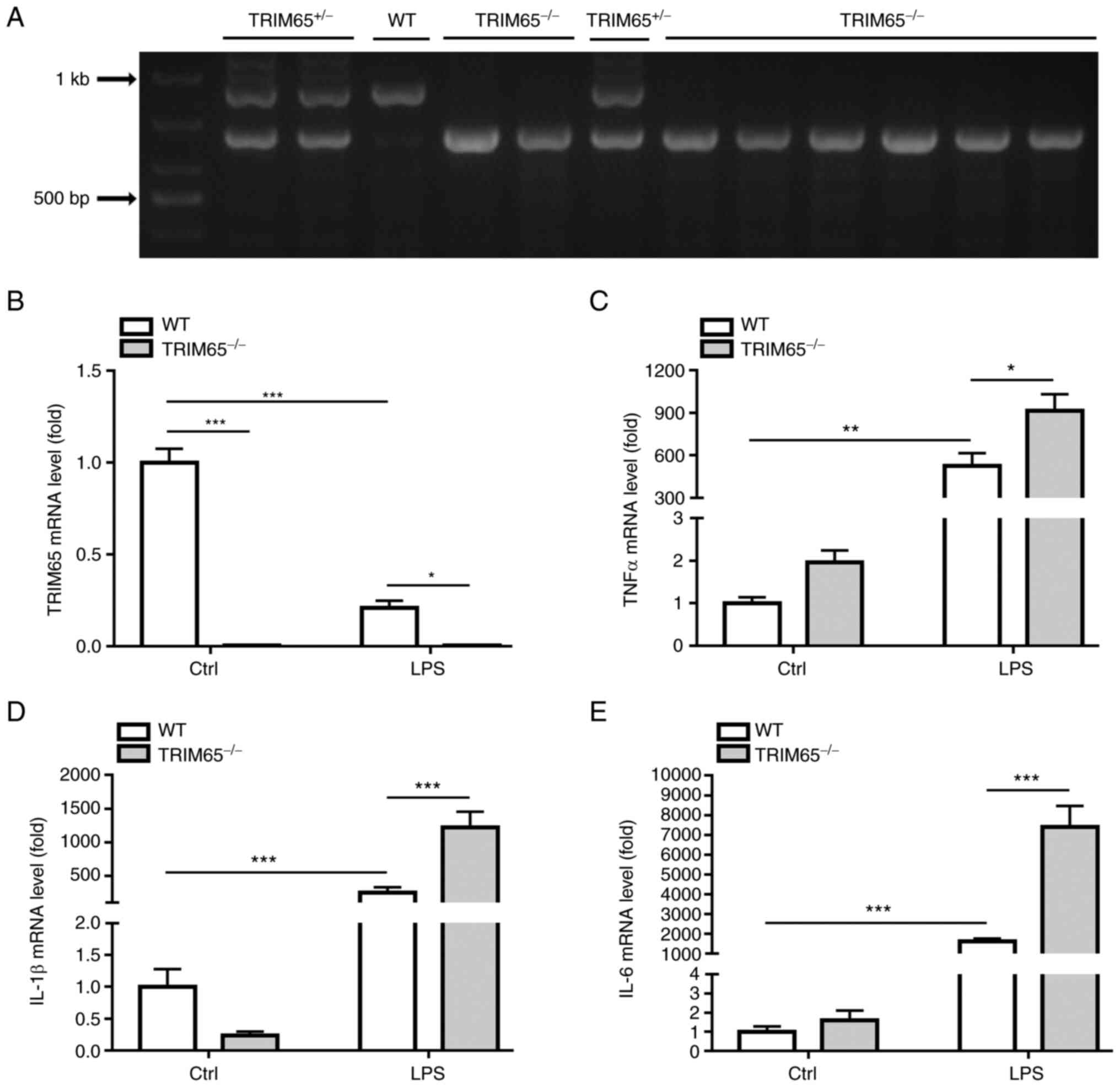
TRIM65 knockout promotes LPS-induced
macrophage activation
Next, the possible role of TRIM65 in macrophage
activation was examined using knockout mice. The genotype of WT and
TRIM65-/- mice were identified by PCR (Fig. 5A) and then PMs isolated from WT and
TRIM65-/- mice were treated with 10 ng/ml LPS for 4 h
followed by the measurement of TRIM65, TNFα, IL-1β and IL-6 mRNA
expression. As shown in Fig. 5B,
TRIM65 mRNA expression was found to be completely abolished in PMs
isolated from TRIM65-/- mice. In addition, LPS treatment
significantly decreased the expression of TRIM65 mRNA in WT-PMs
(Fig. 5B). LPS treatment also
significantly increased the expression of TNFα, IL-1β and IL-6
mRNA, whilst TRIM65-knockout significantly potentiated the
LPS-induced increases in these inflammatory cytokines (Fig. 5C-E).
