Introduction
Pulmonary emphysema is a major component of chronic
obstructive pulmonary disease (COPD) (1-2).
Cigarette smoke (CS) exposure and aging are the leading causes of
COPD, but the molecular mechanism that mediate the pathogenesis of
this disease remains poorly understood (1). It has been previously reported that
cell senescence is implicated in the pathogenesis of COPD, which is
frequently accompanied with the accumulation of damaged cellular
components (1). An age-related
increase in neutrophil myeloperoxidase activity and
caspase-3/7-related apoptotic cell death was observed that is
anticipated to further augment the effect of smoke exposure
(1). This data indicates that
CS-impaired autophagy serves as a mechanism for severe emphysema
progression by inducing aggresome formation that impacts innate and
adaptive immune responses, leading to chronic inflammation
(1). Previous studies have
suggested that CS-induced senescence and autophagy in airway
epithelial cells are closely associated with the pathogenesis of
COPD (1,3,4).
However, the detailed mechanism remains unclear (1).
As a key member of the cAMP-dependent,
cGMP-dependent and protein kinase C protein kinase family,
phosphoinositide dependent protein kinase 1 (PDK1) serves an
important role in a variety of cellular functions, leading to the
activation of the PI3K signaling pathway, an event often associated
with the onset and progression of several human cancers (5). A previous study has shown that PDK1
is an important component of the PI3K/AKT signalling pathway, which
can regulate cell cycle progression, proliferation, metabolism,
autophagy and senescence by activating AKT in mammalian cells
(6). However, the role of this
PDK1-mediated AKT signalling in autophagy and senescence due to
emphysema remains unclear (6). It
has been previously reported that the PI3K/AKT/mTOR signalling
pathway serves a key regulatory role in autophagy (7), where the inhibition of PI3K
contributes to the inhibition of autophagy in mice with COPD
(8). AKT can suppress TSC1/2 and
activate mTOR to promote protein synthesis and cell growth and
regulate cell proliferation by inactivating cell cycle inhibitor
(9,10). The PI3K/AKT/mTOR pathway regulates
autophagy to induce apoptosis of alveolar epithelial cells in a
mouse model of COPD (8,9). This suggests a possible relationship
between the PI3K/AKT pathway and cell autophagy associated with
emphysema. Since activation of the PI3K/AKT/mTOR signalling pathway
is one of the main mechanisms of cell senescence (11,12),
it was speculated that activation of PI3K/AKT may result in both
cell senescence and autophagy during emphysema (7,8,11,12).
LC3B is a structural protein of the autophagosome membrane that is
widely used as a biomarker of autophagy (13,14).
In addition, cyclin-dependent kinase inhibitor 2A (p16) is a key
marker of cellular senescence that has been previously observed to
be induced by CS in lung epithelial cells and fibroblasts (15). Since p16 and LC3B proteins are
involved in cell aging and autophagy, respectively, it was
hypothesized that CS exposure combined with intraperitoneal
injections of CS extract (CSE) in mice may induce the expression of
LC3B and p16 in airway epithelial cells through the PI3K/PDK1/AKT
signalling pathway to promote emphysema-like changes associated
with COPD.
In the present study, a mouse model of emphysema was
induced with CS + CSE. Expression levels of the PI3K, PDK1, AKT
proteins, as well as cell aging marker (p16 protein) and autophagy
marker (LC3B protein) were analysed in the normal control group
(control), emphysema (CS + CSE), PI3K inhibitor (CS3) and PDK1
inhibitor-treated (CS1) groups. The aim of this was to explore the
influence of CS + CSE on the PI3K/AKT signaling pathway and the
cell aging and autophagy, as well as their role in the pathogenesis
of COPD. In parallel, the impact of PDK inhibitor on cell aging and
autophagy in emphysema mice was evaluated.
Materials and methods
Mouse experiments
All experiments were approved by the Animal Ethics
Committee of Guizhou Provincial People's Hospital. A total of 35
C57BL/6J mice (4-week-old male; weight, 14-16 g) were purchased
from the Sibefu (Beijing) Biotechnology Co., Ltd. All mice were
housed in a pathogen-free animal facility, which was maintained at
22-24˚C at 50-60% humidity with 12:12 h light-dark cycle. The mice
had free access to food and water before the experiments. As
previously described (16), mice
were randomly divided into the following four groups: i) Control
(n=8); ii) CS + CSE (n=9); iii) CS3 (CS + CSE + PI3K inhibitor;
n=9); and iv) CS1 (CS + CSE + PDK1 inhibitor; n=9). During the
study, the animals were monitored twice a day by the same
researchers. Body condition, posture, coat and grooming, in
addition to the visible mucosa, eyes, ears, whiskers and mental
status were evaluated. The mental state of the mouse was determined
by measuring the autonomic activity, movement, response to the
outside world (sluggish or overactive) and gait of the mouse.
Animals were individually weighed. Food and water intake were also
determined. Criteria used to determine the humane endpoints for the
animals were the following: i) Extreme or prolonged weight
loss/emaciation (body-weight decrease ≥20%); ii) cramps; iii)
paralysis; iv) breathing difficulties; v) diarrhoea (>48 h); vi)
prolonged lack of activity; vii) prolonged decreased food and water
intake; viii) bleeding from an orifice; ix) repeated or severe
self-mutilation; or x) unconsciousness. Any one of these signs
would be a criterium for immediate euthanasia (17,18).
CS exposure was combined with intraperitoneal
injections of CSE to build an emphysema model (16). The smoke exposure box was
66.3x51.8x36.5 cm. For the first 2 days the mice were exposed to
the smoke of 8 cigarettes (China Tobacco Guizhou Industrial Co.,
Ltd.) smoked in sequence twice a day; then, from day 3 onwards,
they were exposed to 10 cigarettes. A single cigarette contained 13
mg tar, 1.2 mg nicotine and 14 mg carbon monoxide. After the last
lit cigarette was placed on the burning rack and the lid of the box
was closed, timing was started and continued for 40 min each time.
After the exposure, the mice were put back into the rearing box and
raised normally, before the experiment was performed again after a
4 h interval to administer the second CS exposure of the day. Mice
were exposed to CS 40 min/time, twice a day, 6 days per week for 4
weeks. CS exposure was not performed on the day of CSE
intraperitoneal injection. During exposure to tobacco smoke, the
smoke exhaust equipment was used to divert the tobacco smoke
outside the laboratory to avoid poisoning the other laboratory
animals and polluting the indoor environment. The normal control
group was given fresh air at the same time and frequency.
The CS + CSE, CS3 and CS1 groups received an
intraperitoneal injection of 100% CSE on days 1, 12 and 23 of
modelling. The dose (PBS; 0.3 ml/20 g) was injected into the
abdominal cavity of mice, where the injection was completed within
30 min after the preparation of CSE. Mice in the normal control
group were intraperitoneally injected with PBS at the same dose and
frequency.
The mice in CS3 group were given the PI3K inhibitor
PF-04691502 (10 mg/kg per mouse; APExBio Technology LLC) through
oral gavage every day. By contrast, mice in the CS1 group were
injected intraperitoneally with a PDK1 inhibitor GSK-2334470 (80
mg/kg per mouse; APExBio Technology LLC) three times per week. The
control and the CS + CSE groups were given the same frequency and
dose of vehicle intervention.