Atherosclerosis-associated cardiovascular diseases
are among the leading causes of mortality worldwide, in which
macrophages have been documented to serve an important role in the
development of atherosclerosis (1,25).
Therefore, the expression of TRIM65 in mouse aortas collected from
ApoE-/- mice fed a ND or WD for 16 weeks was next
measured. As shown in Fig. S1,
the expression of TRIM65 mRNA was found to be significantly
downregulated in the aorta after feeding on a WD. Taken together,
these results indicate that TRIM65 is a negative regulator of
macrophage activation, which may serve a potential role in the
development of atherosclerosis.
Discussion
Macrophages serve a pivotal role in the progression
of inflammatory diseases, such as atherosclerosis (26-30),
cancer (30-32)
and diabetes (30,33,34).
Macrophages are closely associated with the inflammatory response,
with M1 macrophages mainly facilitating the proinflammatory
response and M2 macrophages mainly promoting the anti-inflammatory
response (28,31). Therefore, improving the
inflammatory environment by regulating the activation status of
macrophages has been proposed to be an effective method for
treating diseases such as cancer and atherosclerosis (35). As an ubiquitin E3 ligase, TRIM65
has been reported to regulate various processes, such as
carcinogenesis including hepatocellular carcinoma, bladder
urothelial carcinoma and colorectal cancer (12-15,36)
and innate immunity (16,17). Although the significance of TRIM65
in inflammation has been acknowledged (19,20),
the regulatory mechanism that controls its expression and activity
in macrophages under inflammatory conditions remain unknown. In the
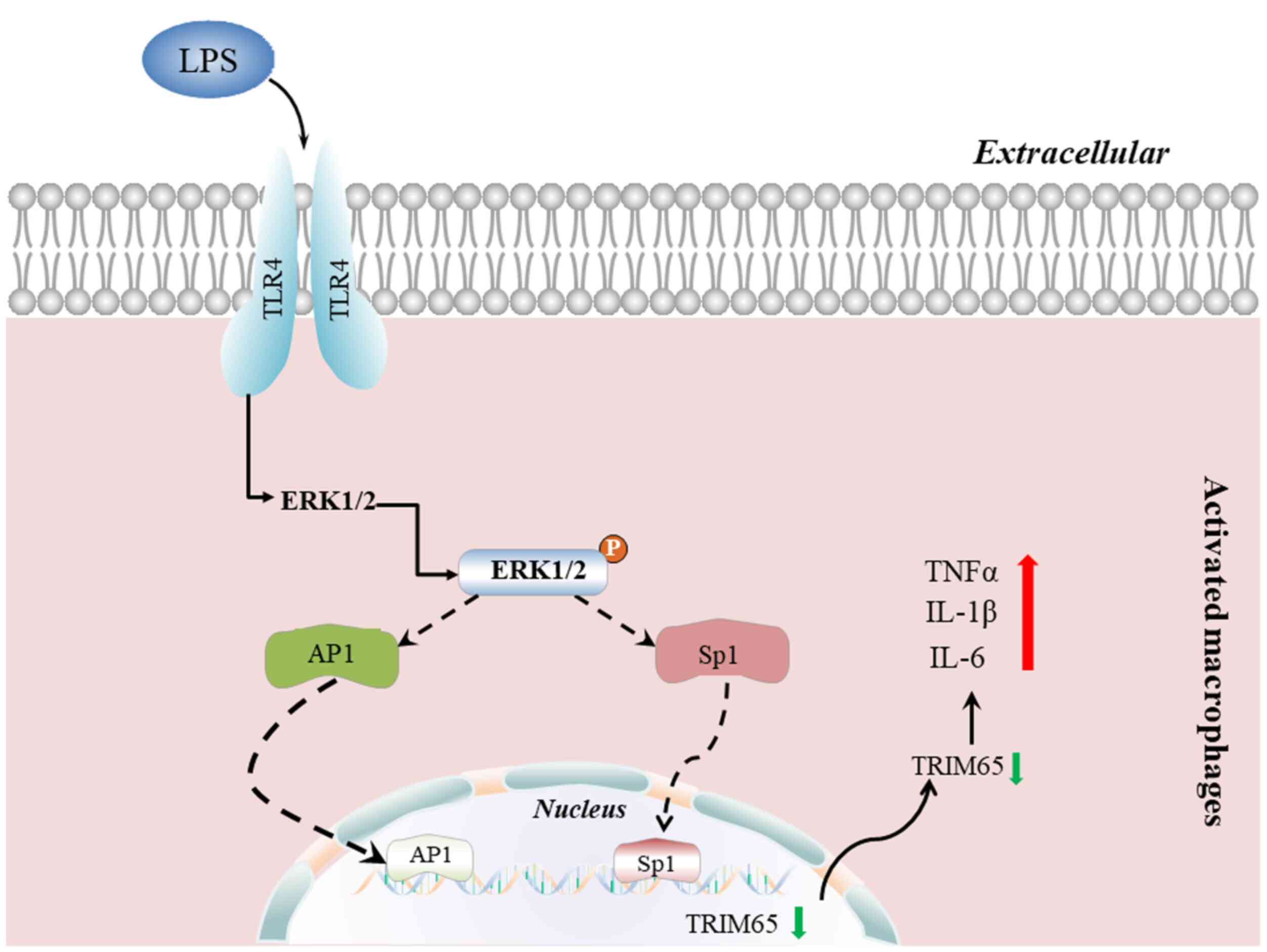
present study, TRIM65 expression was found to be decreased by LPS
treatment in macrophages and C57BL/6J mice through the ERK1/2
signaling pathway, whilst TRIM65 knockout potentiated macrophage
activation by increasing the production of inflammatory cytokines
induced by LPS (Fig. 6). In
addition, it was found that TRIM65 mRNA expression in the aorta of
ApoE-/- mice fed with WD was decreased. Taken together
with the previous findings of anti-inflammatory characteristics of
TRIM65 on LPS-induced lung inflammation by Li et al
(19) and on MSU-induced
peritonitis and gouty arthritis by Tang et al (20), TRIM65 may serve a particularly key
role in the progression of atherosclerosis and be a potential
therapeutic target for human atherosclerotic diseases.
Regarding the potential anti-inflammatory properties
of TRIM65, TRIM65 is likely to be expressed highly in the immune
system. Therefore, in the present study the expression profile of
TRIM65 in mouse tissues was first examined. TRIM65 was found to be
highly expressed in immune tissues, including the spleen, lymph
node and thymus. It was also demonstrated that TRIM65 is highly
expressed in monocytes/macrophages and ECs, both of which have been
reported to serve important roles in inflammatory diseases, such as
atherosclerosis (37). However,
the expression level of TRIM65 was as high as BMDMs in mouse SMCs
but negligible in human SMCs, which may be attributed to the
species differences. Subsequently, possible changes in the
expression of TRIM65 following inflammatory stimuli was examined
further in vitro. Macrophages serve an important role in the
inflammatory response because they can synthesize and release large
quantities of inflammatory mediators and factors following
stimulation by external factors, such as LPS (38). LPS can mimic the inflammatory
response in vivo and have been frequently used as a model of
inflammatory stimulation for study (38). LPS was used for treating a wide
variety of macrophages in the present study, including RAW264.7,
peritoneal macrophages, THP-1-derived macrophages and hBMDMs, LPS
treatment was found to significantly decrease TRIM65 expression in
both murine and human macrophages. This suggest that this
LPS-induced inhibition of macrophage TRIM65 expression is not
species dependent. To further examine whether this can be
replicated in vivo during inflammation, male C57BL/6 mice
were injected with LPS. The results then demonstrate that TRIM65
expression was also reduced by LPS treatment in vivo. Taken
together, these data suggest that LPS can inhibit TRIM65 expression
both in vitro and in vivo.
LPS primarily interacts with monocytes and/or
macrophages through the Toll-like receptor 4 (TLR4) to activate
MAPKs (p38, ERK1/2, c-JNK) and PI3K signaling pathways. These in
turn activate NF-κB or AP-1 to regulate the expression of
downstream targets (1,4,39,40).
Furthermore, activated MAPKs can phosphorylate a number of
transcription factors such as Sp1 to regulate target gene
expression (1,8). Therefore, to clarify the molecular
mechanism underlying LPS-inhibited TRIM65, macrophages were
pre-treated with inhibitors of these aforementioned signaling
pathways before the extent of LPS-inhibited TRIM65 expression was
measured. It was observed that LPS-decreased TRIM65 expression was
reversed by ERK1/2 inhibitors but none of the other inhibitors. It
was also observed that the phosphorylation level of ERK1/2 was
potently inhibited by the ERK1/2 inhibitor but was increased by p38
and PI3K inhibitors, suggesting that the signaling pathway of
ERK1/2, p38 and PI3K-Akt may interact with each other. Although LPS
was found to inhibit macrophage TRIM65 expression by activating the
ERK1/2 signaling pathway, the mechanism downstream of this
activated ERK1/2 signaling in the regulation of TRIM65 expression
in macrophages remains unknown. According to sequence alignment
analysis, several putative AP1 or specificity protein 1 (SP1)
binding sites were observed in the proximal region of the TRIM65
promoter (-1,000 to +123). AP-1 is an important transcription
factor downstream of the ERK1/2 signaling pathway, which can
regulate the expression of target genes such as IL-2 and JunB
(41). By contrast, Sp1 is a
C2H2-type zinc finger protein and an ubiquitous transcription
factor belonging to the Sp/Krüppel-like factor 4 family of proteins
(42). Sp1 has been reported to
regulate a number of cellular processes, such as immune responses,
cell differentiation, cell proliferation and apoptosis (1,43,44)
Post-translational modifications, such as phosphorylation,
sumoylation, acetylation, glycosylation and proteolytic processing
can significantly alter Sp1 activity, resulting in either
activation or repression (43,44).
For example, Sp1 phosphorylation by JNK1/2 kinases increases
protein stability (45). Sp1
sumoylation at Lys16 increases Sp1 degradation (46). In addition, various kinases,
including ERK1/2, JNK and p38, have been found to phosphorylate Sp1
(45,47,48).
A previous study (1) demonstrated
that TRIM59 expression can be regulated by Sp1 and nuclear factor
erythroid 2-related factor-1 in LPS-activated macrophages, which
may be dependent on the activation of JNK signaling. Therefore, LPS
may likely inhibit TRIM65 expression in macrophages by activating
the ERK1/2 signal pathway to regulate AP1 or Sp1 activity, though
this requires further investigation. In future studies, different
lengths of the TRIM65 promoter according to the binding sites of
AP-1 or Sp1 will need to be constructed, before preparing the Ap-1
and Sp1 expression plasmid to further examine the effect of Ap-1
and Sp1 on TRIM65 promoter activity. Additionally, the nuclear
translocation of Ap-1 and Sp1 would need to be measured by western
blotting and immunofluorescent staining assays. The possible
interaction between Ap-1 and Sp1 on the corresponding binding sites
will also need examination by chromatin immunoprecipitation assay.