A total of three mice died during the modelling for
unknown reasons. In total, one died on day 21 in the CS + CSE
group, one died on day 25 in the CS1 group and one died on day 23
in the CS3 group. The mortality rate (2-7.6%) observed in the
present study was similar to previous studies (19,20).
Lung biopsies were performed on the dead mice, which suggested that
the cause of death may be associated with damage to the lungs
caused by smoke. The observed pulmonary histopathological changes
of the mice were compromised of alveolar enlargement, alveolar wall
thinning, tissue hyperplasia in alveolar septum at different
degrees, alveolar sac and alveolar tube enlargement or damage and
bronchiole wall injury.
Preparation of CSE
CSE was prepared as previously described (16). Each non-filter Huangguoshu
cigarette was first burned and the smoke of one cigarette was
bubbled through 10 ml PBS, which was designated as 100% CSE. The
solution was utilized within 30 min after preparation.
Tissue processing
Tissue processing was performed as previously
described (20). Lung tissue
samples were obtained from the right middle lobe of each mouse on
day 29 after CS + CSE exposure. Mice were first injected with 3%
chloral hydrate into the abdominal cavity at 300 mg/kg to induce
anaesthesia, followed by euthanasia through intraperitoneal
injections of 100 mg/kg sodium pentobarbital resulting in breathing
inhibition (21). Death was
confirmed when mice had no breathing or heartbeat and did not
respond to gentle stimulation, such as those in the back or
abdomen. The mouse was placed on the operating table, before the
skin of the operation area was disinfected with alcohol and the
superficial skin was cut from the umbilicus along the midline of
the abdomen to the neck and mandible. Toothless forceps were then
used to bluntly separate the subcutaneous tissue whilst ophthalmic
scissors were used to cut the sternum slightly to the right from
the xiphoid process. After exposing and severing the femoral artery
or internal carotid artery, 5 ml normal saline was slowly injected
into the heart for the perfusion of the lung tissue. Straight
forceps were used to clamp the right lung and the hilar tissue was
cut off by ophthalmology. The stump of the right main bronchus was
then ligated using a silk thread. A small incision was cut under
the cartilage ring of the trachea and a self-made bronchoalveolar
lavage needle was used to insert the needle into the bronchus. A
1-ml syringe was used to take 0.5 ml 4˚C pre-cooled PBS for the
pulse lavage of the left lung. Each left lung specimen was washed
three times (10-sec rest after each injection of PBS) before the
lavage liquid was placed in the same Eppendorf tube. The right
middle lobe of each mouse was inflated with 4% paraformaldehyde at
a constant pressure of 2.5 KPa and subsequently fixed with 4%
paraformaldehyde for 24 h at room temperature, after which it was
dehydrated in graded alcohol and embedded in paraffin.
H&E staining of lung tissues and
morphological assessment
H&E staining of lung tissues was performed as
previously described (16).
Paraffin sections (4-µm) were successively added into xylene I (20
min), xylene II (20 min), xylene II (20 min), anhydrous ethanol I
(5 min), anhydrous ethanol II (5 min), 95% alcohol (5 min), 90%
alcohol (5 min), 80% alcohol (5 min), 70% alcohol (5 min) at room
temperature. Then soaked in distilled water for 5 min to dewax.
Paraffin-embedded tissues were stained with hematoxylin for 5 min
and eosin for 5 min (HE) at room temperature to evaluate the
severity of inflammation using a Olympus BX53 biological microscope
(magnification, x200). To assess emphysematous changes in mice,
H&E-stained sections were collected to measure the mean linear
intercept (MLI) and destructive index (DI) (22). The MLI was measured by dividing the
length of a line drawn across the lung section by the total number
of intercepts counted within this line. The SurePathTM PrepStain
automatic liquid-based thin layer cell filmmaking machine was used
for preparation and Pap staining. The cell nucleus was observed at
400 times of light microscope for cell classification. The DI was
calculated by dividing the defined destructive alveoli (enlarged
alveolar space, thinner alveolar septum and destroyed alveolar
wall) by the total number of alveoli. ImageJ Software v1.8.0
(National Institutes of Health) was used to analyse MLI.
Bronchoalveolar lavage fluid (BALF),
cytokine analysis and measurements of the total and differential
cell counts
BALF cell counting was performed as previously
described (20). The lavage liquid
previously collected was centrifuged at 111 g for 5 min at 4˚C
within 2 h and the cell-free supernatants were stored at -80˚C for
subsequent cytokine analysis. The pellet was smeared onto slides
for cell classification and the counting of BALF. The SurePathTM
PrepStain automatic liquid-based thin layer cell filmmaking machine
(BD TriPath) was used for preparation and Pap staining. The cell
nucleus was observed at 400 times of light microscope for cell
classification. Concentrations of IL-6 were measured in the BALF
supernatant using the IL-6 ELISA kit (cat. no. E-EL-M0044c;
Elabscience Biotechnology, Inc.).
Immunohistochemistry (IHC)
The levels of PDK1, PI3K and AKT expression in the
airway epithelial tissues were evaluated by IHC staining of
paraffin-embedded tissues as previously described (23). Briefly, paraffin-embedded tissues
were cut into 4-µm thick sections, dewaxed and rehydrated, as
aforementioned. The slices were treated with 3%
H2O2 in methanol for 15 min at room
temperature to inhibit endogenous peroxidase activity. Antigen
retrieval was performed in a pressure cooker with 10 mM sodium
citrate buffer (pH 6.0) at 100˚C for 15 min. Slices was blocked
with normal 5% goat serum (cat. no. AR1009; Wuhan Boster Biological
Technology, Ltd.) for 30 min at room temperature. Slices were
incubated overnight at 4˚C with anti-PDK1 (1:100; cat. no.
10026-1-Ig; ProteinTech Group, Inc), anti-PI3K (1:200; cat. no.
67121-1-Ig; ProteinTech Group, Inc) and anti-AKT (1:100; cat. no.
ab23509; Abcam) primary antibodies. After washing with PBS (cat.no.
P1020; Beijing Solarbio Science & Technology Co., Ltd.) and
0.05% Tween-20 (Beijing Solarbio Science & Technology Co.,
Ltd.), the specimens were incubated with CY3-labeled goat
anti-mouse IgG (1:100; cat. no. BA1031; Wuhan Boster Biological
Technology) and goat anti-rabbit IgG (1:100; cat. no. BA1032; Wuhan
Boster Biological Technology) secondary antibodies at room
temperature for 1 h. Subsequently, slides were stained with DAB
(cat. no. K5007; Dako; Agilent Technologies, Inc.) for 3 to 10 min
at room temperature for color development, and the protein was
counterstained with hematoxylin for 2 min at room temperature.
Images were captured using a light microscope (magnification,
x400). Image J Software v1.8.0 (National Institutes of Health) was
used to analyse the average optical density of each protein in the
airway epithelial tissue.
Immunofluorescence (IF)
The levels of LC3B and p16 protein expression in the
airway epithelial tissues were evaluated using IF staining as
previously described (21).
Paraffin-embedded tissues were cut into 4-µm thick sections, before
they were dewaxed and rehydrated as aformentioned. Slices was
blocked with normal 5% goat serum (cat. no. AR1009; Wuhan Boster
Biological Technology, Ltd.) for 30 min at room temperature. Slices
were incubated with anti-LC3B (1:100; cat. no. APG8B; Abcepta) and
anti-p16 antibodies (1:100; cat. no. Bs-11652R; BIOSS) overnight at
4˚C. The samples were then washed (PBST; Tween-20 0.05%; cat. no.