Finally, the possible effects of Ap-1 and Sp1 siRNA on the
expression of TRIM65 in LPS-activated macrophages by will require
assessment by western blotting and/or RT-qPCR.
Since LPS inhibited the expression of TRIM65 in
macrophages, its role in activated macrophages was therefore
explored further. TRIM65 knockout was observed to markedly enhance
LPS-induced expression of proinflammatory cytokines TNFα, IL-1β and
IL-6, suggesting that TRIM65 is a negative regulator of macrophage
activation. These results are consistent with the previously
reported anti-inflammatory characteristics of TRIM65 by Li et
al (19). However, the
mechanism by which TRIM65 can negatively regulate macrophage
activation remain unclear and further investigations are
required.
Collectively, the present study revealed that LPS
decreased the expression of TRIM65 not only in murine and human
macrophages in vitro, but also in spleen, lung, aorta and
bone marrow of mice in vivo. Mechanistically, it was found
that the LPS-reduced TRIM65 expression was blocked by ERK1/2
inhibitors but not by other inhibitors. Moreover, TRIM65 was found
to negatively regulate macrophage activation. Taken together, these
findings suggest that LPS can inhibit TRIM65 expression in
vitro and in vivo by activating the ERK1/2 signaling
pathway. However, reducing TRIM65 expression can potentiate
macrophage activation by increasing the production of inflammatory
cytokines. These data may provide an insight into the regulation of
TRIM65-mediated immune responses, where TRIM65 may serve to be a
potential target for the treatment of various inflammatory
diseases.
Supplementary Material
Expression of TRIM65 mRNA expression
is decreased in the aorta after feeding with WD. Apolipoprotein
E-/- mice were randomly divided into two groups and fed on ND or WD
for 16 weeks. At the end of the experiment, the aortas of the mice
were collected before TRIM65 mRNA expression was measured by
reverse transcription-quantitative PCR. Data are presented as the
mean ± SEM (n=4); *P<0.05, unpaired Student’s t-test.
ND, normal diet; WD, western diet; TRIM65, Tripartite
motif-containing protein 65.
Acknowledgements
The authors would like to thank Professor Yuanli
Chen (College of Food and Biological Engineering, Hefei University
of Technology, Hefei, China), who donated the cell lines.
Funding
Funding: The present study was supported by National Natural
Science Foundation of China grants (grant nos. 82160094, 82200509
and 81901546) and Natural Science Foundation of Jiangxi Province,
China (grant nos. 20202BAB206087, 20212BCJ23043 and
20212BAB206087).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XFZ, XXD and YQN conducted the experiments. HFB, MLJ
and DW searched the literature and performed data analysis. PZD and
YFX performed data analysis. MXJ designed the study. All authors
have read and approved the final manuscript. MXJ and XFZ confirm
the authenticity of all the raw data.
Ethics approval and consent to
participate
Studies with human samples were approved by the
Ethics Committee of Second Affiliated Hospital of Nanchang
University (Nanchang, China; approval no. 2019002) and complied
strictly with the 2008 Declaration of Helsinki Principle. Written
informed consents were signed by the one participant prior to the
collection of samples. The protocols for animal experiments were
approved by the Ethics Committee of Nanchang University (approval
nos. 20X049 and X20200703) and complied with the Guide for the Care
and Use of Laboratory Animals published by NIH.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
An Y, Ni Y, Xu Z, Shi S, He J, Liu Y, Deng
K, Fu M, Jiang M and Xin HB: TRIM59 expression is regulated by Sp1
and Nrf1 in LPS-activated macrophages through JNK signaling
pathway. Cell Signal. 67(109522)2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Jiang MX, Hong X, Liao BB, Shi SZ, Lai XF,
Zheng HY, Xie L, Wang Y, Wang XL, Xin HB, et al: Expression
profiling of TRIM protein family in THP1-derived macrophages
following TLR stimulation. Sci Rep. 7(42781)2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Liu Z, Zhu H, Dai X, Wang C, Ding Y, Song
P and Zou MH: Macrophage liver kinase B1 inhibits foam cell
formation and atherosclerosis. Circ Res. 121:1047–1057.