P1031; Beijing Solarbio Science & Technology Co., Ltd.)
extensively and incubated with the appropriate CY3-labeled goat
anti-mouse IgG (1:100; cat. no. BA1031; Wuhan Boster Biological
Technology) and goat anti-rabbit IgG (1:100; cat. no. BA1032; Wuhan
Boster Biological Technology) HRP-conjugated secondary antibodies
for 1 h at room temperature in the dark. Subsequently, the slices
were counterstained with DAPI (1:100; cat. no. C1002; Beyotime
Institute of Biotechnology) for 5 min at 25˚C in the dark.
Immunofluorescence images were captured using a fluorescence
microscope (magnification, x400; Leica Microsystems GmbH). The mean
fluorescence intensity (MFI) of proteins in the airway epithelial
tissues was calculated by the Image J Software v1.8.0 (National
Institutes of Health).
Western blotting
PI3K, PDK1, AKT, p16 and LC3B protein expression
levels were detected and semi-quantified using western blotting as
previously described (21). The
lung tissues were lysed at 4˚C in RIPA buffer (CoWin Biosciences)
and extracts were clarified at 10,000 x g at 4˚C for 20 min.
Protein concentration was determined using a bicinchoninic acid
assay kit (CoWin Biosciences). Subsequently, total protein was
separated using SDS-PAGE (80-µg protein/well) on an 10% gel.
Separated protein was then transferred onto a PVDF membrane. After
the PVDF membrane was blocked with 5% BSA (cat. no. A8020; Beijing
Solarbio Science & Technology Co., Ltd.) for 1 h at room
temperature, it was incubated overnight at 4˚C with the following
primary antibodies: PI3K (1:5,000; cat. no. 60225-1-1g); PDK1
(1:500; cat. no. 18262-1-AP); AKT (1:5,000; cat. no. 60203-1-1g);
LC3B (1:500; cat. no. 18725-1-AP) (ProteinTech Group, Inc.) and p16
(1:2,000; cat. no. R22878; Zen BioScience Co., Ltd.). Separated
proteins were then transferred onto a PVDF membrane, which was
incubated at room temperature for 1 h with the anti-GAPDH
(1:10,000; cat. no. ab181602; Abcam). After three washes (TBST;
Tween-20 0.05%; cat. no. T1085; Beijing Solarbio Science &
Technology Co., Ltd.) for 10 min each, the membrane was incubated
with polyclonal anti-rabbit/mouse HRP-conjugated secondary
antibodies overnight at 4˚C (1:200; cat. no. CW02029 and CW02030;
CoWin Biosciences) and developed with the Pierce ECL Western
Blotting substrate (cat. no. CW0049C; CoWin Biosciences). Equal
loading of the samples was determined by the semi-quantitation of
proteins, as well as by using GAPDH as the loading control. Bands
were semi-quantified using optical densities that were analysed
using Gel-Pro analyser 4.0 (Media Cybernetics).
Statistical analysis
All data were analysed by SPSS 22.0 statistical
software (IBM Corp.). Data from each group are expressed as the
mean ± standard deviation. Differences between two groups were
compared using the unpaired Student's t-test. Comparisons among
multiple groups were performed using one-way ANOVA followed by
Tukey's post hoc test. Each experiment was repeated ≥ three times.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Model evaluation
CS exposure was combined with an intraperitoneal
injection of CSE to build an emphysema model. Histomorphological
changes in lung tissues from emphysematous mice induced by CS
exposure combined with intraperitoneal injection of CSE included
enlarged alveolar space, thinner alveolar septum and destroyed
alveolar wall. Mouse lung histological results showed that this
emphysema model was successfully established in the mice (Fig. 1A and B). The levels of IL-6, number of total
cells, the number of neutrophils and the number of macrophages in
BALF were significantly increased in the CS + CSE group compared
with those in the control (P<0.05; Fig. 1E-H). To assess changes in emphysema
in mice, H&E-stained sections were analysed to measure the MLI
and the DI. The MLI and the DI were significantly greater in the CS
+ CSE group compared with those in the control group (P<0.05;
Fig. 1I and J). These results suggest that the
combined CS exposure with an intraperitoneal injection of CSE
successfully induced lung emphysema. In addition, histomorphology
improvements were observed in lung tissues from mice in the CS3
group and the CS1 group, which included improvements in alveolar
wall damage (Fig. 1C and D). The levels of IL-6, number of total
cells, the number of neutrophils and the number of macrophages in
BALF were significantly decreased in the CS3 group and the CS1
group compared with those in the CS + CSE group (P<0.05;
Fig. 1E-H). The MLI and the DI
were significantly decreased in the CS3 group and the CS1 group
compared with those in the CS + CSE group (P<0.05; Fig. 1I and J).
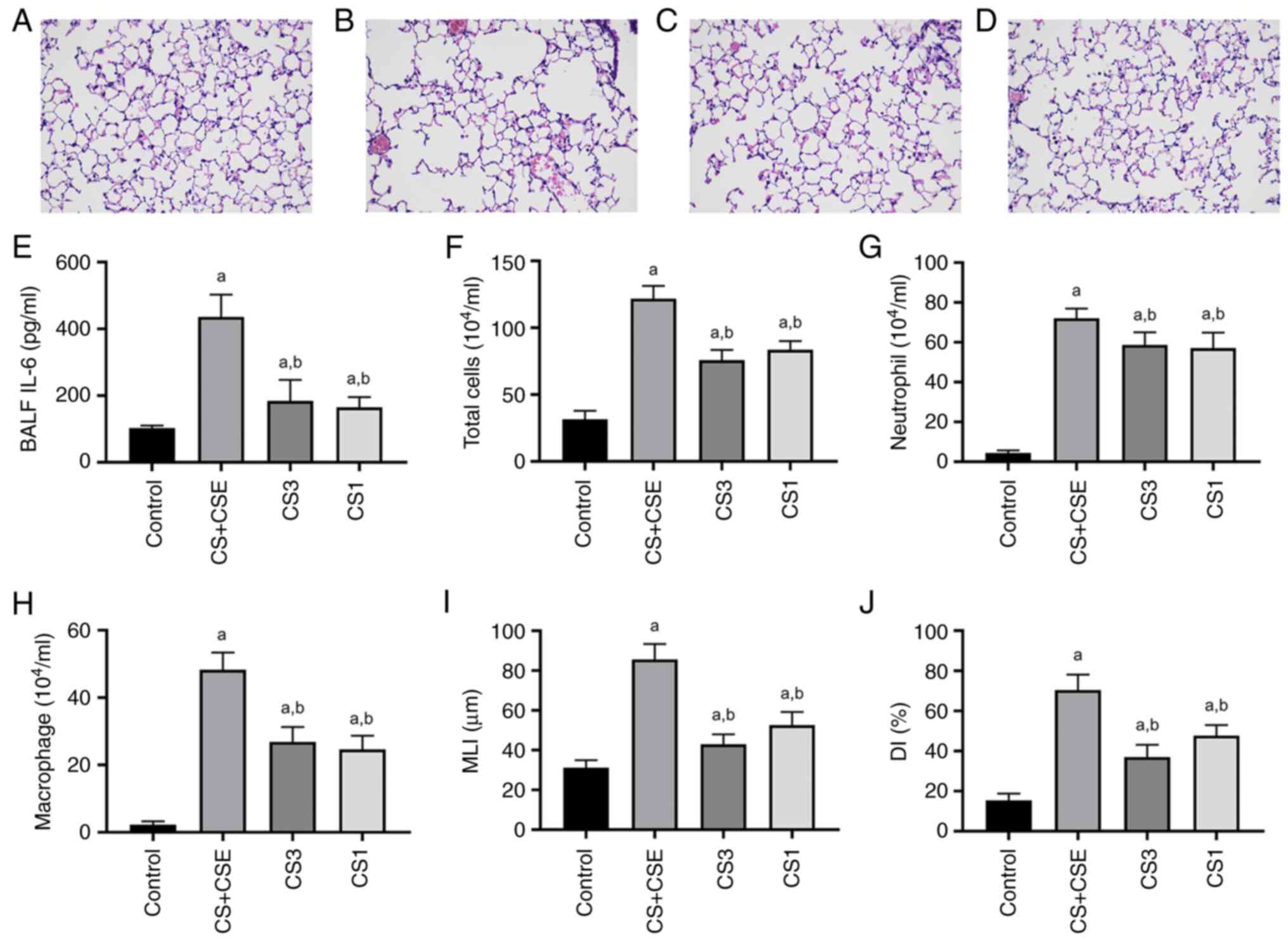 | Figure 1Evaluation of the mouse emphysema
model. H&E staining of lung tissues from the (A) control, (B)
emphysema, (C) PI3K inhibitor and (D) PDK1 inhibitor groups.
Magnification, x400. (E) IL-6 protein levels and (F) total cell
count in BALF. Numbers of (G) neutrophils and (H) macrophages in
BALF. (I) MLI and (J) DI were measured show the extent of airway
remodelling in lung tissues. aP<0.05 vs. control.
bP<0.05 vs. emphysema. BALF, bronchoalveolar lavage
fluid; MLI, mean linear intercept; DI, destructive index; CS + CSE,
emphysema group; CS3, PI3K inhibitor group; CS1, PDK1 inhibitor
group; PI3K, phosphatidylinositol-3-kinase; PDK1, phosphoinositide
dependent protein kinase 1. |
CS + CSE upregulates PDK1 and the
PI3K/AKT signalling pathways in airway epithelial tissues
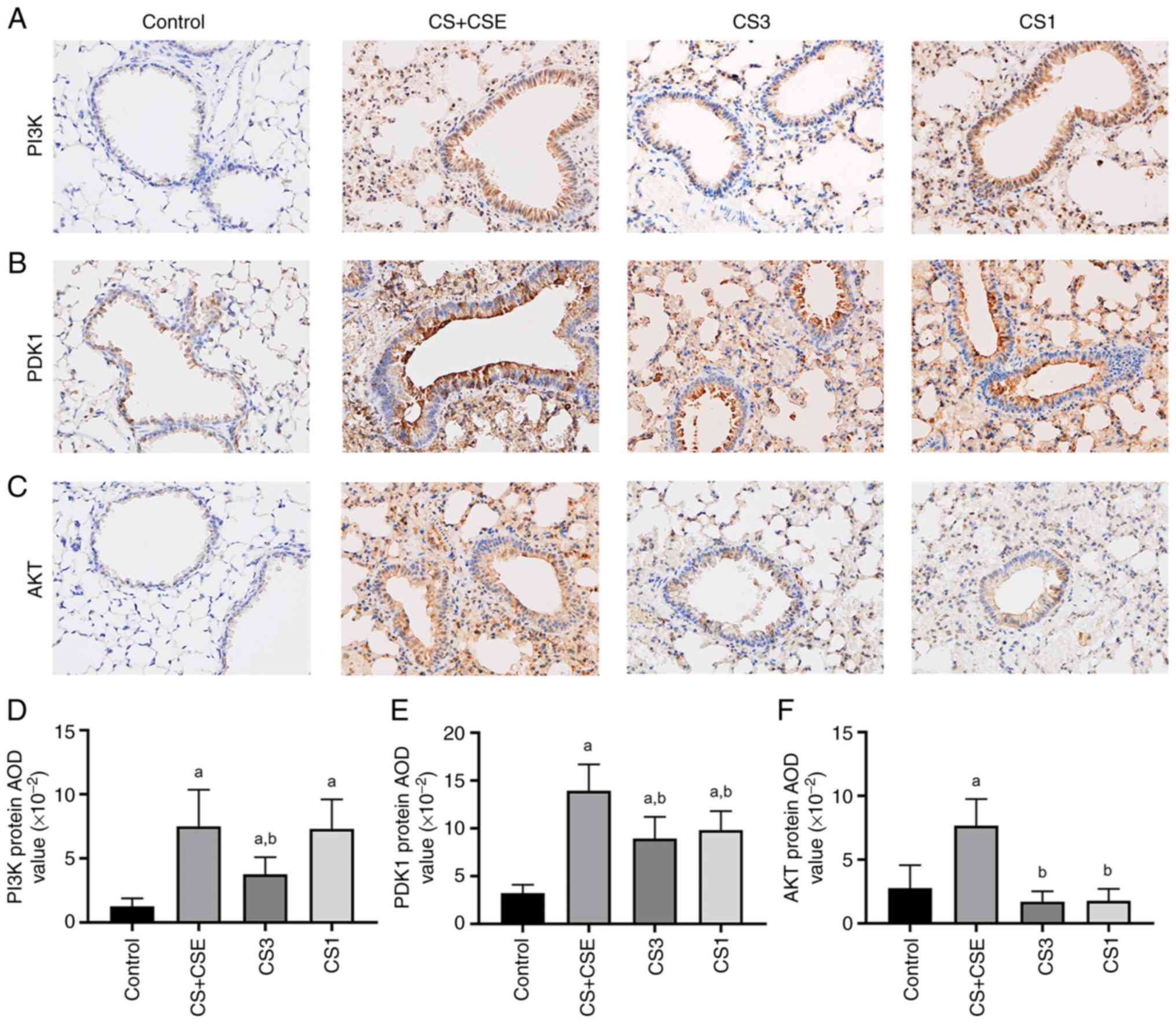
After IHC staining of the lung tissues, the
expression of PI3K, PDK1 and AKT proteins in the airway epithelial
tissues presented as brown staining (Fig. 2A-C). Compared with those in the
control group, the levels of PI3K, PDK1 and AKT protein expression
in the CS + CSE group were significantly increased (P<0.05;
Fig. 2D-F). Overall, these results
suggest that PI3K, PDK1 and AKT protein expression was upregulated
in the CS + CSE-induced emphysema mouse airway epithelial
tissues.