2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Shi Q, Cao J, Fang L, Zhao H, Liu Z, Ran
J, Zheng X, Li X, Zhou Y, Ge D, et al: Geniposide suppresses
LPS-induced nitric oxide, PGE2 and inflammatory cytokine by
downregulating NF-κB, MAPK and AP-1 signaling pathways in
macrophages. Int Immunopharmacol. 20:298–306. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Yang J, Wise L and Fukuchi KI: TLR4
cross-talk with NLRP3 inflammasome and complement signaling
pathways in Alzheimer's disease. Front Immunol.
11(724)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Calder PC, Albers R, Antoine JM, Blum S,
Bourdet-Sicard R, Ferns GA, Folkerts G, Friedmann PS, Frost GS,
Guarner F, et al: Inflammatory disease processes and interactions
with nutrition. Br J Nutr. 101 (Suppl 1):S1–S45. 2009.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zhang L, Huang C, Guo Y, Gou X, Hinsdale
M, Lloyd P and Liu L: MicroRNA-26b modulates the NF-κB pathway in
alveolar macrophages by regulating PTEN. J Immunol. 195:5404–5414.
2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Cargnello M and Roux PP: Activation and
function of the MAPKs and their substrates, the MAPK-activated
protein kinases. Microbiol Mol Biol Rev. 75:50–83. 2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hatakeyama S: TRIM proteins and cancer.
Nat Rev Cancer. 11:792–804. 2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sang Y, Li Y, Song L, Alvarez AA, Zhang W,
Lv D, Tang J, Liu F, Chang Z, Hatakeyama S, et al: TRIM59 promotes
gliomagenesis by inhibiting TC45 dephosphorylation of STAT3. Cancer
Res. 78:1792–1804. 2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ozato K, Shin DM, Chang TH and Morse HC
III: TRIM family proteins and their emerging roles in innate
immunity. Nat Rev Immunol. 8:849–860. 2008.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Li Y, Ma C, Zhou T, Liu Y, Sun L and Yu Z:
TRIM65 negatively regulates p53 through ubiquitination. Biochem
Biophys Res Commun. 473:278–282. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yang YF, Zhang MF, Tian QH and Zhang CZ:
TRIM65 triggers β-catenin signaling via ubiquitylation of Axin1 to
promote hepatocellular carcinoma. J Cell Sci. 130:3108–3115.
2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wei WS, Chen X, Guo LY, Li XD, Deng MH,
Yuan GJ, He LY, Li YH, Zhang ZL, Jiang LJ, et al: TRIM65 supports
bladder urothelial carcinoma cell aggressiveness by promoting ANXA2
ubiquitination and degradation. Cancer Lett. 435:10–22.
2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Chen D, Li Y, Zhang X, Wu H, Wang Q, Cai
J, Cui Y, Liu H, Lan P, Wang J, et al: Ubiquitin ligase TRIM65
promotes colorectal cancer metastasis by targeting ARHGAP35 for
protein degradation. Oncogene. 38:6429–6444. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Meng J, Yao Z, He Y, Zhang R, Zhang Y, Yao
X, Yang H, Chen L, Zhang Z, Zhang H, et al: ARRDC4 regulates
enterovirus 71-induced innate immune response by promoting K63
polyubiquitination of MDA5 through TRIM65. Cell Death Dis.