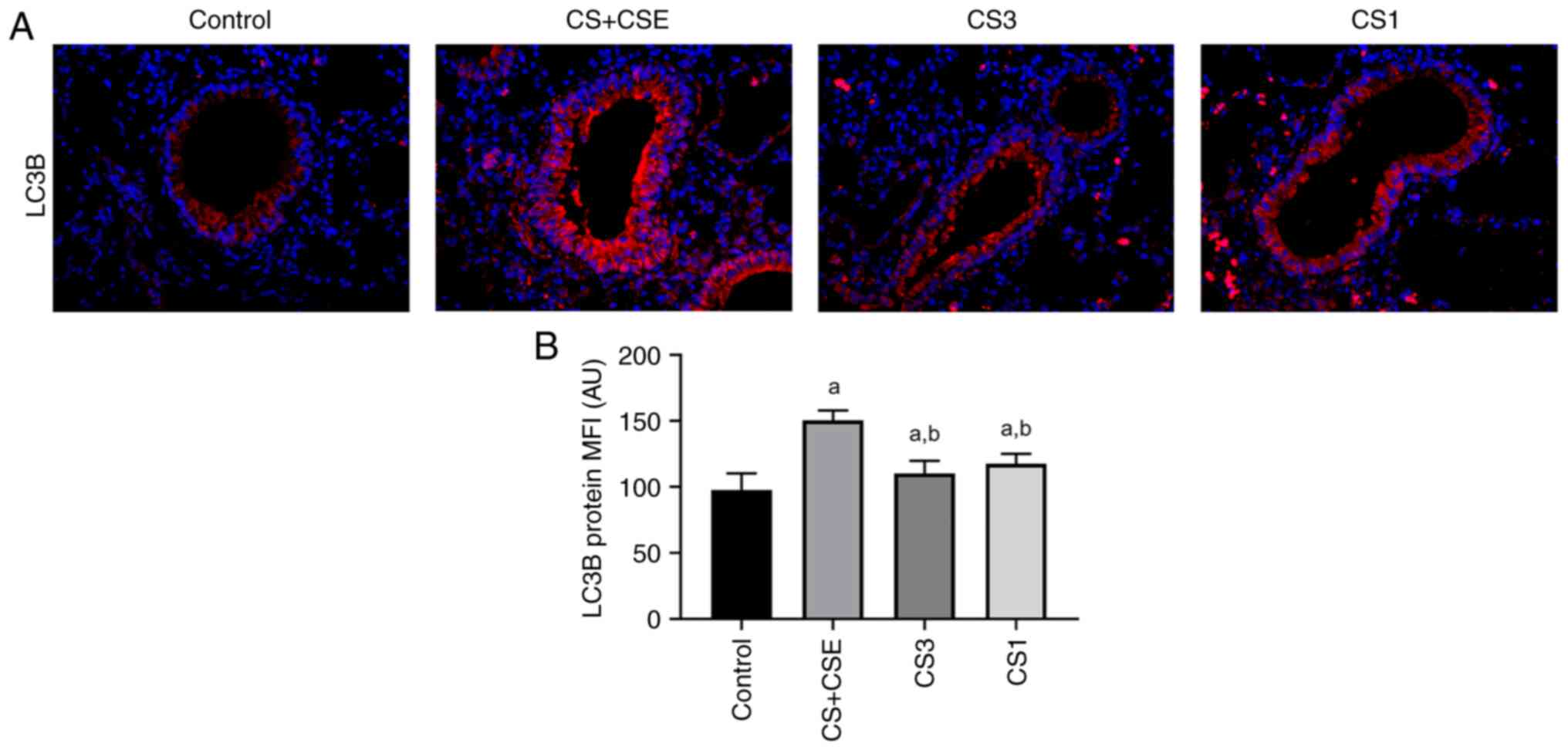
Effects of CS + CSE on autophagy
IF staining was used to detect the levels of LC3B
protein expression in the airway epithelial tissues (Fig. 3A). Under fluorescence microscopy
(magnification, x400), the airway epithelial tissues were observed,
where red represented the positive expression of the LC3B protein
(Fig. 3A). Image J was used to
semi-quantitatively analyse the MFI of the LC3B proteins in the
airway epithelial tissues. Compared with those in the control
group, the expression levels of the LC3B in the CS + CSE group were
significantly increased (P<0.05; Fig. 3B).
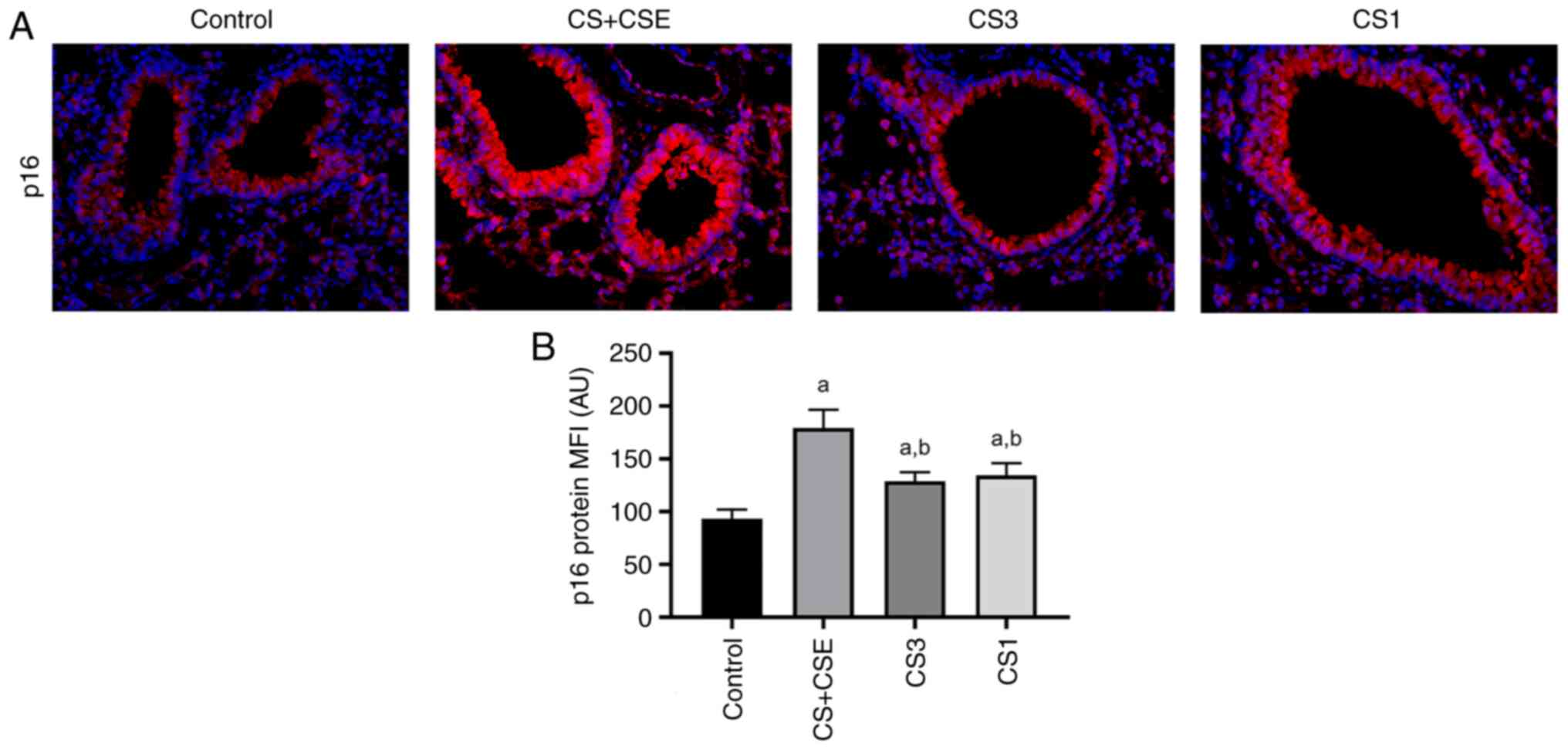
Effects of CS + CSE on cell
senescence
IF staining was used to measure the levels of p16
protein expression in airway epithelial tissues (Fig. 4A). The area of positive p16 protein
expression was stained in red (Fig.