8(e2866)2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Lang X, Tang T, Jin T, Ding C, Zhou R and
Jiang W: TRIM65-catalized ubiquitination is essential for
MDA5-mediated antiviral innate immunity. J Exp Med. 214:459–473.
2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Li S, Wang L, Fu B, Berman MA, Diallo A
and Dorf ME: TRIM65 regulates microRNA activity by ubiquitination
of TNRC6. Proc Natl Acad Sci USA. 111:6970–6975. 2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Li Y, Huang X, Guo F, Lei T, Li S,
Monaghan-Nichols P, Jiang Z, Xin HB and Fu M: TRIM65 E3 ligase
targets VCAM-1 degradation to limit LPS-induced lung inflammation.
J Mol Cell Biol. 12:190–201. 2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Tang T, Li P, Zhou X, Wang R, Fan X, Yang
M and Qi K: The E3 ubiquitin ligase TRIM65 negatively regulates
inflammasome activation through promoting ubiquitination of NLRP3.
Front Immunol. 12(741839)2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Yu M, Jiang M, Chen Y, Zhang S, Zhang W,
Yang X, Li X, Li Y, Duan S, Han J and Duan Y: Inhibition of
macrophage CD36 expression and cellular oxidized low density
lipoprotein (oxLDL) accumulation by tamoxifen: A peroxisome
proliferator-activated receptor (PPAR)γ-dependent mechanism. J Biol
Chem. 291:16977–16989. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Lv XF, Zhang YJ, Liu X, Zheng HQ, Liu CZ,
Zeng XL, Li XY, Lin XC, Lin CX, Ma MM, et al: TMEM16A ameliorates
vascular remodeling by suppressing autophagy via inhibiting
Bcl-2-p62 complex formation. Theranostics. 10:3980–3993.
2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Adhikari N, Shekar KC, Staggs R, Win Z,
Steucke K, Lin YW, Wei LN, Alford P and Hall JL: Guidelines for the
isolation and characterization of murine vascular smooth muscle
cells. A report from the international society of cardiovascular
translational research. J Cardiovasc Transl Res. 8:158–163.
2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Björkegren JLM and Lusis AJ:
Atherosclerosis: Recent developments. Cell. 185:1630–1645.
2022.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Chistiakov DA, Bobryshev YV, Nikiforov NG,
Elizova NV, Sobenin IA and Orekhov AN: Macrophage phenotypic
plasticity in atherosclerosis: The associated features and the
peculiarities of the expression of inflammatory genes (vol 184, pg
436, 2015). Int J Cardiol. 325(186)2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Chinetti-Gbaguidi G, Colin S and Staels B:
Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 12:10–17.
2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Yang S, Yuan HQ, Hao YM, Ren Z, Qu SL, Liu
LS, Wei DH, Tang ZH, Zhang JF and Jiang ZS: Macrophage polarization
in atherosclerosis. Clin Chim Acta. 501:142–146. 2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Barrett TJ: Macrophages in atherosclerosis
regression. Arterioscler Thromb Vasc Biol. 40:20–33.
2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Shapouri-Moghaddam A, Mohammadian S,
Vazini H, Taghadosi M, Esmaeili SA, Mardani F, Seifi B, Mohammadi
A, Afshari JT and Sahebkar A: Macrophage plasticity, polarization,
and function in health and disease. J Cell Physiol. 233:6425–6440.
2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ruffell B and Coussens LM: Macrophages and
therapeutic resistance in cancer. Cancer Cell. 27:462–472.
2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Chittezhath M, Dhillon MK, Lim JY, Laoui
D, Shalova IN, Teo YL, Chen JM, Kamaraj R, Raman L, Lum J, et al:
Molecular profiling reveals a tumor-promoting phenotype of
monocytes and macrophages in human cancer progression. Immunity.
41:815–829. 2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Meshkani R and Vakili S: Tissue resident
macrophages: Key players in the pathogenesis of type 2 diabetes and
its complications. Clin Chim Acta. 462:77–89. 2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Kraakman MJ, Murphy AJ, Jandeleit-Dahm K
and Kammoun HL: Macrophage polarization in obesity and type 2
diabetes: Weighing down our understanding of macrophage function?
Front Immunol. 5(470)2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Yunna C, Mengru H, Lei W and Weidong C:
Macrophage M1/M2 polarization. Eur J Pharmacol.
877(173090)2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wang XY, Mao HW, Guan XH, Huang QM, Yu ZP,
Wu J, Tan HL, Zhang F, Huang X, Deng KY and Xin HB: TRIM65 promotes
cervical cancer through selectively degrading p53-mediated
inhibition of autophagy and apoptosis. Front Oncol.
12(853935)2022.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Libby P, Buring JE, Badimon L, Hansson GK,
Deanfield J, Bittencourt MS, Tokgözoğlu L and Lewis EF:
Atherosclerosis. Nat Rev Dis Primers. 5(56)2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Tanikawa T, Kitamura M, Hayashi Y, Tomida
N, Uwaya A, Isami F and Inoue Y: Anti-inflammatory effects of
morinda citrifolia extract against lipopolysaccharide-induced
inflammation in RAW264 cells. Medicines (Basel).
8(43)2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Zhao K, Song X, Huang Y, Yao J, Zhou M, Li
Z, You Q, Guo Q and Lu N: Wogonin inhibits LPS-induced tumor
angiogenesis via suppressing PI3K/Akt/NF-κB signaling. Eur J
Pharmacol. 737:57–69. 2014.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Fu SP, Li SN, Wang JF, Li Y, Xie SS, Xue
WJ, Liu HM, Huang BX, Lv QK, Lei LC, et al: BHBA suppresses
LPS-induced inflammation in BV-2 cells by inhibiting NF-κB
activation. Mediators Inflamm. 2014(983401)2014.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Atsaves V, Leventaki V, Rassidakis GZ and
Claret FX: AP-1 transcription factors as regulators of immune
responses in cancer. Cancers (Basel). 11(1037)2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Bouwman P and Philipsen S: Regulation of
the activity of Sp1-related transcription factors. Mol Cell
Endocrinol. 195:27–38. 2002.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Chu S: Transcriptional regulation by
post-transcriptional modification-role of phosphorylation in Sp1
transcriptional activity. Gene. 508:1–8. 2012.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Young MJ, Chen YC, Wang SA, Chang HP, Yang
WB, Lee CC, Liu CY, Tseng YL, Wang YC, Sun HS, et al:
Estradiol-mediated inhibition of Sp1 decreases miR-3194-5p
expression to enhance CD44 expression during lung cancer
progression. J Biomed Sci. 29(3)2022.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Chuang JY, Wang YT, Yeh SH, Liu YW, Chang
WC and Hung JJ: Phosphorylation by c-Jun NH2-terminal kinase 1
regulates the stability of transcription factor Sp1 during mitosis.
Mol Biol Cell. 19:1139–1151. 2008.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Wang YT, Chuang JY, Shen MR, Yang WB,
Chang WC and Hung JJ: Sumoylation of specificity protein 1 augments
its degradation by changing the localization and increasing the
specificity protein 1 proteolytic process. J Mol Biol. 380:869–885.
2008.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Kim HS and Lim IK: Phosphorylated
extracellular signal-regulated protein kinases 1 and 2
phosphorylate Sp1 on serine 59 and regulate cellular senescence via
transcription of p21Sdi1/Cip1/Waf1. J Biol Chem. 284:15475–15486.
2009.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Lin HH, Lai SC and Chau LY: Heme
oxygenase-1/carbon monoxide induces vascular endothelial growth
factor expression via p38 kinase-dependent activation of Sp1. J
Biol Chem. 286:3829–3838. 2011.PubMed/NCBI View Article : Google Scholar
|