4A). Compared with those in the control group, the expression
levels of p16 in the airway of the CS + CSE group were
significantly increased (P<0.05; Fig. 4B).
The western blotting were used to detect the levels
of PI3K, PDK1, AKT, LC3B and p16 protein expression. The same
results were obtained by western blot. Compared with those in the
control group, the levels of PI3K, PDK1 and AKT protein expression
in the CS + CSE group were significantly increased (P<0.05;
5A-C). Compared with those in the control group, the expression
levels of the LC3B in the CS + CSE group were significantly
increased (P<0.05; Fig. 5D).
Compared with those in the control group, the expression levels of
p16 in the airway of the CS + CSE group were significantly
increased (P<0.05; Fig.
5E).
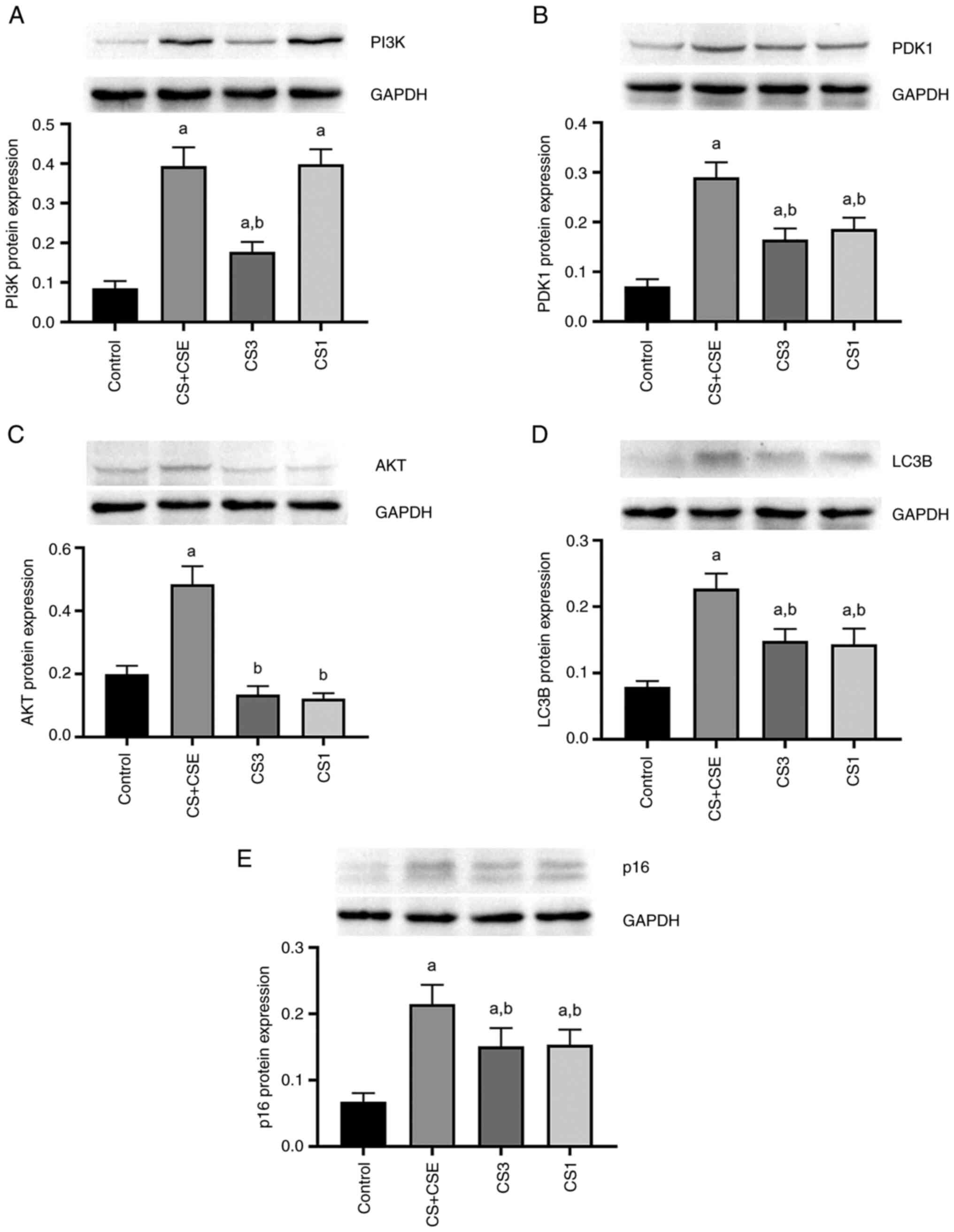 | Figure 5PI3K, PDK1, AKT, LC3B II and p16
protein expression levels. Western blotting for (A) PI3K, (B) PDK1,
(C) AKT, (D) LC3B and (E) p16 expression in the airway epithelial
tissues. GAPDH as an internal control. Semi-quantification of PI3K,
PDK1, AKT, LC3B and p16 protein expression in airway epithelial
tissues in each group. aP<0.05 vs. control.
bP<0.05 vs. emphysema. The protein expression levels
were expressed as the ratio of band intensity for the target
protein relative to that for the internal control GAPDH. Values are
expressed as the mean ± standard deviation. CS + CSE, emphysema
group; CS3, PI3K inhibitor group; CS1, PDK1 inhibitor group; PDK1,
phosphoinositide dependent protein kinase 1; p16, cyclin-dependent
kinase inhibitor 2A. |
Inhibition of PDK1 reduces the
autophagy and cell senescence through the PI3K/AKT signalling
pathway in CS + CSE-induced emphysema mice
To explore the role of PDK1 protein expression and
PI3K/AKT signalling in the present animal model, small molecule
inhibitors were applied. PDK1 inhibitor GSK-2334470 was applied in
the CS1 group. PI3K inhibitor PF-04691502 was applied in the CS1
group. Compared with those in the CS + CSE group, the protein
expression levels of PI3K, PDK1 and AKT in airway epithelial
tissues were significantly decreased in the CS3 group (P<0.05;
Figs. 2D-F and 5A-C). In addition, the protein expression
levels of LC3B and p16 were also significantly decreased in the CS3
group (P<0.05; Figs. 3B,
4B and 5D-E). The protein expression levels of
PDK1, AKT, p16 and LC3B in the airway epithelial tissues of the CS1
group were decreased compared with those in the CS + CSE group
(P<0.05; Figs. 2E-F, 3B, 4B
and 5B-E). However, there was no
significant difference in the expression level of PI3K between the
CS1 and the CS + CSE groups (P>0.05; Figs. 2D and 5A). PI3K is the upstream protein of
PI3K/PDK1/AKT signalling pathway. PDK1 inhibitor does not inhibit
the expression of PI3K, so PI3K does not decrease in CS1 group.
Discussion
CS exposure is the leading cause of emphysema in the
world, though the exact mechanism of pathogenesis remain unclear
(1). In the present study, CS
exposure + short-term CSE intraperitoneal injections were applied
to establish an emphysema model in mice to investigate the impact
of CS exposure and the pathophysiological changes associated with
emphysema progression, in order to explore novel potential
therapeutic options for the prevention of this disease. The levels
of IL-6, numbers of total cells, neutrophils and macrophages in
BALF were found to be significantly higher in the CS + CSE group
compared with those in the control group. In addition, the H&E
staining of lung tissues revealed histologically advanced emphysema
in the CS + CSE group compared with that in the control group,
where the MLI and DI were significantly greater, demonstrating the
validity of this emphysema model in mice.
PDK1 is the major transducer of PI3K downstream
(24). The binding of PDK1 to PIP3
activates the PI3K/AKT signalling pathway, which serves an
essential role in regulating several important physiological
processes, such as cell proliferation, survival and metabolism
(25). A previous study
demonstrated that activation of the PTEN/PI3K/AKT pathway lead to
macrophage M2 polarization in a mouse model of emphysema generated
using CS exposure combined with CSE (22). This study combined CS exposure with
intraperitoneal injection of CS extract (CSE) to build an emphysema
model (22). Activation of the
PI3K pathway controls production of transcriptional factors that
regulate key inflammatory cytokines (26). The results also showed that there
was notably increased p-Akt expression and decreased PTEN
expression in the lung tissue of emphysematous mice, and inhibiting
PI3K/Akt with LY294002 downregulated the expression of M2
macrophage polarization markers and cytokines (22). Therefore, the present study
hypothesized that PDK1 participation in the PI3K/AKT pathway may be
involved in the pathogenesis of this type of emphysema. The present
results concerning the enhanced expression of PI3K, PDK1 and AKT in
the airway epithelial tissues of mice with emphysema were
consistent with those observed in a previous study by Lu et
al (22), which demonstrated
the important role of PI3K, PDK1 and AKT in emphysema of mice.
Autophagy is a cellular degradation and recycling
process that is highly conserved in all eukaryotes (27). It serves an important role in cell
survival and maintenance, the dysfunction of which may contribute
to the pathogenesis of a number of diseases, including lung, liver
and heart disease, neurodegeneration, myopathies, cancer, ageing
and metabolic diseases such as diabetes and COPD (27-29).
As previously described by Koukourakis et al (13), the most extensively studied
endogenous autophagic marker is LC3B, one of the structural
proteins of autophagosome membranes, has also been reported to be
involved in COPD (14). The
present study showed that the expression of LC3B in the airway
epithelial tissues of emphysematous mice was significantly
increased compared with those in the control group, which is
consistent with the previous findings by Chen et al
(14). This suggests that
autophagy is implicated in the pathogenesis of emphysema. In
addition, previous studies also showed that the activation of
PI3K/AKT signalling can promote cell autophagy and aging (30), whereby CS exposure can increase the
expression of senescence marker p16 in the lung tissues of mice
(31), consistent with the present
results. Therefore, it was hypothesized that activation of the
PI3K/AKT signalling pathway in airway epithelial tissues can cause
cell autophagy and aging associated with emphysema. Since PDK1 can
contribute to the activation of the PI3K/AKT pathway (32), it was speculated that its
inhibition may suppress cell autophagy and senescence in
emphysematous mice by blocking PI3K/AKT sigalling. However, further
studies are needed to clarify the exact mechanisms.
GSK-2334470 is an effective PDK1 inhibitor (32) that can regulate the activity of the
PI3K/AKT signalling pathway by inhibiting PDK1, T-loop
phosphorylation and AKT activation (33). The PI3K/AKT signaling pathway plays
a key regulatory role in autophagy (7). Suppression of PI3K can greatly block
the downstream signaling pathways AKT (34). As previously described by Zhang
et al (8) in mice with
COPD, the PI3K inhibitor suppresses protein expression of PI3K,
p-AKT and p-mTOR to reduce autophagy. Previous studies demonstrate
a pivotal role for the autophagic protein LC3B in CS-induced
apoptosis and emphysema (8,14),
autophagic protein microtubule-associated protein 1 light chain-3B
(LC3B) as a positive regulator of CS-induced lung epithelial cell
death (14). In the present study,
the levels of PDK1, AKT, p16 and LC3B expression in the airway
epithelial tissues of mice treated with GSK-2334470 were lower
compared with those in the CS + CSE group. This suggests that
GSK-2334470 may suppress the expression of AKT, p16 and LC3B by
partially blocking PDK1. Since PI3K is the upstream activator of
PDK1, there was no significant difference in the expression of PI3K
between these two groups, consistent with this theoretical
prediction. The present study also showed that PI3K, PDK1, AKT, p16
and LC3B expression were lower in the CS3 group compared with those
in CS + CSE group, which further confirmed the important role of
PI3K in emphysema and the relationship between PI3K, PDK1 and AKT.
It was therefore concluded that PDK1 inhibitors may reduce the
expression of p16 and LC3B proteins in airway epithelial cells by
regulating the PI3K/AKT signalling pathway, thereby protecting
against CS + CSE-induced emphysema in mice. PDK1 inhibitors may
represent a potentially novel therapeutic option for the prevention
of emphysema in humans.
However, further in vitro experiments will be
required. The PDK1-specific short hairpin RNA (shRNA) with the
highest inhibitory efficiency should be screened out with its
interference efficiency verified. Human airway epithelial cells
should be treated with different concentrations of CSE (0, 2.5, 5,
10 and 20%) or fixed concentrations of CSE for different times (0,
0.5, 2, 4, 8, 12 and 24 h). Cell proliferation detection kits, such
as Cell Counting Kit-8, should be used to measure the proliferation
rate of human airway epithelial cells. The optimal concentration
and time of CSE should be selected. Therefore, future studies
should aim to investigate the effect of PDK1 knockdown on the
PI3K/AKT signalling pathway, autophagy and cellular senescence.
They will contribute to an in-depth understanding of the molecular
regulation mechanism of COPD.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by the National Natural
Science Foundation of China (grant no. 1760013; 2018) and the
Non-Profit Central Research Institute Fund of Chinese Academy of
Medical Sciences (grant no. 2019pt320003).
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
PZ, YJ and XY designed and performed the
experiments, wrote the manuscript, read and approved it. YJ, CZ and
YT collected and analysed the data. PZ and YJ confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The project design was conducted in line with
scientific and ethical principles. The institutional review board
of Guizhou Provincial People's Hospital (Guiyang, China) approved
the present study (approval no. 2017034). The present study was
conducted according to the revised Declaration of Helsinki and the
experiments were performed following the Guide for the Care and Use
of Laboratory Animals published by the National Institutes of
Health, eighth edition (35).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Vij N, Chandramani-Shivalingappa P, Van
Westphal C, Hole R and Bodas M: Cigarette smoke-induced autophagy
impairment accelerates lung aging, COPD-emphysema exacerbations and
pathogenesis. Am J Physiol Cell Physiol. 314:C73–C87.
2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Janssen R, Piscaer I, Franssen FME and
Wouters EFM: Emphysema: Looking beyond alpha-1 antitrypsin
deficiency. Expert Rev Respir Med. 13:381–397. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bodas M and Vij N: Augmenting autophagy
for prognosis based intervention of COPD-pathophysiology. Respir
Res. 18(83)2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Bodas M, Van Westphal C,
Carpenter-Thompson R, K Mohanty D and Vij N: Nicotine exposure
induces bronchial epithelial cell apoptosis and senescence via ROS
mediated autophagy-impairment. Free Radic Biol Med. 97:441–453.
2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Barile E, De SK and Pellecchia M: PDK1
inhibitors. Pharm Pat Anal. 1:145–163. 2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Alessi DR, James SR, Downes CP, Holmes AB,
Gaffney PR, Reese CB and Cohen P: Characterization of a
3-phosphoinositide-dependent protein kinase which phosphorylates
and activates protein kinase Balpha. Curr Biol. 7:261–269.
1997.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wu N, Zhu Y, Xu X, Zhu Y, Song Y, Pang L
and Chen Z: The anti-tumor effects of dual PI3K/mTOR inhibitor
BEZ235 and histone deacetylase inhibitor Trichostatin A on inducing
autophagy in esophageal squamous cell carcinoma. J Cancer.
9:987–997. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zhang F, Ma H, Wang ZL, Li WH, Liu H and
Zhao YX: The PI3K/AKT/mTOR pathway regulates autophagy to induce
apoptosis of alveolar epithelial cells in chronic obstructive
pulmonary disease caused by PM2.5 particulate matter. J Int Med
Res. 48(300060520927919)2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Joassard OR, Amirouche A, Gallot YS,
Desgeorges MM, Castells J, Durieux AC, Berthon P and Freyssenet DG:
Regulation of Akt-mTOR, ubiquitin-proteasome and autophagy-lysosome
pathways in response to formoterol administration in rat skeletal
muscle. Int J Biochem Cell Biol. 45:2444–2455. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wang Y, Liu J, Zhou JS, Huang HQ, Li ZY,
Xu XC, Lai TW, Hu Y, Zhou HB, Chen HP, et al: MTOR suppresses
cigarette smoke-induced epithelial cell death and airway
inflammation in chronic obstructive pulmonary disease. J Immunol.
200:2571–2580. 2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Barnes PJ: Mechanisms of development of
multimorbidity in the elderly. Eur Respir J. 45:790–806.
2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Johnson SC, Rabinovitch PS and Kaeberlein
M: mTOR is a key modulator of ageing and age-related disease.
Nature. 493:338–345. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Koukourakis MI, Kalamida D, Giatromanolaki
A, Zois CE, Sivridis E, Pouliliou S, Mitrakas A, Gatter KC and
Harris AL: Autophagosome proteins LC3A, LC3B and LC3C have distinct
subcellular distribution kinetics and expression in cancer cell
lines. PLoS One. 10(e0137675)2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Chen ZH, Lam HC, Jin Y, Kim HP, Cao J, Lee
SJ, Ifedigbo E, Parameswaran H, Ryter SW and Choi AM: Autophagy
protein microtubule-associated protein 1 light chain-3B (LC3B)
activates extrinsic apoptosis during cigarette smoke-induced
emphysema. Proc Natl Acad Sci USA. 107:18880–18885. 2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Sundar IK, Rashid K, Gerloff J, Li D and
Rahman I: Genetic ablation of p16INK4a does not protect against
cellular senescence in mouse models of chronic obstructive
pulmonary disease/emphysema. Am J Respir Cell Mol Biol. 59:189–199.
2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
He S, Li L, Sun S, Zeng Z, Lu J and Xie L:
A novel murine chronic obstructive pulmonary disease model and the
pathogenic role of microRNA-21. Front Physiol.
9(503)2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zieglowski L, Kümmecke AM, Ernst L, Palme
R, Weiskirchen R, Talbot SR and Tolba RH: Assessing the severity of
laparotomy and partial hepatectomy in male rats-A multimodal
approach. PLoS One. 16(e0255175)2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Nemzek JA, Xiao HY, Minard AE, Bolgos GL
and Remick DG: Humane endpoints in shock research. Shock. 21:17–25.
2004.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Cai S, Chen P, Zhang C, Chen JB and Wu J:
Oral N-acetylcysteine attenuates pulmonary emphysema and alveolar
septal cell apoptosis in smoking-induced COPD in rats. Respirology.
14:354–359. 2009.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhang XY, Zhang C, Sun QY, Li D, Luo RR,
Wan ZF, Ye XW, Liu WJ, Rao SS and Han J: Infliximab protects
against pulmonary emphysema in smoking rats. Chin Med J (Engl).
124:2502–2506. 2011.PubMed/NCBI
|
|
21
|
Lee H, Park JR, Kim WJ, Sundar IK, Rahman
I, Park SM and Yang SR: Blockade of RAGE ameliorates
elastase-induced emphysema development and progression via
RAGE-DAMP signaling. FASEB J. 31:2076–2089. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Lu J, Xie L, Liu C, Zhang Q and Sun S:
PTEN/PI3k/AKT regulates macrophage polarization in emphysematous
mice. Scand J Immunol. 85:395–405. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Lin L, Hou G, Han D, Yin Y, Kang J and
Wang Q: Ursolic acid alleviates airway-vessel remodeling and muscle
consumption in cigarette smoke-induced emphysema rats. BMC Pulm
Med. 19(103)2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Bayascas JR: PDK1: the major transducer of
PI3-kinase actions. Curr Top Microbiol Immunol. 346:9–29.
2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Lin JX, Xie XS, Weng XF, Qiu SL, Yoon C,
Lian NZ, Xie JW, Wang JB, Lu J, Chen QY, et al: UFM1 suppresses
invasive activities of gastric cancer cells by attenuating the
expres7sion of PDK1 through PI3K/AKT signaling. J Exp Clin Cancer
Res. 38(410)2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kaneda MM, Messer KS, Ralainirina N, Li H,
Leem CJ, Gorjestani S, Woo G, Nguyen AV, Figueiredo CC, Foubert P,
et al: PI3Kγ is a molecular switch that controls immune
suppression. Nature. 539:437–442. 2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Parzych KR and Klionsky DJ: An overview of
autophagy: Morphology, mechanism, and regulation. Antioxid Redox
Signal. 20:460–473. 2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Mizumura K, Maruoka S, Shimizu T and Gon
Y: Autophagy, selective autophagy, and necroptosis in COPD. Int J
Chron Obstruct Pulmon Dis. 13:3165–3172. 2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wirawan E, Vanden Berghe T, Lippens S,
Agostinis P and Vandenabeele P: Autophagy: for better or for worse.
Cell Res. 22:43–61. 2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Barnes PJ, Baker J and Donnelly LE:
Cellular senescence as a mechanism and target in chronic lung
diseases. Am J Respir Crit Care Med. 200:556–564. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Sorrentino JA, Krishnamurthy J, Tilley S,
Alb JG Jr, Burd CE and Sharpless NE: p16INK4a reporter mice reveal
age-promoting effects of environmental toxicants. J Clin Invest.
124:169–173. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
32
|
Najafov A, Sommer EM, Axten JM, Deyoung MP
and Alessi DR: Characterization of GSK2334470, a novel and highly
specific inhibitor of PDK1. Biochem J. 433:357–369. 2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Lamming DW, Ye L, Sabatini DM and Baur JA:
Rapalogs and mTOR inhibitors as anti-aging therapeutics. J Clin
Invest. 123:980–989. 2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Ito K, Caramori G and Adcock IM:
Therapeutic potential of phosphatidylinositol 3-kinase inhibitors
in inflammatory respiratory disease. J Pharmacol Exp Ther. 321:1–8.
2007.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Council NR: Guide for the care and use of
laboratory animals: Eighth Edition. Washington, DC, The National
Academies Press 246, 2011.
|